Massive Axial and Appendicular Skeletal Deformities in Connection with Gorham-Stout Syndrome

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Ethics
2.2. Clinical Examination
2.3. Laboratory Measurements
3. Results
3.1. Group I: Patients with Progressive Painful Tilting of the Spine
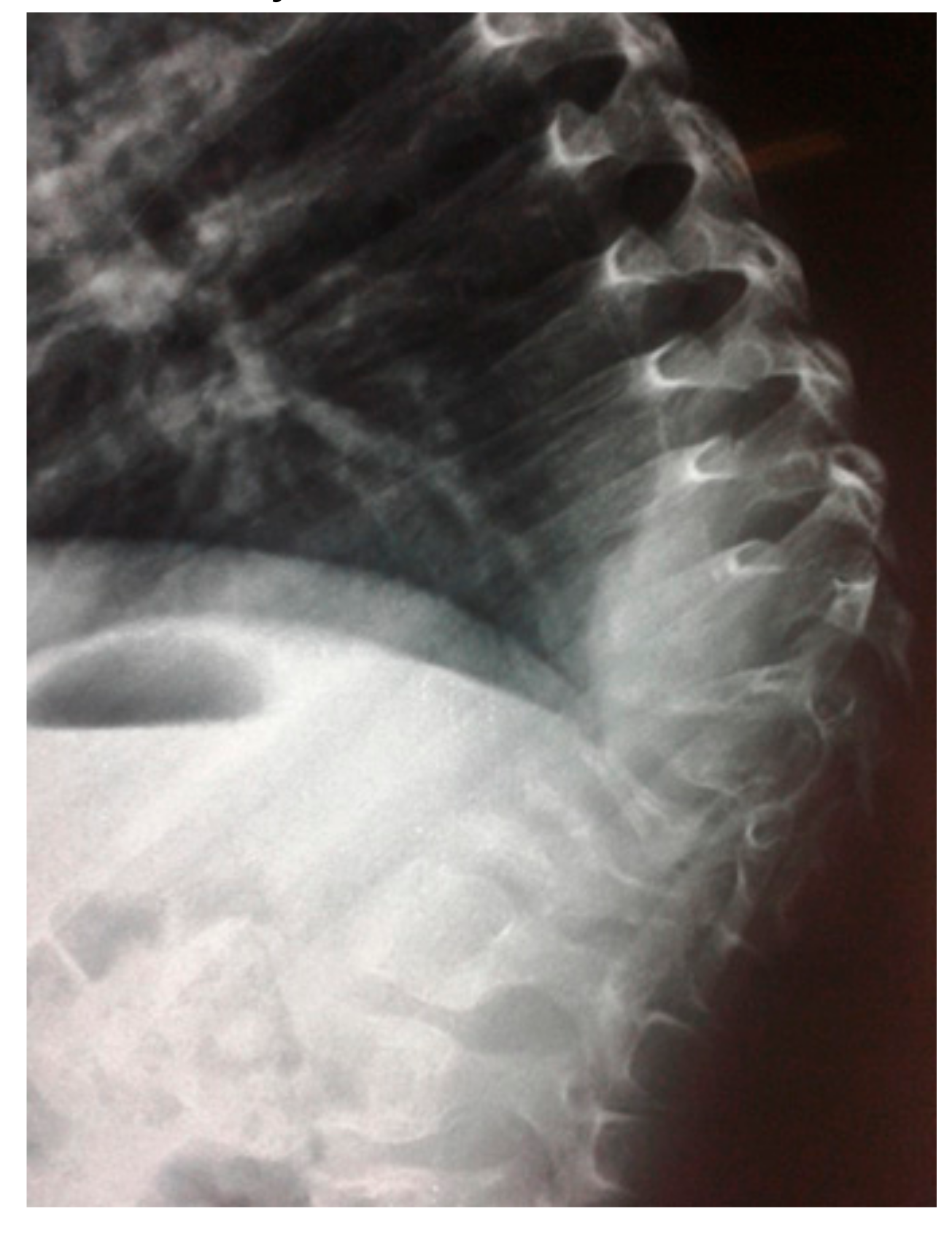
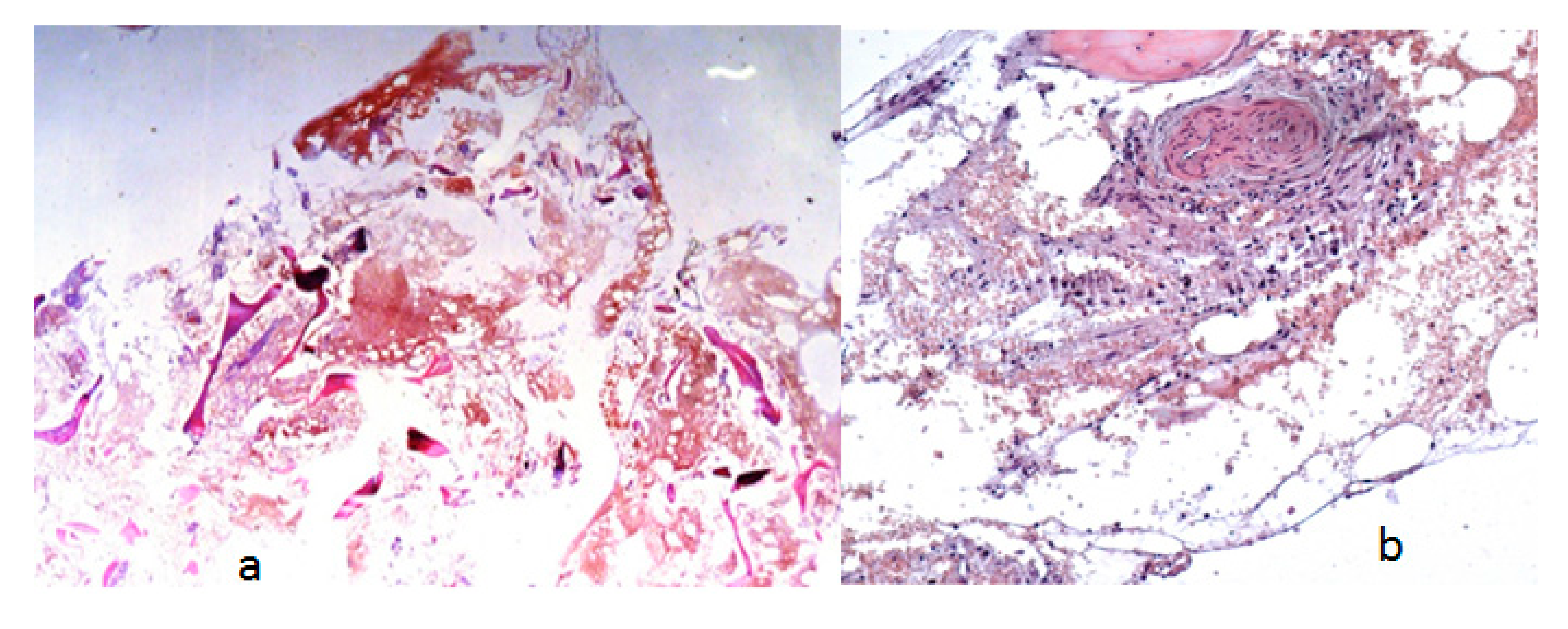
- A seven-year-old-boy was seen for the first time at the age of three years because of painful thoraco-lumbar kyphosis resulted from osteolysis of T9-L1. The left iliac crest was also involved in the pathological process of osteolysis (Figure 1). Back pain associated with Trendelenburg gait were the most bothersome symptomatology. Bone biopsy confirmed the diagnosis of Gorham-Stout disease. The kyphosis has been treated by a brace. Follow-ups showed stability of the kyphosis and dormancy of the osteolysis.
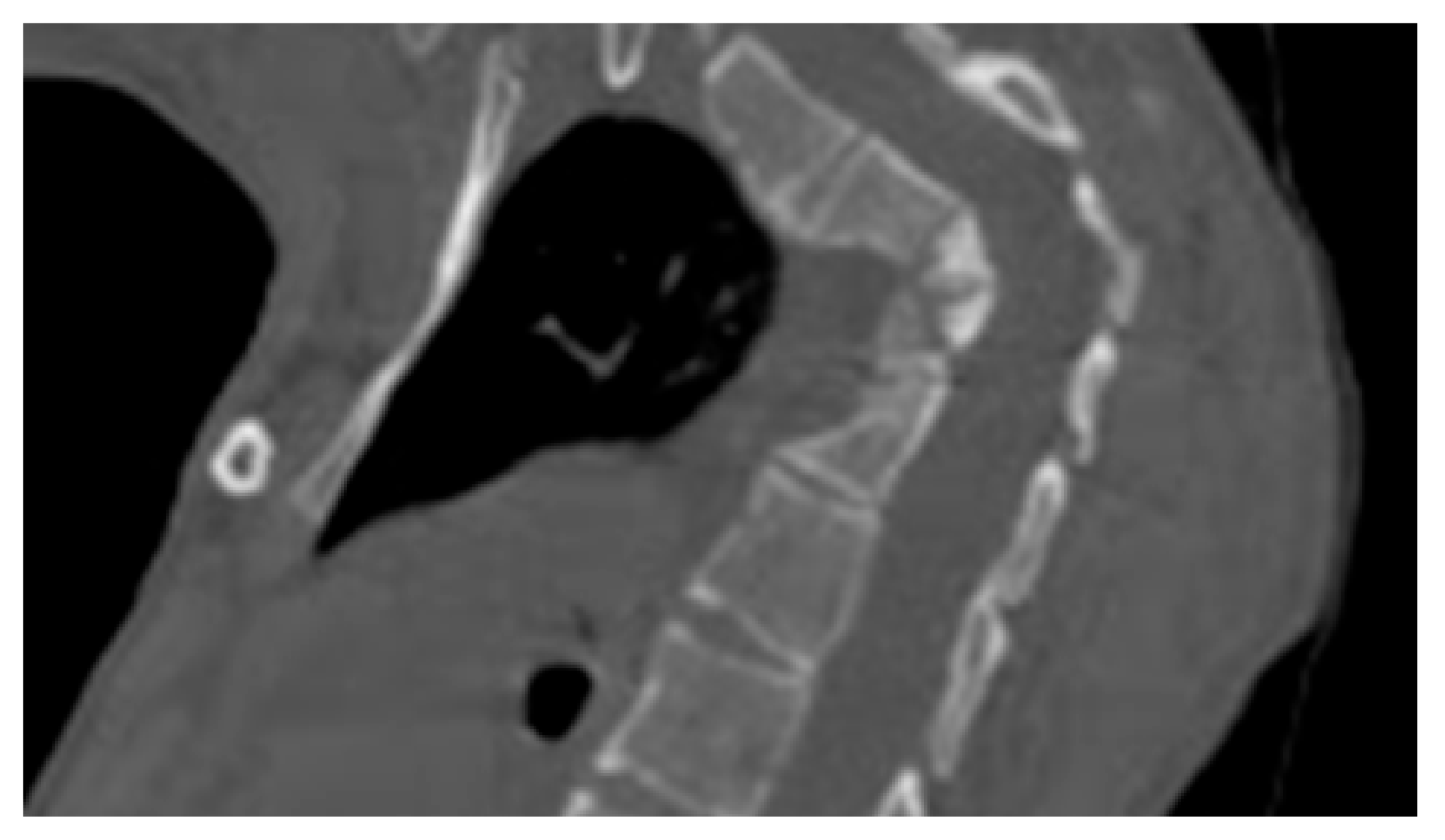
- A ten-year-old-girl presented with painful acute upper thoracic kyphosis. Radiographs of the spine were difficult to assess. 3D reformatted CT scan of the thoracic spine showed progressive osteolysis of T3-T6 (Figure 2). Skeletal survey did not show any other involved areas.
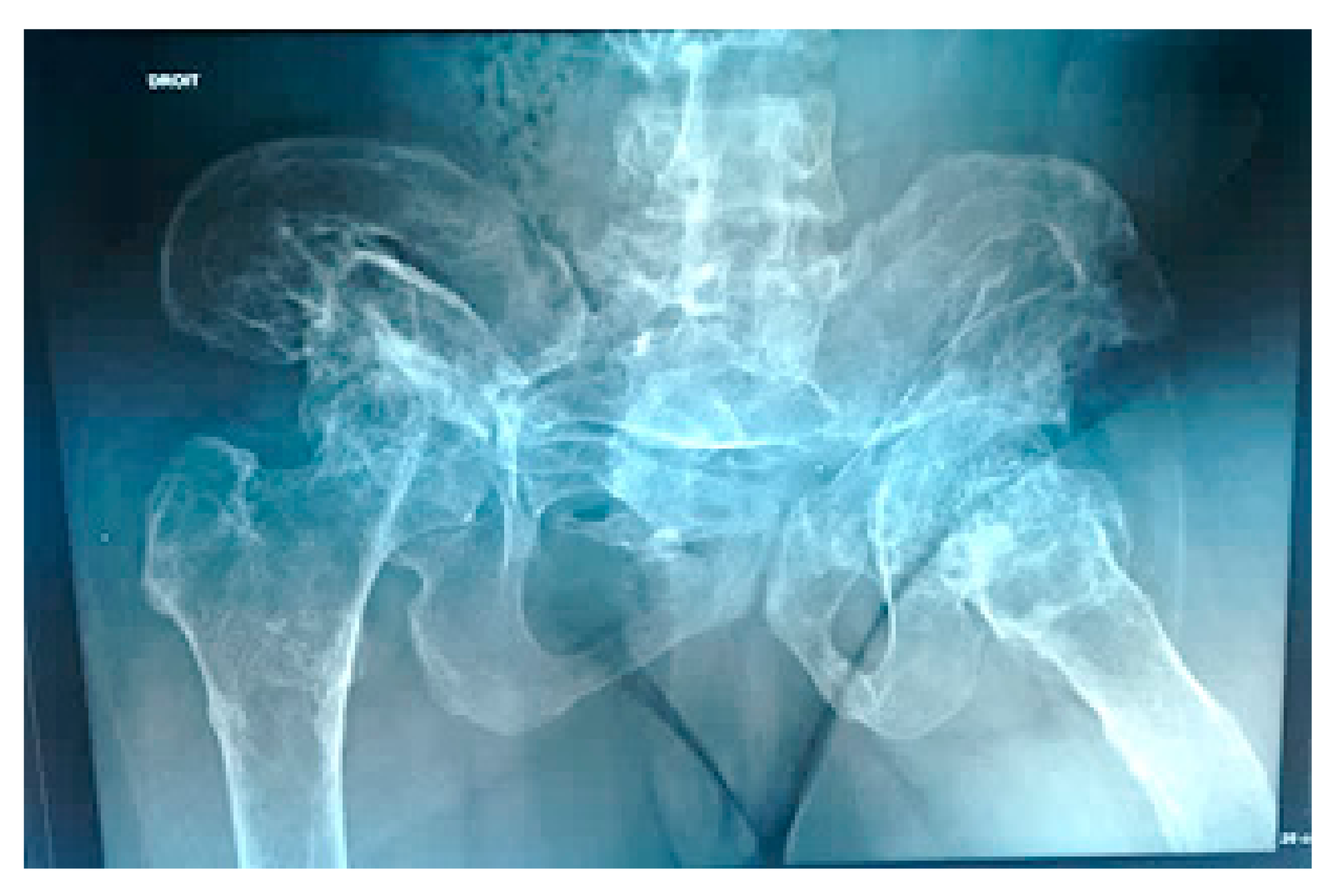
- 3D reconstruction CT scan in a-20-years-old-girl showed severe flattening, fusion, shrinkage and compression of the vanished thoracic spine T3-T9 causing effectively the development of painful kyphoscoliosis. Vanishing bone extended to involve the right shoulder joint resulted in total drop of the right upper limb (Figure 3).
3.2. Group II: Patients with Progressive Disorganization of the Weight Bearing Joints Associated with Painful Dissolution of the Long Bones
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jackson, J.B.S. A boneless armle. Bost. Med. Surg. J. 1938, 18, 368–369. [Google Scholar]
- Gorham, L.W.; Stout, A.P. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone); its relation to hemangiomatosis. J. Bone Joint Surg. Am. 1955, 37, 985–1004. [Google Scholar] [PubMed]
- Deveci, M.; İnan, N.; Çorapçıoğlu, F.; Ekingen, G. Gorham-Stout Syndrome with Chylothorax in a Six-Year-Old Boy. Indian J. Pediatr. 2011, 78, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, M.T.; Garg, N.; Olsen, B.R. Viewpoints on vessels and vanishing bones in Gorham–Stout disease. Bone 2014, 63, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, H.; Zhou, X.; Li, X.; Sun, W.; Dellinger, M.; Boyce, B.F.; Xing, L. Lymphatic Endothelial Cells Produce M-CSF, Causing Massive Bone Loss in Mice. J. Bone Miner. Res. 2017, 32, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Hagendoorn, J.; Padera, T.P.; Yock, T.I.; Nielsen, G.P.; di Tomaso, E.; Duda, D.G.; Delaney, T.F.; Gaissert, H.A.; Pearce, J.; Rosenberg, A.E.; et al. Platelet-derived growth factor receptor-beta in Gorham’s disease. Nat. Clin. Pract. Oncol. 2006, 3, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Franchi, A.; Bertoni, F.; Bacchini, P.; Mourmouras, V.; Miracco, C. CD105/endoglin expression in Gorham disease of bone. J. Clin. Pathol. 2009, 62, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Hominick, D.; Silva, A.; Khurana, N.; Liu, Y.; Dechow, P.C.; Feng, J.Q.; Pytowski, B.; Rutkowski, J.M.; Alitalo, K.; Dellinger, M.T. VEGF-C promotes the development of lymphatics in bone and bone loss. Elife 2018, 7, e34323. [Google Scholar] [CrossRef]
- Dupond, J.-L.; Bermont, L.; Runge, M.; de Billy, M. Plasma VEGF determination in disseminated lymphangiomatosis–Gorham–Stout syndrome: A marker of activity? A case report with a 5-year follow-up. Bone 2010, 46, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Hagendoorn, J.; Yock, T.I.; Rinkes, I.H.M.B.; Padera, T.P.; Ebb, D.H. Novel molecular pathways in Gorham disease: Implications for treatment. Pediatr. Blood Cancer 2014, 61, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Hardegger, F.; Simpson, L.A.; Segmueller, G. The syndrome of idiopathic osteolysis. Classification, review, and case report. J. Bone Joint Surg. Br. 1985, 67, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Florchinger, E.; Bottger, E.; Claass-Bottger, F.; Georgi, M.; Harms, J. Gorham-Stout syndrome of the spine. Case report and review of the literature. Rofo 1998, 168, 68–76. [Google Scholar] [PubMed]
- Ross, J.L.; Schinella, R.; Shenkman, L. Massive osteolysis. An unusual cause of bone destruction. Am. J. Med. 1978, 65, 367–372. [Google Scholar] [CrossRef]
- Stöve, J.; Reichelt, A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch. Orthop. Trauma Surg. 1995, 114, 207–210. [Google Scholar] [CrossRef]
- Tie, M.L.; Poland, G.A.; Rosenow, E.C. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest 1994, 105, 208–213. [Google Scholar] [CrossRef]
- Bode-Lesniewska, B.; von Hochstetter, A.; Exner, G.U.; Hodler, J. Gorham-Stout disease of the shoulder girdle and cervico-thoracic spine: Fatal course in a 65-year-old woman. Skeletal Radiol. 2002, 31, 724–729. [Google Scholar]
- Nikolaou, V.S. Vanishing bone disease (Gorham-Stout syndrome): A review of a rare entity. World J. Orthop. 2014, 5, 694–698. [Google Scholar] [CrossRef]
- Heffez, L.; Doku, H.C.; Carter, B.L.; Feeney, J.E. Perspectives on massive osteolysis. Report of a case and review of the literature. Oral Surg. Oral Med. Oral Pathol. 1983, 55, 331–343. [Google Scholar] [CrossRef]
- Zhu, X.; Gao, J.J.; Landao-Bassonga, E.; Pavlos, N.J.; Qin, A.; Steer, J.H.; Zheng, M.H.; Dong, Y.; Cheng, T.S. Thonzonium bromide inhibits RANKL-induced osteoclast formation and bone resorption in vitro and prevents LPS-induced bone loss in vivo. Biochem. Pharmacol. 2016, 15, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Kiran, D.N.; Anupama, A. Vanishing Bone Disease: A Review. J. Oral Maxillofac. Surg. 2011, 69, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Ohla, V.; Bayoumi, A.B.; Hefty, M.; Anderson, M.; Kasper, E.M. Complex single step skull reconstruction in Gorham’s disease—A technical report and review of the literature. BMC Surg. 2015, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Heyd, R.; Micke, O.; Surholt, C.; Berger, B.; Martini, C.; Füller, J.; Schimpke, T.; Seegenschmiedt, M.H. Radiation therapy for Gorham-Stout syndrome: Results of a national patterns-of-care study and literature review. Int. J. Radiat. Oncol. Biol. Phys. 2011, 18, e179–e185. [Google Scholar] [CrossRef]
- Ruggieri, P.; Montalti, M.; Angelini, A.; Alberghini, M.; Mercuri, M. Gorham–Stout disease: The experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011, 40, 1391–1397. [Google Scholar] [CrossRef]
- Foster, B.L.; Ramnitz, M.S.; Gafni, R.I.; Burke, A.B.; Boyce, A.M.; Lee, J.S.; Wright, J.T.; Akintoye, S.O.; Somerman, M.J.; Collins, M.T. Rare Bone Diseases and Their Dental, Oral, and Craniofacial Manifestations. J. Dent. Res. 2014, 93, 7S–19S. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Ogawa, C.; Kanazawa, T.; Watanabe, H.; Suzuki, M.; Suzuki, N.; Tsuchida, Y.; Morikawa, A.; Kuwano, H. Remission induced by interferon alfa in a patient with massive osteolysis and extension of lymph-hemangiomatosis: A severe case of Gorham-Stout syndrome. J. Pediatr. Surg. 2005, 40, E47–E50. [Google Scholar] [CrossRef]
- Liu, M.; Liu, W.; Qiao, C.; Han, B. Mandibular Gorham-Stout disease: A case report and literature review. Medicine (Baltimore) 2017, 96, e8184. [Google Scholar] [CrossRef]
- Remia, L.F.; Richolt, J.; Buckley, K.M.; Donovan, M.J.; Gebhardt, M.C. Pain and weakness of the shoulder in a 16-year-old boy. Clin. Orthop. Relat. Res. 1998, 268–271, 287–290. [Google Scholar] [CrossRef]
- Foult, H.; Goupille, P.; Aesch, B.; Valat, J.P.; Burdin, P.; Jan, M. Massive osteolysis of the cervical spine. A case report. Spine (Phila. 1976) 1995, 20, 1636–1639. [Google Scholar] [CrossRef]
- Kulenkampff, H.A.; Richter, G.M.; Hasse, W.E.; Adler, C.P. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int. Orthop. 1990, 14, 361–366. [Google Scholar] [CrossRef]
- Ng, L.C.; Sell, P. Gorham Disease of the Cervical Spine—A Case Report and Review of the Literature. Spine (Phila. 1976) 2003, 28, E355–E358. [Google Scholar]
- Der Linden-van der Zwaag, H.; Onvlee, G.J. Massive osteolysis (Gorham’s disease) affecting the femur. Acta Orthop. Belg. 2006, 72, 261–268. [Google Scholar]
- Drewry, G.R.; Sutterlin, C.E.; Martinez, C.R.; Brantley, S.G. Gorham disease of the spine. Spine (Phila. 1976) 1994, 19, 2213–2222. [Google Scholar] [CrossRef]
- Patel, D.V. Gorham’s disease or massive osteolysis. Clin. Med. Res. 2005, 3, 65–74. [Google Scholar] [CrossRef]
- McNeil, K.D.; Fong, K.M.; Walker, Q.J.; Jessup, P.; Zimmerman, P.V. Gorham’s syndrome: A usually fatal cause of pleural effusion treated successfully with radiotherapy. Thorax 1996, 51, 1275–1276. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.S.; Parisi, M.T.; Osborn, R.E. Gorham’s disease involving the thoracic skeleton—Plain films and CT in two cases. Pediatr. Radiol. 1993, 23, 543–544. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al Kaissi, A.; Bouchoucha, S.; Shboul, M.; Kenis, V.; Grill, F.; Ganger, R.; Kircher, S.G. Massive Axial and Appendicular Skeletal Deformities in Connection with Gorham-Stout Syndrome. Medicines 2019, 6, 54. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020054
Al Kaissi A, Bouchoucha S, Shboul M, Kenis V, Grill F, Ganger R, Kircher SG. Massive Axial and Appendicular Skeletal Deformities in Connection with Gorham-Stout Syndrome. Medicines. 2019; 6(2):54. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020054
Chicago/Turabian StyleAl Kaissi, Ali, Sami Bouchoucha, Mohammad Shboul, Vladimir Kenis, Franz Grill, Rudolf Ganger, and Susanne Gerit Kircher. 2019. "Massive Axial and Appendicular Skeletal Deformities in Connection with Gorham-Stout Syndrome" Medicines 6, no. 2: 54. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6020054