Early Stage Glycosylation Biomarkers in Alzheimer’s Disease

1
Institute of Technology Sligo, Ash Lane, F91 YW50 Sligo, Ireland
2
Cellular Health and Toxicology Research Group, Institute of Technology Sligo, Ash Lane, F91 YW50 Sligo, Ireland
3
Northern Ireland Centre for Stratified Medicine, Biomedical Sciences Research Institute, Clinical Translational Research and Innovation Centre, Altnagelvin Area Hospital, Glenshane Road, Derry BT47 6SB, UK
*
Author to whom correspondence should be addressed.
Medicines 2019, 6(3), 92; https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6030092
Submission received: 30 June 2019
/
Revised: 29 August 2019
/
Accepted: 30 August 2019
/
Published: 3 September 2019
(This article belongs to the Special Issue Application of Glycobiology in the Treatment of Diseases)
Abstract
:Alzheimer’s disease (AD) is of great cause for concern in our ageing population, which currently lacks diagnostic tools to permit accurate and timely diagnosis for affected individuals. The development of such tools could enable therapeutic interventions earlier in the disease course and thus potentially reducing the debilitating effects of AD. Glycosylation is a common, and important, post translational modification of proteins implicated in a host of disease states resulting in a complex array of glycans being incorporated into biomolecules. Recent investigations of glycan profiles, in a wide range of conditions, has been made possible due to technological advances in the field enabling accurate glycoanalyses. Amyloid beta (Aβ) peptides, tau protein, and other important proteins involved in AD pathogenesis, have altered glycosylation profiles. Crucially, these abnormalities present early in the disease state, are present in the peripheral blood, and help to distinguish AD from other dementias. This review describes the aberrant glycome in AD, focusing on proteins implicated in development and progression, and elucidates the potential of glycome aberrations as early stage biomarkers of AD.
1. Introduction
1.1. Alzheimer’s Disease (AD)—A Cause for Concern
There are currently over 50 million cases of dementia in the world, of which AD is the predominant form, possibly making up 60–70% of cases [1,2]. Development of AD is related to advanced age and, as life expectancy increases, it is expected that AD cases will increase four-fold by the year 2050 [3,4]. Sufferers of the disease experience deterioration of memory and cognition, which slowly worsens as the disease progresses until they are eventually unable to care for themselves [5]. From initial diagnosis, death typically occurs after 8–10 years [6].
1.2. AD Pathogenesis
AD is characterised by the formation of plaques composed of amyloid beta (Aβ) peptides, which are formed from abnormally processed amyloid precursor protein (APP) protein, and the presence of neurofibrillary tangles derived predominantly from hyperphosphorylated tau protein, in the brain [7]. Hyperphosphorylated tau (AD P-tau) aggregates, causing paired helical filaments (PHFs) to form, accumulation of which leads to neurofibrillary tangle formation [8]. Intraneuronal tau and extraneuronal amyloid plaques, contribute to neuronal cell death and loss of synaptic function [9,10].
Neurofibrillary tangles progressively spread from the entorhinal cortex to other parts of the brain such as the hippocampus and cerebral cortex, and these brain regions are associated with significant neuronal loss and physical shrinkage [11,12,13]. The cortex and hippocampus are strongly associated with cognition, thought, memory, learning and awareness [14,15]. Hippocampal atrophy is severe and has been used as a diagnostic and prognostic marker of AD [16,17]. Earlier diagnostic markers in these brain regions would be invaluable.
1.3. Treatments for AD and the Urgency of An Early Diagnosis
A deficit in the cholinergic system is a major characteristic of AD [18]. AD treatments currently available aim to correct the altered neurotransmission, but these are symptomatic as the individual will still eventually succumb to the disease [19,20]. Disease modifying treatments under investigation aim to reduce Aβ plaque formation by inhibition or modulation of secretases involved in the amyloid pathway [21,22], or increase Aβ clearance by immunological approaches [23,24]. Immunological removal of tau aggregates is also under investigation [25], in addition to prevention of tau hyperphosphorylation using kinase inhibitors, as kinases induce the hyperphosphorylation of tau [26,27]. However, the success of interventions in clinical trials has been very poor to date [28,29]. To compound matters, often current treatments are only significantly effective in mild AD sufferers, but not in moderate AD sufferers [30,31]. There is therefore a critical need to diagnose patients as early as possible so that they may derive maximum benefit from existing therapies and initiate disease modifying therapies, when they become available, as early in the disease course as possible.
1.4. AD Diagnosis—Invasive and Inconclusive
Aside from advanced age, the apolipoprotein E4 gene is the biggest risk factor for late onset AD; some 64% of sporadic AD sufferers and 80% of familial AD sufferers have at least one copy of the gene [32]. Mutations in APP, presenilin (PS)1 and PS2 genes almost guarantee that an individual will develop early onset AD, which occurs before 65 years of age [33]. However, early onset AD accounts for only 1–2% of AD cases [34].
At present, AD is routinely diagnosed via cognitive and learning assessments, however, this merely identifies AD as the most probable cause of symptoms which have presented. Distinguishing AD from other types of dementias is difficult as memory-related symptoms and physical changes in the brain are also observed in other neurodegenerative disorders, as well as in the normal, ageing brain [35,36,37,38]. Characteristic whole-brain and hippocampal tissue atrophy seen in AD [11] is identified using magnetic resonance imaging (MRI) and computed tomography (CT) [39], however, this atrophy is present in other forms of dementia [40,41,42]. Post mortem identification of pathological defects in the AD brain is necessary for definitive diagnosis [43].
Positron emission tomography (PET) is another imaging technique which uses radiotracers of biomarkers in an AD diagnosis [44]. 2-[18F]fluoro-2-Deoxy-D-glucose (FDG) is a common PET tracer which identifies the reduction of cerebral metabolic rates of glucose (CMRglc) seen in AD [45]. However, this disturbed CMRglc metabolism is seen in other types of dementias, making it non-specific to the disease state [46]. PET tracers of Aβ peptides, such as Pittsburg compound B, have emerged which bind specifically to amyloid plaques in the brain [47]. However, it appears that Aβ plaque formation does not correlate directly with cognitive decline [48], making it unreliable as a disease progression indicator. Promising tau-specific PET tracers have recently been developed but will require further clinical validation studies [49].
The difficulties in AD diagnosis indicates a significant clinical need to develop non-invasive diagnostic tests, ideally from peripheral blood. The popular approaches to biomarker testing today are costly neuroimaging, as discussed, and cerebrospinal fluid (CSF) analysis. CSF may be assessed for elevated total tau and AD P-tau levels, and decreased Aβ peptide levels, which requires patients to undergo a painful lumbar puncture [50]. As this review will explore, there are many reported changes in the AD glycome occurring early in the disease pathogenesis, many of which are present in the periphery and could be exploited as biomarkers of AD. Before these changes are discussed, it is important to understand glycosylation and the role it plays in human health and disease.
2. Glycosylation Overview
2.1. What is Glycosylation?
Glycosylation is an enzymatic process where glycosyltransferase enzymes use activated sugar donor molecules to attach monosaccharides to growing glycans via a glycosidic linkage [51,52,53]. Glycosidases and glycosyltransferases, involved in the synthesis of glycans, are each specific to a particular sugar and to their type of linkage [54,55]. N- and O-linked glycosylation are the most common types of protein glycosylation. O-linked glycans are linked to the hydroxyl group of threonine or serine in a protein, whereas N-linked glycans are linked to the nitrogen atom of an asparagine residue [56,57]. However there is no site specific addition of O-glycans to serine or threonine residues and the core structure of an O-glycan is variable, therefore O-glycan synthesis is not always initiated by the same glycosyltransferase [58]. The focus of this review is on N-glycans which are present in 90% of glycoproteins [59], are the most characterised of all glycans and strongly implicated in the AD pathogenesis [60,61,62,63,64]. A single enzyme known as oligosaccharyltransferase initiates glycan synthesis in the N-linked pathway. All N-glycans have a common core, and oligosaccharyltransferase specifically catalyzes the addition of the growing N-glycan to asparagine at the sequences asparagine-X-serine or asparagine-X-threonine, X being any amino acid except proline [52]. The synthesis of N-glycans begins in the endoplasmic reticulum and continues into the Golgi, occurring co- and post-translationally [52,65]. N-glycans may be either high mannose, complex or hybrid types depending on the additional sugar modifications extending from their core structure of two N-acetyl glucosamine (GlcNAc) monosaccharides and three mannoses [66,67,68,69].
2.2. Approaches to Glycoanalysis
Glycans may be analysed after cleavage from the respective protein, or glycoproteins may be analysed intact [70]. N-glycan cleavage may be achieved chemically by hydrazinolysis [71] or enzymatically by peptide-N4-(N-acetyl-β-glucosaminyl) asparagine amidase, typically known as PNGase F, digestion which cleaves glycans at asparagine residues, the exclusive site of N-glycosylation [72]. Since the attachment sites of glycans to the peptide backbone are different depending on the type of glycosylation, there is no single method which may be used to cleave glycans from the respective glycoproteins. Liquid chromatography (LC), porous graphitic carbon chromatography (PGC), and capillary electrophoresis are commonly used chromatographic separation methods for released glycan analysis [73].
Glycans are typically fluorescently labelled prior to LC analysis as they lack natural chromophores [74]. Hydrophilic interaction LC offers the advantage of identifying structural isomers as well as determining linkage information in some cases, which other LC modes cannot [75]. Weak anion exchange high performance liquid chromatography (HPLC) is sometimes used as an orthogonal method for the separation of similar glycans or those containing negatively charged sialic acids [76]. Databases such as GlycoStore have been established to give information related to the separated glycan structures [77].
The advent of ultra-high performance liquid chromatography (UPLC) and sub-2 µm particle size columns has permitted quicker separation and improved separation capacity [78,79,80]. Separation of glycan mixtures is still difficult because isomeric glycans, that are commonly present [81], will co-elute. Exoglycosidase sequencing is therefore often used to delineate isomeric glycans identified by the initial separation, while also providing conclusive linkage information of the monosaccharides present [82,83,84]. Figure 1 illustrates a typical N-glycan profiling approach employing PNGase F digestion, fluorescent labelling, exoglycosidase sequencing and UPLC analysis of a glycoprotein mixture.
Lectins are commonly employed in glycoprofiling studies [85,86,87] and are useful in the analysis of intact glycoproteins, as well as the localisation of these glycoproteins in cells and tissues [88,89]. Mass spectrometry (MS) is another widely used and accurate method to analyse glycan structure without derivatization, which allows for analysis of intact glycoproteins [90]. MS is often used as an orthogonal technique in LC/MS or LC/MS-MS, to give an overall comprehensive analysis of glycoproteins by providing greater separation of structural or geometric glycan isomers [91,92,93,94], in addition to confirming structural linkages identified using exoglycosidase approaches [95,96]. Nuclear magnetic resonance spectroscopy is another excellent method for detailed structural analysis of glycans, but requires a relatively large quantity of purified glycans [70].
2.3. Glycosylation and Disease
N-glycans are vital for a multicellular organism’s survival [97,98]. Glycoproteins are on the extracellular surface of the plasma membrane, the extracellular matrix of cells, and are also secreted into bodily fluids. Almost all proteins in human serum and on mammalian cell membranes are glycosylated [59]. Glycans ensure proper protein folding, trafficking, and functionality in addition to facilitating cell signaling and cell-cell communication [99,100,101]. Changes in the physiological state may alter glycans found on glycoproteins at the same location [102,103]. Glycosylation changes also occur in many disease states. Changes in glycosylation patterns is a hallmark of cancer [104]. Breast [105], stomach [106], and liver [107] cancers all have altered serum glycoprofiles. Rheumatoid arthritis is associated with reduced IgG galactosylation and sialic acid termini [108]. Congenital disorders of glycosylation (CDGs) are a group of illnesses caused by aberrant glycosylation rooted in genetic defects in the enzymes and biomolecules involved in glycan synthesis. Over 100 CDGs exist, common examples being ALG1-CDG, PMM2-CDG and MPI-CDG, each named after the defective gene in the respective CDG [109]. Such disorders result in symptoms affecting multiple organs, commonly the brain and nervous system, due to the far reaching impact of glycosylation on the functionality of the human proteome [110,111].
3. Glycosylation and AD
3.1. Glycosylation of Proteins Implicated in AD in the Brain
Glycosylation has been implicated in AD pathology in many studies. Numerous changes have been detected in the AD brain. Immunoprecipitation and lectin blotting of β-site APP-cleaving enzyme 1 (BACE1), the N-glycosylated enzyme [112] responsible for the toxic β-secretase cleavage of APP [113], from the brains of AD patients revealed increased levels of bisecting GlcNAc, and crucially this increase was observed in both early and late stage patients [112]. A later study found that this increase in bisecting GlcNAc on BACE1 stabilises the protein under oxidative stress conditions [114], a feature characteristic of AD pathology [115].
APP is modified with O-GlcNAc [116] and studies have indicated that the mucin-type O-glycosylation of APP has a negative impact on Aβ production [117,118]. One of these studies revealed an increase in expression levels of transferases responsible for this mucin-type O-glycosylation in the early and late stage AD brain versus non-AD controls through quantitative real-time polymerase chain reaction (RT-PCR) analysis of human brain tissue [117]. N-acetylglucosaminyltransferase III (GnT-III) is the glycosyltransferase responsible for adding bisecting GlcNAc [119]. RT-PCR analysis of GnT-III mRNA revealed an increase in its expression in AD patient brains [119].
Findings suggest N-glycosylation occurs before hyperphosphorylation of tau, where non-hyperphosphorylated tau in AD brain exhibited N-glycan specific lectin staining versus an observed absence of this in normal tau in healthy controls [120]. In fact glycosylation of tau may induce hyperphosphorylation as the N-glycosylated form of the protein proved to be a better substrate for kinase than its native form [120]. The N-glycans found on phosphorylated tau and PHFs also differ, with more truncated glycans found on PHF-tau, identified in human AD brain tissue using complimentary analyses by HPLC, exoglycosidase digestions and MS [121].
Polysialylated neural cell adhesion molecule (PSA-NCAM) is recognized for its role in central nervous system development but is also reported to function in the connectivity of interneurons in the adult cerebral cortex [122]. A study revealed significantly decreased immunostaining of PSA-NCAM in the entorhinal cortex of AD brain tissue taken post mortem, relative to healthy controls. The same study found the decrease in PSA-NCAM load in the AD entorhinal cortex to be inversely proportional to hyperphosphorylated tau load [123]. This indicates a reduction in PSA-NCAM load specific to a brain region known to be severely affected in the AD pathogenesis [11,12,13], and shows that this change is potentially related in some way to increases in hyperphosphorylated tau load. Despite these findings, the identification of glycosylation changes in the AD brain is clearly not diagnostically viable.
3.2. Glycosylation Biomarkers of AD in the CSF and Blood
A study of the glycosylation changes in both pre-dementia and AD patient CSF using matrix-assisted laser desorption/ionization-MS showed an increase in bisect type N-glycans and a decrease in sialylated species in both pre-dementia and AD cases, indicating that AD related glycosylation changes occur before clinical symptoms manifest [124].
Detection of alterations in the AD glycome via peripheral serum and plasma would be advantageous diagnostically, given the invasive nature of CSF sampling. An obstacle in the analysis of serum is that many brain derived proteins are not present in the blood due to their inability to cross the blood brain barrier [125]. However, the N-glycoprofiles of a mouse model of neurodegeneration revealed decreases in specific glycans in the serum and cerebral cortex, and the changes correlated to learning and memory deficiencies [126]. In humans, AD patient serum exhibited increased bisect-type and highly branched glycoforms in a study which employed glycoblotting and MS analysis of patient serum samples [127]. The same study similarly showed an increase in these particular species of glycan in AD patient CSF versus non-AD controls [127]. Of note, bisect type glycans are atypical of serum, but are common “brain-type” glycans [128]. Reduced sialyltransferase activity has been observed in AD patient serum using a radio-enzymatic assay [129]. It is interesting to note that the gene for a sialic acid binding receptor, known as Siglec 33, is implicated in late-onset AD [130].
Sera levels of a bi-galactosylated core fucosylated bi-antennary glycan were shown to decrease with age in a study employing a glycoanalytical tool known as DNA sequencer-assisted, fluorophore-assisted carbohydrate electrophoresis (DSA-FACE) [131]. However, in another study employing DSA-FACE, sera levels of the same glycan were shown to decrease significantly more in AD patients in comparison to healthy controls and could additionally distinguish AD patients from other non-AD patients [132]. This is significant from a diagnostic standpoint, due to the difficulty in distinguishing AD from other neurodegenerative disorders, if it could be identified in a clinical setting using a higher throughput approach.
O-glycan changes in AD are widely reported [133,134,135,136,137,138,139,140]. Recently it was observed that neuroinflammation induced by Aβ modifies mucin-type O-glycosylation in rat hippocampus, which was observed by injection of Aβ into rat brain hippocampus followed by lectin analysis [141]. Interestingly, Aβ immunopurified from AD and non-AD CSF revealed a unique O-glycosylation of a specific tyrosine residue upon LC-MS/MS analysis, and the ratio of this tyrosine glycosylation in Aβ to the respective unglycosylated form was found to be up to 2.5 times more prevalent in the CSF of the AD patients compared to healthy and non-AD controls [142]. Aside from these general glycomic changes at play, glycosylation changes have been observed in all of the major AD related proteins, and crucially some changes are occurring at early stages in the disease state [60,61,62].
3.3. The Direct and Indirect Impact of BACE1 Glycosylation in AD Pathology
Cleavage of APP in neurons by α-secretase followed by γ-secretase yields a soluble peptide under normal physiological conditions [143]. Soluble APP has neuroprotective functions against excitotoxicity [144] and Aβ toxicity [145] in neurons. In AD, β-secretase and γ-secretase cleave APP sequentially, resulting in the formation of toxic Aβ peptide [146]. As stated previously, BACE1 is the enzyme responsible for the toxic β-secretase cleavage of APP [113] and is N-glycosylated [112]. Alteration of N-glycosylation sites in aspartyl protease 2, now known to be BACE1, was shown to reduce its proteolytic activity [147]. GnT-III knockout in mice resulted in reduced Aβ deposition and improved short term memory [112]. It therefore appears that aberrant N-glycosylation of BACE1 has a direct role in AD pathology. β-secretase additionally functions in protein glycosylation, specifically in cleaving sialyltransferases [148,149]. An in vitro study where a particular sialyltransferase was overexpressed in order to induce increased sialylation of APP, resulted in increased production of Aβ peptides [150]. Another in vitro study showed that overexpression of BACE1 enhances sialylation of soluble secreted glycoproteins through lectin analysis [151]. Elucidating the effects of BACE1 on the glycoprofiles of specific AD related glycoproteins may make its role more clear in the AD pathology.
3.4. APP Glycans as Protective Mechanisms in AD
Several studies indicate that APP sorting, secretion, transport and localisation are affected by its N-glycosylation [150,152,153,154]. Therefore glycoforms of this major AD related protein are likely to have a significant effect on the pathology of the disease. An in vitro study where mouse neuroblastoma cells were transfected with either wild-type APP or APP mutants related to AD, revealed increased levels of bisecting GlcNAc and core fucose N-glycan alterations on APP mutants compared to wild-type APP upon analysis of isolated glycans by HPLC [155]. GnT-III may act as a protective mechanism in the presence of amyloid plaques, as a study additionally revealed a decrease in Aβ production in GnT-III transfected mouse neuroblastoma cells when Aβ concentrations in culture supernatants were determined using enzyme-linked immunosorbent assay (ELISA). What is more, an increased activity of the neuroprotective α-secretase was also observed in GnT-III transfected cells [119]. A disintegrin and metalloproteinase 10 (ADAM10) is the major α-secretase in neurons responsible for normal, non-amyloidogenic cleavage of APP [156]. ADAM10 is key to neurodevelopment [157] and synaptic plasticity [158]. It interacts with APP and other proteins, and levels of ADAM10 are reduced in AD patient blood [159]. Although N-glycosylation of this glycoprotein has been shown to be crucial to its functionality by mutation of its second N-glycosylation site in vitro which caused both reduced enzymatic activity and proteolytic activation [160], its glycoprofile in AD remains elusive and warrants investigation.
3.5. Tau Phosphorylation is Directed by its Glycosylation
A disruption to O-GlcNAc modification of proteins in AD has been reported [134,135,136,137,138,139,140]. Tau is ordinarily O-glycosylated by single GlcNAc residues [161]. Tau protein is involved in microtubule assembly and stabilisation [162], and is hyperphosphorylated in AD [163,164]. Decreased wheat germ agglutinin (WGA) lectin blotting of tau was observed in phosphatase-inhibited human neuroblastoma cells [165]. WGA is a lectin with a high affinity for GlcNAc [166]. This indicates that hyperphosphorylation of tau, which is characteristic of AD, results in downregulation of O-linked tau glycans. As O-linked glycans attach to threonine or serine, they occupy the principal sites of phosphorylation [167]. Another study where decreased O-GlcNAcylation of tau was induced by glucose starvation of mice to mimic AD pathology resulted in increased phosphorylation of tau upon western blot analysis. This indicates that O-GlcNAcylation acts as a protective mechanism against hyperphosphorylation of tau in AD, and that the impaired glucose metabolism may be causing the reduced O-GlcNAc modification of proteins [168].
It appears that tau N-glycans are key to the stabilisation of PHF structures. The dephosphorylation and deglycosylation of tau in PHF structures increased the release of tau and restored its microtubule polymerization activity, further implicating the role of tau N-glycans in AD [169]. Tau is normally a cytosolic protein [170] and because N-glycosylation normally only occurs in secreted or membrane bound proteins [171], this may indicate a subcellular relocation of tau in AD pathogenesis.
3.6. Presenilin and Transmembrane Protein 59 (TMEM59) are Regulators of Protein Glycosylation
γ-secretase is partly composed of the protein presenilin [172]. Mutations in PS1 and PS2 genes are associated with early onset familial AD [33]. Acetylcholinesterase (AChE) is known to complex with presenilin, and a study conducted in transgenic mice with a PS1 mutation revealed altered mannose termini and disturbed maturation of AChE, indicating PS1 affects AChE functionality [86]. Nicastrin is the only protein in the γ-secretase complex that is N-glycosylated [173,174], and presenilin knockout cells revealed a reduction in nicastrin N-glycosylation and maturation [175]. An in vitro study where AD-related PS1 was overexpressed in a human neuroblastoma cell line identified a reduction in sialylation of neural cell adhesion molecule (NCAM) using lectin analysis [176].
TMEM59 expression is increased in late-onset AD [177]. An in vitro study showed that TMEM59 expression affects Golgi localised complex glycosylation, reducing galactosylation and sialylation of key AD related proteins such as APP, BACE1 and nicastrin using western blot analysis. The study also showed that α- and β-secretase shedding of APP and APP cell surface expression were both suppressed by TMEM59 expression [178]. These findings indicate that presenilin [86,175,176] and TMEM59 [178] regulate protein glycosylation. Further study of the effects of abnormal presenilin and upregulated TMEM59 on relevant glycoproteins may be worth investigation.
3.7. Apolipoprotein Glycosylation Changes in AD
Clusterin, also known as apolipoprotein J (ApoJ), is known to be associated with AD pathogenesis [179] and increased levels of clusterin are present in AD patient blood [180]. Specific glycans on clusterin were decreased in abundance in AD patients with high hippocampal atrophy relative to those with low hippocampal atrophy, analysed using LC-MS/MS analysis [181]. Apolipoprotein D (ApoD) has been shown to modulate amyloid pathology in an AD mouse model [182]. ApoD in human plasma is N-glycosylated [183]. Overall the N-glycoprofiles of the different apolipoproteins have not been investigated in detail and may be worth exploring given the indicated implications this group of proteins have in AD pathology.
3.8. AD-Associated Glycosylation Changes to Transferrin are Different in CSF to Serum
Increased iron concentration and disturbed iron metabolism are associated with AD [184,185]. Transferrin was identified from AD patient CSF as having reduced sialic acid termini present by lectin binding analysis and isoelectric focusing, confirmed by comparable levels of transferrin in the CSF of AD patients and controls. Combination of the reduced transferrin lectin binding with phosphorylated tau ELISA detection, this biomarker was determined to be highly specific and sensitive at distinguishing AD from other dementias [186]. Conversely, transferrin sialylation was found to be increased in AD patient serum using isoelectric focusing and immunoblotting [187]. As such, the glycoprofile of transferrin in AD appears to be altered differently in the periphery to the central nervous system.
3.9. Other Glycoproteins Associated with AD Pathology
Glycosylation is further implicated in other proteins associated with AD pathology. Acetylcholine has various functions in the nervous system, one of which is related to short term memory and learning [188]. A deficit in acetylcholine is associated with AD [189]. Several studies indicate glycoforms of AChE may present early in the disease state [190,191], however, conflicting findings indicate that the changes are in fact not present at early stages [192], meaning it may not be reliable early marker of AD. Reelin is a glycoprotein found in the extracellular matrix which has a role in maintaining synaptic plasticity and can reverse Aβ synaptic dysfunction [193]. It is involved in embryonic brain development [194] and adult brain functionality [195]. A change in mannose specific lectin binding to reelin has been seen in AD patient CSF in comparison to controls [196]. Neprilysin is a metalloproteinase [197] which is known to break down Aβ in the brain [198]. Neprilysin expression is downregulated in AD [199], and mutation of the N-glycosylation sites of neprilysin was shown to reduce its activity and cell surface expression [200,201]. The R47H variant of triggering receptor expressed on myeloid cells 2 (TREM2), a transmembrane protein that is reported to be associated with late-onset AD progression [202], was recently found to have an altered N-glycoprofile in comparison to the wild type. The study determined that the R47H variant has increased complex oligosaccharide alterations in comparison to the wild type, in addition to reduced stability [203].
A study in which the glycoprofiles of IgG in human blood were assessed using LC-MS/MS analysis revealed significant glycan changes in the AD group relative to controls, with a lower abundance of specific complex galactosylated and sialylated glycans observed in the AD group. Interestingly, the abundance of complex glycans in females reduced steadily from the pre-dementia cases to AD patients, whereas the opposite trend was observed in males just prior to disease onset [204]. The characteristic role of inflammation in AD [205] may explain the observed reduction in sialic acid modification of IgG seen here, as removal of sialic acids from IgG causes a shift in its function from anti-inflammation to pro-inflammation [206]. This is of interest as a readily available marker of AD which is in high abundance in the blood [207], making analyses less complex.
Regarding biomarker discovery, the focus must now turn towards a more comprehensive analysis of the AD glycome, elucidating both the well-known players in the AD pathogenesis in more detail in addition to exploring the lesser known glycoproteins implicated in the AD pathogenesis, as mentioned in this review. Crucially the methods employed should aim to provide a detailed structural analysis of glycan structures and identify their respective proteins, as opposed to the many studies of the AD glycome to date that have popularly employed approaches such as lectin analysis to gain a non-specific insight into the changes at play.
4. Concluding Remarks
This review highlights an alteration to the glycan portion of many proteins involved in AD pathogenesis at early stages in the disease state, some examples of which are illustrated in Figure 2. With improved glycoanalytical technologies more readily available, a more detailed and comprehensive analysis of the AD glycome is the appropriate approach in future studies. Table 1 presents promising human AD biomarkers of the CSF and blood that have been reported here. Efforts of future studies should be founded in the analysis of peripheral plasma or serum samples to make glycan biomarkers of AD a viable diagnostic tool.
Author Contributions
Writing-Original Draft Preparation, P.R.; Writing-Review & Editing, P.R., P.L.M., T.S., M.D.
Funding
This research was funded by the Institute of Technology Sligo President’s Bursary fund. The APC was funded by the Institute of Technology Sligo President’s Bursary fund.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Morris, M.C. The role of nutrition in Alzheimer’s disease: Epidemiological evidence. Eur. J. Neurol. 2009, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dementia. Available online: http://www.who.int/en/news-room/fact-sheets/detail/dementia (accessed on 1 March 2018).
- Brookmeyer, R.; Johnson, E.; Ziegler-Graham, K.; Arrighi, H.M. Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dement. 2007, 3, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. What Is Alzheimer’s? Available online: https://www.alz.org/alzheimers_disease_what_is_alzheimers.asp (accessed on 15 January 2018).
- Montgomery, W.; Goren, A.; Kahle-Wrobleski, K.; Nakamura, T.; Ueda, K. Detection, diagnosis, and treatment of Alzheimer’s disease dementia stratified by severity as reported by caregivers in Japan. Neuropsychiatr. Dis. Treat. 2018, 14, 1843–1854. [Google Scholar] [CrossRef]
- Musicco, M.; Palmer, K.; Salamone, G.; Lupo, F.; Perri, R.; Mosti, S.; Spalletta, G.; di Iulio, F.; Pettenati, C.; Cravello, L.; et al. Predictors of progression of cognitive decline in Alzheimer’s disease: The role of vascular and sociodemographic factors. J. Neurol. 2009, 256, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.G.; Martinez-Marmol, R.; Meunier, F.A.; Gotz, J. Amyloid-beta and tau complexity—Towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Kadowaki, H.; Nishitoh, H.; Urano, F.; Sadamitsu, C.; Matsuzawa, A.; Takeda, K.; Masutani, H.; Yodoi, J.; Urano, Y.; Nagano, T.; et al. Amyloid beta induces neuronal cell death through ROS-mediated ASK1 activation. Cell Death Differ. 2005, 12, 19–24. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Waser, M.; Garn, H.; Schmidt, R.; Benke, T.; Dal-Bianco, P.; Ransmayr, G.; Schmidt, H.; Seiler, S.; Sanin, G.; Mayer, F.; et al. Quantifying synchrony patterns in the EEG of Alzheimer’s patients with linear and non-linear connectivity markers. J. Neural Transm. 2016, 123, 297–316. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Sabatini, B.; Südhof, T.C. Synapses and Alzheimer’s disease. Cold Spring Harb. Perspect. Biol. 2012, 4, a005777. [Google Scholar] [CrossRef] [PubMed]
- Mora-Bermúdez, F.; Badsha, F.; Kanton, S.; Camp, J.G.; Vernot, B.; Köhler, K.; Voigt, B.; Okita, K.; Maricic, T.; He, Z.; et al. Differences and similarities between human and chimpanzee neural progenitors during cerebral cortex development. ELife 2016, 5, e18683. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S. Hippocampus-dependent Task Improves the Cognitive Function after Ovariectomy in Rats. Osong Public Health Res. Perspect. 2017, 8, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ridha, B.H.; Barnes, J.; Bartlett, J.W.; Godbolt, A.; Pepple, T.; Rossor, M.N.; Fox, N.C. Tracking atrophy progression in familial Alzheimer’s disease: A serial MRI study. Lancet Neurol. 2006, 5, 828–834. [Google Scholar] [CrossRef]
- Frisoni, G.B.; Fox, N.C.; Jack, C.R.; Scheltens, P.; Thompson, P.M. The clinical use of structural MRI in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Yiannopoulou, K.G.; Papageorgiou, S.G. Current and future treatments for Alzheimer’s disease. Ther. Adv. Neurol. Disord. 2013, 6, 19–33. [Google Scholar] [CrossRef]
- Malik, G.A.; Robertson, N.P. Treatments in Alzheimer’s disease. J. Neurol. 2017, 264, 416–418. [Google Scholar] [CrossRef]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 99–107. [Google Scholar] [CrossRef]
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 Inhibitors for Alzheimer’s Disease Treatment. Front. Chem. 2018, 6, 1–10. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Black, R.S.; Sperling, R.A.; Safirstein, B.; Motter, R.N.; Pallay, A.; Nichols, A.; Grundman, M. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 44, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.S.; Wang, Y.Y.; Song, J.T.; Yu, J.H. Acylphloroglucinols as kinase inhibitors from Sargassum nigrifoloides. J. Asian Nat. Prod. Res. 2018, 21, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.; Anand, R.; Messina, J., Jr.; Hartman, R.; Veach, J. A 52-Week Study of the Efficacy of Rivastigmine in Patients with Mild to Moderately Severe Alzheimer’s Disease. Eur. Neurol. 2000, 44, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Almkvist, O.; Darreh-Shori, T.; Stefanova, E.; Spiegel, R.; Nordberg, A. Preserved cognitive function after 12 months of treatment with rivastigmine in mild Alzheimer’s disease in comparison with untreated AD and MCI patients. Eur. J. Neurol. 2004, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Meraz-Ríos, M.A.; Franco-Bocanegra, D.; Toral Rios, D.; Campos-Peña, V. Early onset Alzheimer’s disease and oxidative stress. Oxidative Med. Cell. Longev. 2014, 2014, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sassi, C.; Guerreiro, R.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; Medway, C.; et al. Exome sequencing identifies 2 novel presenilin 1 mutations (p. L166V and p. S230R) in British early-onset Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2422-e13. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Correia, S. Alzheimer disease: Time to improve its diagnosis and treatment. Clevel. Clin. J. Med. 2009, 76, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugroschl, J.; Wang, S. Alzheimer’s Disease: Diagnosis and Treatment Across the Spectrum of Disease Severity. Mt. Sinai J. Med. N. Y. 2011, 78, 596–612. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.C.; Schott, J.M. Imaging cerebral atrophy: Normal ageing to Alzheimer’s disease. Lancet 2004, 363, 392–394. [Google Scholar] [CrossRef]
- Szot, P. Common factors among Alzheimer’s disease, Parkinson’s disease, and epilepsy: Possible role of the noradrenergic nervous system. Epilepsia 2012, 53, 61–66. [Google Scholar] [CrossRef]
- Cuttler, J.M.; Moore, E.R.; Hosfeld, V.D.; Nadolski, D.L. Treatment of Alzheimer Disease with CT Scans: A Case Report. Dose Response Int. J. 2016, 14, 1559325816640073. [Google Scholar] [CrossRef]
- van de Pol, L.A.; Hensel, A.; van der Flier, W.M.; Visser, P.J.; Pijnenburg, Y.A.L.; Barkhof, F.; Gertz, H.J.; Scheltens, P. Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2006, 77, 439–442. [Google Scholar] [CrossRef]
- Montoya, A.; Price, B.H.; Menear, M.; Lepage, M. Brain imaging and cognitive dysfunctions in Huntington’s disease. J. Psychiatry Neurosci. 2006, 31, 21–29. [Google Scholar]
- Cruz de Souza, L.; Chupin, M.; Bertoux, M.; Lehéricy, S.; Dubois, B.; Lamari, F.; Le Ber, I.; Bottlaender, M.; Colliot, O.; Sarazin, M. Is Hippocampal Volume a Good Marker to Differentiate Alzheimer’s Disease from Frontotemporal Dementia? J. Alzheimer’s Dis. 2013, 36, 57–66. [Google Scholar] [CrossRef]
- Fodero, L.R.; Sáez-Valero, J.; Barquero, M.S.; Marcos, A.; McLean, C.A.; Small, D.H. Wheat germ agglutinin-binding glycoproteins are decreased in Alzheimer’s disease cerebrospinal fluid. J. Neurochem. 2001, 79, 1022–1026. [Google Scholar] [CrossRef] [PubMed]
- Schilling, L.P.; Zimmer, E.R.; Shin, M.; Leuzy, A.; Pascoal, T.A.; Benedet, A.L.; Borelli, W.V.; Palmini, A.; Gauthier, S.; Rosa-Neto, P. Imaging Alzheimer’s disease pathophysiology with PET. Dement. Neuropsychol. 2016, 10, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Marcus, C.; Mena, E.; Subramaniam, R.M. Brain PET in the diagnosis of Alzheimer’s disease. Clin. Nucl. Med. 2014, 39, e413–e426. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Berti, V.; Glodzik, L.; Pupi, A.; De Santi, S.; de Leon, M.J. Pre-Clinical Detection of Alzheimer’s Disease Using FDG-PET, with or without Amyloid Imaging. J. Alzheimer’s Dis. 2010, 20, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mucke, L. Alzheimer Mechanisms and Therapeutic Strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef] [Green Version]
- Rabinovici, G.D.; Jagust, W.J. Amyloid Imaging in Aging and Dementia: Testing the Amyloid Hypothesis In Vivo. Behav. Neurol. 2009, 21, 117–128. [Google Scholar] [CrossRef]
- Okamura, N.; Harada, R.; Ishiki, A.; Kikuchi, A.; Nakamura, T.; Kudo, Y. The development and validation of tau PET tracers: Current status and future directions. Clin. Transl. Imaging 2018, 6, 305–316. [Google Scholar] [CrossRef]
- Khan, T.; Alkon, D. Alzheimer’s Disease Cerebrospinal Fluid and Neuroimaging Biomarkers: Diagnostic Accuracy and Relationship to Drug Efficacy. J. Alzheimer’s Dis. 2015, 46, 817–836. [Google Scholar] [CrossRef]
- Liu, L.; Xu, Y.-X.; Hirschberg, C.B. The role of nucleotide sugar transporters in development of eukaryotes. Semin. Cell Dev. Biol. 2010, 21, 600–608. [Google Scholar] [CrossRef] [Green Version]
- Aebi, M. N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 2013, 1833, 2430–2437. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Vijayan, M. Influence of glycosidic linkage on the nature of carbohydrate binding in β-prism I fold lectins: An X-ray and molecular dynamics investigation on banana lectin–carbohydrate complexes. Glycobiology 2011, 21, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.; Henrissat, B.; Davies, G.J.; Withers, S. Glycosyltransferases: Structures, Functions, and Mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, D.H. Chapter Two—Structure-Guided Directed Evolution of Glycosidases: A Case Study in Engineering a Blood Group Antigen-Cleaving Enzyme. In Methods in Enzymology; Imperiali, B., Ed.; Academic Press: New York, NY, USA, 2017; Volume 597, pp. 25–53. [Google Scholar]
- An, H.J.; Froehlich, J.W.; Lebrilla, C.B. Determination of Glycosylation Sites and Site-specific Heterogeneity in Glycoproteins. Curr. Opin. Chem. Biol. 2009, 13, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, S.; Shao, F. Sweet Talk: Protein Glycosylation in Bacterial Interaction with the Host. Trends Microbiol. 2015, 23, 630–641. [Google Scholar] [CrossRef]
- Jensen, P.H.; Kolarich, D.; Packer, N.H. Mucin-type O-glycosylation—Putting the pieces together. FEBS J. 2010, 277, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Hermjakob, H.; Sharon, N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1999, 1473, 4–8. [Google Scholar] [CrossRef]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-glycan and Alzheimer’s disease. Biochim. Biophys. Acta 2017, 1861, 2447–2454. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Frenkel-Pinter, M.; Stempler, S.; Tal-Mazaki, S.; Losev, Y.; Singh-Anand, A.; Escobar-Alvarez, D.; Lezmy, J.; Gazit, E.; Ruppin, E.; Segal, D. Altered protein glycosylation predicts Alzheimer’s disease and modulates its pathology in disease model Drosophila. Neurobiol. Aging 2017, 56, 159–171. [Google Scholar] [CrossRef]
- Taniguchi, N.; Takahashi, M.; Kizuka, Y.; Kitazume, S.; Shuvaev, V.V.; Ookawara, T.; Furuta, A. Glycation vs. glycosylation: A tale of two different chemistries and biology in Alzheimer’s disease. Glycoconj. J. 2016, 33, 487–497. [Google Scholar] [CrossRef]
- Lassen, P.S.; Thygesen, C.; Larsen, M.R.; Kempf, S.J. Understanding Alzheimer’s disease by global quantification of protein phosphorylation and sialylated N-linked glycosylation profiles: A chance for new biomarkers in neuroproteomics? J. Proteom. 2017, 161, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Higel, F.; Seidl, A.; Sörgel, F.; Friess, W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur. J. Pharm. Biopharm. 2016, 100, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, P.; Ungar, D. Bridging the Gap between Glycosylation and Vesicle Traffic. Front. Cell Dev. Biol. 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Roth, Z.; Yehezkel, G.; Khalaila, I. Identification and Quantification of Protein Glycosylation. Int. J. Carbohydr. Chem. 2012, 2012, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.Y.; Majewska, N.I.; Wang, Q.; Paul, J.T.; Betenbaugh, M.J. SnapShot: N-Glycosylation Processing Pathways across Kingdoms. Cell 2017, 171, 258. [Google Scholar] [CrossRef]
- Zhang, L.; Luo, S.; Zhang, B. Glycan analysis of therapeutic glycoproteins. MAbs 2016, 8, 205–215. [Google Scholar] [CrossRef]
- Geyer, H.; Geyer, R. Strategies for analysis of glycoprotein glycosylation. Biochim. Biophys. Acta 2006, 1764, 1853–1869. [Google Scholar] [CrossRef]
- Tarentino, A.L.; Gomez, C.M.; Plummer, T.H. Deglycosylation of asparagine-linked glycans by peptide: N-glycosidase F. Biochemistry 1985, 24, 4665–4671. [Google Scholar] [CrossRef]
- Mucha, E.; Stuckmann, A.; Marianski, M.; Struwe, W.B.; Meijer, G.; Pagel, K. In-depth structural analysis of glycans in the gas phase. Chem. Sci. 2019, 10, 1272–1284. [Google Scholar] [CrossRef] [Green Version]
- Mechref, Y. Analysis of glycans derived from glycoconjugates by capillary electrophoresis-mass spectrometry. Electrophoresis 2011, 32, 3467–3481. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Nie, Y.; Boyes, B.; Orlando, R. Resolving Isomeric Glycopeptide Glycoforms with Hydrophilic Interaction Chromatography (HILIC). J. Biomol. Tech. 2016, 27, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bones, J.; McLoughlin, N.; Hilliard, M.; Wynne, K.; Karger, B.L.; Rudd, P.M. 2D-LC Analysis of BRP 3 Erythropoietin N-Glycosylation using Anion Exchange Fractionation and Hydrophilic Interaction UPLC Reveals Long Poly-N-Acetyl Lactosamine Extensions. Anal. Chem. 2011, 83, 4154–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Walsh, I.; Abrahams, J.L.; Royle, L.; Nguyen-Khuong, T.; Spencer, D.; Fernandes, D.L.; Packer, N.H.; Rudd, P.M.; Campbell, M.P. GlycoStore: A database of retention properties for glycan analysis. Bioinformatics 2018, 34, 3231–3232. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Bones, J.; Yu, Y.Q.; Rudd, P.M.; Gilar, M. Separation of 2-aminobenzamide labeled glycans using hydrophilic interaction chromatography columns packed with 1.7 μm sorbent. J. Chromatogr. B 2010, 878, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Grumbach, E.; Diehl, D.; D Neue, U. The application of novel 1.7 μm ethylene bridged hybrid particles for hydrophilic interaction chromatography. J. Sep. Sci. 2008, 31, 1511–1518. [Google Scholar] [CrossRef]
- Wilson, I.D.; Nicholson, J.K.; Castro-Perez, J.; Granger, J.H.; Johnson, K.A.; Smith, B.W.; Plumb, R.S. High Resolution “Ultra Performance” Liquid Chromatography Coupled to oa-TOF Mass Spectrometry as a Tool for Differential Metabolic Pathway Profiling in Functional Genomic Studies. J. Proteome Res. 2005, 4, 591–598. [Google Scholar] [CrossRef]
- Pu, Y.; Ridgeway, M.E.; Glaskin, R.S.; Park, M.A.; Costello, C.E.; Lin, C. Separation and Identification of Isomeric Glycans by Selected Accumulation-Trapped Ion Mobility Spectrometry-Electron Activated Dissociation Tandem Mass Spectrometry. Anal. Chem. 2016, 88, 3440–3443. [Google Scholar] [CrossRef] [Green Version]
- Mauko, L.; Lacher, N.A.; Pelzing, M.; Nordborg, A.; Haddad, P.R.; Hilder, E.F. Comparison of ZIC-HILIC and graphitized carbon-based analytical approaches combined with exoglycosidase digestions for analysis of glycans from monoclonal antibodies. J. Chromatogr. B 2012, 911, 93–104. [Google Scholar] [CrossRef]
- Gotz, L.; Abrahams, J.L.; Mariethoz, J.; Rudd, P.M.; Karlsson, N.G.; Packer, N.H.; Campbell, M.P.; Lisacek, F. GlycoDigest: A tool for the targeted use of exoglycosidase digestions in glycan structure determination. Bioinformatics 2014, 30, 3131–3133. [Google Scholar] [CrossRef]
- Kobata, A. Exo- and endoglycosidases revisited. Proc. Jpn. Acad. Ser. B 2013, 89, 97–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, K.; Bones, J.; Kattla, J.J.; Rudd, P.M. A systematic approach to protein glycosylation analysis: A path through the maze. Nat. Chem. Biol. 2010, 6, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Silveyra, M.X.; Evin, G.; Montenegro, M.F.; Vidal, C.J.; Martínez, S.; Culvenor, J.G.; Sáez-Valero, J. Presenilin 1 Interacts with Acetylcholinesterase and Alters Its Enzymatic Activity and Glycosylation. Mol. Cell. Biol. 2008, 28, 2908–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guevara, J.; Dilhuydy, H.; Espinosa, B.; Delacourte, A.; Quirion, R.; Mena, R.; Joanette, Y.; Zenteno, E.; Robitaille, Y. Coexistence of reactive plasticity and neurodegeneration in Alzheimer diseased brains. Histol. Histopathol. 2004, 19, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Belický, Š.; Katrlík, J.; Tkáč, J. Glycan and lectin biosensors. Essays Biochem. 2016, 60, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Yoshida, M.; Nagai-Okatani, C.; Iwaki, J.; Matsuda, A.; Tan, B.; Hagiwara, K.; Sato, T.; Itakura, Y.; Noro, E.; et al. A standardized method for lectin microarray-based tissue glycome mapping. Sci. Rep. 2017, 7, 43560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Vega, E.; Tengattini, S.; Peintner, C.; van Angeren, J.; Temporini, C.; Haselberg, R.; Massolini, G.; Somsen, G.W. High-resolution glycoform profiling of intact therapeutic proteins by hydrophilic interaction chromatography-mass spectrometry. Talanta 2018, 184, 375–381. [Google Scholar] [CrossRef]
- Nwosu, C.; Yau, H.K.; Becht, S. Assignment of Core versus Antenna Fucosylation Types in Protein N-Glycosylation via Procainamide Labeling and Tandem Mass Spectrometry. Anal. Chem. 2015, 87, 5905–5913. [Google Scholar] [CrossRef]
- Tsai, T.H.; Wang, M.; Di Poto, C.; Hu, Y.; Zhou, S.; Zhao, Y.; Varghese, R.S.; Luo, Y.; Tadesse, M.G.; Ziada, D.H.; et al. LC-MS profiling of N-Glycans derived from human serum samples for biomarker discovery in hepatocellular carcinoma. J. Proteome Res. 2014, 13, 4859–4868. [Google Scholar] [CrossRef]
- Zhou, S.; Wooding, K.; Mechref, Y. Analysis of Permethylated Glycan by Liquid Chromatography (LC) and Mass Spectrometry (MS). Methods Mol. Biol. 2017, 1503, 83–96. [Google Scholar] [CrossRef]
- Leymarie, N.; Zaia, J. Effective Use of Mass Spectrometry for Glycan and Glycopeptide Structural Analysis. Anal. Chem. 2012, 84, 3040–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumer-Bayraktar, Z.; Nguyen-Khuong, T.; Jayo, R.; Chen, D.D.; Ali, S.; Packer, N.H.; Thaysen-Andersen, M. Micro- and macroheterogeneity of N-glycosylation yields size and charge isoforms of human sex hormone binding globulin circulating in serum. Proteomics 2012, 12, 3315–3327. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Nguyen-Khuong, T.; Wongtrakul-Kish, K.; Tay, S.J.; Chew, D.; Tasha, J.; Taron, C.; Rudd, P. GlycanAnalyzer: Software for Automated Interpretation of N-Glycan Profiles after Exoglycosidase Digestions. Bioinformatics 2018, 35, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Ioffe, E.; Stanley, P. Mice lacking N-acetylglucosaminyltransferase I activity die at mid-gestation, revealing an essential role for complex or hybrid N-linked carbohydrates. Proc. Natl. Acad. Sci. USA 1994, 91, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.W.; Vijay, I.K.; Marth, J.D. A recessive deletion in the GlcNAc-1-phosphotransferase gene results in peri-implantation embryonic lethality. Glycobiology 1999, 9, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Helenius, A.; Aebi, M. Intracellular functions of N-linked glycans. Science 2001, 291, 2364–2369. [Google Scholar] [CrossRef] [PubMed]
- Griffin, M.E.; Hsieh-Wilson, L.C. Glycan Engineering for Cell and Developmental Biology. Cell Chem. Biol. 2016, 23, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.Y.; Takahashi, M.; Gu, J.G.; Miyoshi, E.; Matsumoto, A.; Kitazume, S.; Taniguchi, N. Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 2008, 99, 1304–1310. [Google Scholar] [CrossRef]
- Ding, N.; Nie, H.; Sun, X.; Sun, W.; Qu, Y.; Liu, X.; Yao, Y.; Liang, X.; Chen, C.C.; Li, Y. Human serum N-glycan profiles are age and sex dependent. Age Ageing 2011, 40, 568–575. [Google Scholar] [CrossRef] [Green Version]
- Selman, M.H.; Derks, R.J.; Bondt, A.; Palmblad, M.; Schoenmaker, B.; Koeleman, C.A.; van de Geijn, F.E.; Dolhain, R.J.; Deelder, A.M.; Wuhrer, M. Fc specific IgG glycosylation profiling by robust nano-reverse phase HPLC-MS using a sheath-flow ESI sprayer interface. J. Proteom. 2012, 75, 1318–1329. [Google Scholar] [CrossRef]
- Adamczyk, B.; Tharmalingam, T.; Rudd, P.M. Glycans as cancer biomarkers. Biochim. Biophys. Acta 2012, 1820, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Saldova, R.; Asadi Shehni, A.; Haakensen, V.D.; Steinfeld, I.; Hilliard, M.; Kifer, I.; Helland, A.; Yakhini, Z.; Borresen-Dale, A.L.; Rudd, P.M. Association of N-glycosylation with breast carcinoma and systemic features using high-resolution quantitative UPLC. J. Proteome Res. 2014, 13, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Bones, J.; Byrne, J.C.; O’Donoghue, N.; McManus, C.; Scaife, C.; Boissin, H.; Nastase, A.; Rudd, P.M. Glycomic and glycoproteomic analysis of serum from patients with stomach cancer reveals potential markers arising from host defense response mechanisms. J. Proteome Res. 2011, 10, 1246–1265. [Google Scholar] [CrossRef] [PubMed]
- Block, T.M.; Comunale, M.A.; Lowman, M.; Steel, L.F.; Romano, P.R.; Fimmel, C.; Tennant, B.C.; London, W.T.; Evans, A.A.; Blumberg, B.S.; et al. Use of targeted glycoproteomics to identify serum glycoproteins that correlate with liver cancer in woodchucks and humans. Proc. Natl. Acad. Sci. USA 2005, 102, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, S.; Unwin, L.; Muniyappa, M.; Rudd, P. Glycosylation as a marker for inflammatory arthritis. Cancer Biomark. 2014, 14, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jaeken, J.; Peanne, R. What is new in CDG? J. Inherit. Metab. Dis. 2017, 40, 569–586. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Quelhas, D.; Critchley, A.J.; Carchon, H.; Hebestreit, H.F.; Hibbert, R.G.; Vilarinho, L.; Teles, E.; Matthijs, G.; Schollen, E.; et al. Detailed glycan analysis of serum glycoproteins of patients with congenital disorders of glycosylation indicates the specific defective glycan processing step and provides an insight into pathogenesis. Glycobiology 2003, 13, 601–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A. Human total serum N-glycome. Adv. Clin. Chem. 2008, 46, 51–85. [Google Scholar]
- Kizuka, Y.; Kitazume, S.; Fujinawa, R.; Saito, T.; Iwata, N.; Saido, T.C.; Nakano, M.; Yamaguchi, Y.; Hashimoto, Y.; Staufenbiel, M.; et al. An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer’s disease. EMBO Mol. Med. 2015, 7, 175. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease β-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef]
- Kizuka, Y.; Nakano, M.; Kitazume, S.; Saito, T.; Saido, T.C.; Taniguchi, N. Bisecting GlcNAc modification stabilizes BACE1 protein under oxidative stress conditions. Biochem. J. 2016, 473, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-beta precursor protein at threonine 576 residue regulates trafficking and processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Endo, T.; Manya, H.; Kawamura, M.; Hisanaga, S.I.; Tsumoto, H.; Miura, Y.; Hatsuta, H.; Murayama, S.; Saito, Y.; et al. Excess APP O-glycosylation by GalNAc-T6 decreases Aβ production. J. Biochem. 2016, 161, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Tachida, Y.; Kato, M.; Yamaguchi, Y.; Honda, T.; Hashimoto, Y.; Wada, Y.; Saito, T.; Iwata, N.; Saido, T.; et al. Brain endothelial cells produce amyloid {beta} from amyloid precursor protein 770 and preferentially secrete the O-glycosylated form. J. Biol. Chem. 2010, 285, 40097–40103. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Kozutsumi, Y.; Saito, Y.; Taniguchi, N.; Murayama, S.; Spitalnik, S.L.; Endo, T. Protective effect of N -glycan bisecting GlcNAc residues on β-amyloid production in Alzheimer’s disease. Glycobiology 2010, 20, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zaidi, T.; Iqbal, K.; Grundke-Iqbal, I.; Merkle, R.K.; Gong, C.X. Role of glycosylation in hyperphosphorylation of tau in Alzheimer’s disease. FEBS Lett. 2002, 512, 101–106. [Google Scholar] [CrossRef]
- Sato, Y.; Naito, Y.; Grundke-Iqbal, I.; Iqbal, K.; Endo, T. Analysis of N-glycans of pathological tau: Possible occurrence of aberrant processing of tau in Alzheimer’s disease. FEBS Lett. 2001, 496, 152–160. [Google Scholar] [CrossRef]
- Crespo, C.; García-Mompó, C.; Sanchez-Mataredona, D.; Varea, E.; Castillo-Gómez, E.; Blasco-Ibáñez, J.M.; Nacher, J.; Gómez-Climent, M.Á.; Guirado, R.; Hernández, S.; et al. The Polysialylated Form of the Neural Cell Adhesion Molecule (PSA-NCAM) Is Expressed in a Subpopulation of Mature Cortical Interneurons Characterized by Reduced Structural Features and Connectivity. Cereb. Cortex 2010, 21, 1028–1041. [Google Scholar] [CrossRef] [Green Version]
- Murray, H.C.; Low, V.F.; Swanson, M.E.; Dieriks, B.V.; Turner, C.; Faull, R.L.; Curtis, M.A. Distribution of PSA-NCAM in normal, Alzheimer’s and Parkinson’s disease human brain. Neuroscience 2016, 330, 359–375. [Google Scholar] [CrossRef]
- Palmigiano, A.; Barone, R.; Sturiale, L.; Sanfilippo, C.; Bua, R.O.; Romeo, D.A.; Messina, A.; Capuana, M.L.; Maci, T.; Le Pira, F.; et al. CSF N-glycoproteomics for early diagnosis in Alzheimer’s disease. J. Proteom. 2016, 131, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Henriksen, K.; O’Bryant, S.E.; Hampel, H.; Trojanowski, J.Q.; Montine, T.J.; Jeromin, A.; Blennow, K.; Lönneborg, A.; Wyss-Coray, T.; Soares, H.; et al. The future of blood-based biomarkers for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Cheng, X.; Zeng, J.; Yuan, J.; Wang, Z.; Zhou, W.; Zhang, Y. LW-AFC Effects on N-glycan Profile in Senescence-Accelerated Mouse Prone 8 Strain, a Mouse Model of Alzheimer’s Disease. Aging Dis. 2017, 8, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Gizaw, S.T.; Ohashi, T.; Tanaka, M.; Hinou, H.; Nishimura, S. Glycoblotting method allows for rapid and efficient glycome profiling of human Alzheimer’s disease brain, serum and cerebrospinal fluid towards potential biomarker discovery. Biochim. Biophys. Acta 2016, 1860, 1716–1727. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Sturiale, L.; Palmigiano, A.; Zappia, M.; Garozzo, D. Glycomics of pediatric and adulthood diseases of the central nervous system. J. Proteom. 2012, 75, 5123–5139. [Google Scholar] [CrossRef] [PubMed]
- Maguire, T.M.; Gillian, A.M.; O’Mahony, D.; Coughlan, C.M.; Dennihan, A.; Breen, K.C. A decrease in serum sialyltransferase levels in Alzheimer’s disease. Neurobiol. Aging 1994, 15, 99–102. [Google Scholar] [CrossRef]
- Bertram, L.; Lange, C.; Mullin, K.; Parkinson, M.; Hsiao, M.; Hogan, M.F.; Schjeide, B.M.M.; Hooli, B.; Divito, J.; Ionita, I.; et al. Genome-wide association analysis reveals putative Alzheimer’s disease susceptibility loci in addition to APOE. Am. J. Hum. Genet. 2008, 83, 623–632. [Google Scholar] [CrossRef]
- Vanhooren, V.; Desmyter, L.; Liu, X.E.; Cardelli, M.; Franceschi, C.; Federico, A.; Libert, C.; Laroy, W.; Dewaele, S.; Contreras, R.; et al. N-glycomic changes in serum proteins during human aging. Rejuvenation Res. 2007, 10, 521–531a. [Google Scholar] [CrossRef]
- Chen, C.C.; Engelborghs, S.; Dewaele, S.; Le Bastard, N.; Martin, J.J.; Vanhooren, V.; Libert, C.; De Deyn, P.P. Altered serum glycomics in Alzheimer disease: A potential blood biomarker? Rejuvenation Res. 2010, 13, 439–444. [Google Scholar] [CrossRef]
- Martínez-Cairo, S.; Zenteno, E.; Guzmán, A.; Espinosa, B.; Guevara, J.; Slomianny, M.C.; Hernández, P. Characterization of an O-Glycosylated Plaque-Associated Protein from Alzheimer Disease Brain. J. Neuropathol. Exp. Neurol. 2003, 62, 34–41. [Google Scholar] [CrossRef]
- Dos Santos, J.P.A.; Vizuete, A.; Hansen, F.; Biasibetti, R.; Goncalves, C.A. Early and Persistent O-GlcNAc Protein Modification in the Streptozotocin Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shan, X.; Yuzwa, S.A.; Vocadlo, D.J. The emerging link between O-GlcNAc and Alzheimer disease. J. Biol. Chem. 2014, 289, 34472–34481. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.; Welleford, A.S.; Triplett, J.C.; Sultana, R.; Schmitz, B.; Butterfield, D.A. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim. Biophys. Acta 2014, 1842, 1333–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.W.; Purvine, S.O.; Wang, Z.; Camp, D.G., 2nd; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.J.; Gong, C.X.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 2017, 243, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Akan, I.; Olivier-Van Stichelen, S.; Bond, M.R.; Hanover, J.A. Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 2018, 144, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Wani, W.Y.; Chatham, J.C.; Darley-Usmar, V.; McMahon, L.L.; Zhang, J. O-GlcNAcylation and neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Martinez, I.; Martinez-Loustalot, P.; Lozano, L.; Issad, T.; Limon, D.; Diaz, A.; Perez-Torres, A.; Guevara, J.; Zenteno, E. Neuroinflammation induced by amyloid beta25-35 modifies mucin-type O-glycosylation in the rat’s hippocampus. Neuropeptides 2018, 67, 56–62. [Google Scholar] [CrossRef]
- Halim, A.; Brinkmalm, G.; Ruetschi, U.; Westman-Brinkmalm, A.; Portelius, E.; Zetterberg, H.; Blennow, K.; Larson, G.; Nilsson, J. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid beta-peptides in human cerebrospinal fluid. Proc. Natl. Acad. Sci. USA 2011, 108, 11848–11853. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Culwell, A.R.; Esch, F.S.; Lieberburg, I.; Rydel, R.E. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the β-amyloid precursor protein. Neuron 1993, 10, 243–254. [Google Scholar] [CrossRef]
- Goodman, Y.; Mattson, M.P. Secreted Forms of β-Amyloid Precursor Protein Protect Hippocampal Neurons against Amyloid β-Peptide-Induced Oxidative Injury. Exp. Neurol. 1994, 128, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Charlwood, J.; Dingwall, C.; Matico, R.; Hussain, I.; Johanson, K.; Moore, S.; Powell, D.J.; Skehel, J.M.; Ratcliffe, S.; Clarke, B.; et al. Characterization of the glycosylation profiles of Alzheimer’s beta -secretase protein Asp-2 expressed in a variety of cell lines. J. Biol. Chem. 2001, 276, 16739–16748. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Tachida, Y.; Oka, R.; Nakagawa, K.; Takashima, S.; Lee, Y.C.; Hashimoto, Y. Screening a series of sialyltransferases for possible BACE1 substrates. Glycoconj. J. 2006, 23, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Kitazume, S.; Nakagawa, K.; Oka, R.; Tachida, Y.; Ogawa, K.; Luo, Y.; Citron, M.; Shitara, H.; Taya, C.; Yonekawa, H.; et al. In Vivo Cleavage of α2,6-Sialyltransferase by Alzheimer β-Secretase. J. Biol. Chem. 2005, 280, 8589–8595. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Kitazume, S.; Oka, R.; Maruyama, K.; Saido, T.C.; Sato, Y.; Endo, T.; Hashimoto, Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J. Neurochem. 2006, 96, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, I.; Futakawa, S.; Oka, R.; Ogawa, K.; Marth, J.D.; Miyoshi, E.; Taniguchi, N.; Hashimoto, Y.; Kitazume, S. Beta-galactoside alpha2,6-sialyltransferase I cleavage by BACE1 enhances the sialylation of soluble glycoproteins. A novel regulatory mechanism for alpha2,6-sialylation. J. Biol. Chem. 2007, 282, 34896–34903. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, I.; Breen, K.C.; Di Giamberardino, L.; Moya, K.L. Inhibition of N-glycan processing alters axonal transport of synaptic glycoproteins in vivo. Neuroreport 2000, 11, 1543–1547. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, I.; Georgopoulou, N.; Coughlan, C.M.; Gillian, A.M.; Breen, K.C. The role of the protein glycosylation state in the control of cellular transport of the amyloid beta precursor protein. Neuroscience 1999, 90, 15–25. [Google Scholar] [CrossRef]
- Tienari, P.J.; De Strooper, B.; Ikonen, E.; Simons, M.; Weidemann, A.; Czech, C.; Hartmann, T.; Ida, N.; Multhaup, G.; Masters, C.L.; et al. The beta-amyloid domain is essential for axonal sorting of amyloid precursor protein. EMBO J. 1996, 15, 5218–5229. [Google Scholar] [CrossRef] [PubMed]
- Akasaka-Manya, K.; Manya, H.; Sakurai, Y.; Wojczyk, B.S.; Spitalnik, S.L.; Endo, T. Increased bisecting and core-fucosylated N-glycans on mutant human amyloid precursor proteins. Glycoconj. J. 2008, 25, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef] [PubMed]
- Malinverno, M.; Carta, M.; Epis, R.; Marcello, E.; Verpelli, C.; Cattabeni, F.; Sala, C.; Mulle, C.; Di Luca, M.; Gardoni, F. Synaptic Localization and Activity of ADAM10 Regulate Excitatory Synapses through N-Cadherin Cleavage. J. Neurosci. 2010, 30, 16343. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. α-Secretase ADAM10 as well as αAPPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Escrevente, C.; Morais, V.A.; Keller, S.; Soares, C.M.; Altevogt, P.; Costa, J. Functional role of N-glycosylation from ADAM10 in processing, localization and activity of the enzyme. Biochim. Biophys. Acta 2008, 1780, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.S.; Johnson, G.V.W.; Cole, R.N.; Dong, D.L.Y.; Lee, M.; Hart, G.W. The Microtubule-associated Protein Tau Is Extensively Modified with O-linked N-acetylglucosamine. J. Biol. Chem. 1996, 271, 28741–28744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, T.; Ferreira, S.; Dupont-Wallois, L.; Bussière, T.; Dupire, M.J.; Delacourte, A.; Michalski, J.C.; Caillet-Boudin, M.L. Evidence of a balance between phosphorylation and O-GlcNAc glycosylation of Tau proteins—A role in nuclear localization. Biochim. Biophys. Acta 2003, 1619, 167–176. [Google Scholar] [CrossRef]
- Hsu, Y.P.; Meng, X.; VanNieuwenhze, M.S. Chapter 1—Methods for visualization of peptidoglycan biosynthesis. In Methods in Microbiology; Harwood, C., Jensen, G.J., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 43, pp. 3–48. [Google Scholar]
- Saraswathy, N.; Ramalingam, P. 15—Phosphoproteomics. In Concepts and Techniques in Genomics and Proteomics; Woodhead Publishing: Cambridge, UK, 2012; pp. 203–211. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Z.; Grundke-Iqbal, I.; Iqbal, K. Glycosylation of microtubule–associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat. Med. 1996, 2, 871. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Stanley, P.; Taniguchi, N.; Aebi, N. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2017. [Google Scholar]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Tandon, A.; Chen, F.; Yu, G.; Yu, H.; Arawaka, S.; Hasegawa, H.; Duthie, M.; Schmidt, S.D.; Ramabhadran, T.V.; et al. Mature glycosylation and trafficking of nicastrin modulate its binding to presenilins. J. Biol. Chem. 2002, 277, 28135–28142. [Google Scholar] [CrossRef]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and betaAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef]
- Herreman, A.; Van Gassen, G.; Bentahir, M.; Nyabi, O.; Craessaerts, K.; Mueller, U.; Annaert, W.; De Strooper, B. Gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 2003, 116, 1127–1136. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Gray, C.W.; Breen, K.C. The over-expression of the wild type or mutant forms of the presenilin-1 protein alters glycoprotein processing in a human neuroblastoma cell line. Neurosci. Lett. 2003, 346, 53–56. [Google Scholar] [CrossRef]
- Bakulski, K.M.; Dolinoy, D.C.; Sartor, M.A.; Paulson, H.L.; Konen, J.R.; Lieberman, A.P.; Albin, R.L.; Hu, H.; Rozek, L.S. Genome-wide DNA methylation differences between late-onset Alzheimer’s disease and cognitively normal controls in human frontal cortex. J. Alzheimer’s Dis. 2012, 29, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Münch, A.; Neumann, S.; Kremmer, E.; Tatzelt, J.; Lichtenthaler, S.F. The novel membrane protein TMEM59 modulates complex glycosylation, cell surface expression, and secretion of the amyloid precursor protein. J. Biol. Chem. 2010, 285, 20664–20674. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.T.; Tan, L. The role of clusterin in Alzheimer’s disease: Pathways, pathogenesis, and therapy. Mol. Neurobiol. 2012, 45, 314–326. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.B.; Hone, E.; Pedrini, S.; Doecke, J.; O’Bryant, S.; James, I.; Bush, A.I.; Rowe, C.C.; Villemagne, V.L.; Ames, D.; et al. Altered levels of blood proteins in Alzheimer’s disease longitudinal study: Results from Australian Imaging Biomarkers Lifestyle Study of Ageing cohort. Alzheimer’s Dement. 2017, 8, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.C.; Russell, C.; Mitra, V.; Chung, R.; Hye, A.; Bazenet, C.; Lovestone, S.; Pike, I.; Ward, M. Glycosylation of Human Plasma Clusterin Yields a Novel Candidate Biomarker of Alzheimer’s Disease. J. Proteome Res. 2015, 14, 5063–5076. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruberu, K.; Munoz, S.S.; Jenner, A.M.; Spiro, A.; Zhao, H.; Rassart, E.; Sanchez, D.; Ganfornina, M.D.; Karl, T.; et al. Apolipoprotein D modulates amyloid pathology in APP/PS1 Alzheimer’s disease mice. Neurobiol. Aging 2015, 36, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ruberu, K.; Karl, T.; Garner, B. Cerebral Apolipoprotein-D Is Hypoglycosylated Compared to Peripheral Tissues and Is Variably Expressed in Mouse and Human Brain Regions. PLoS ONE 2016, 11, e0148238. [Google Scholar] [CrossRef]
- Berg, D.; Youdim, M.B. Role of iron in neurodegenerative disorders. Top. Magn. Reson. Imaging 2006, 17, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Menzies, S.L.; St Martin, S.M.; Mufson, E.J. A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J. Neurosci. Res. 1992, 31, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Okayama, Y.; Hashimoto, Y.; Kitaura, M.; Jimbo, D.; Wakutani, Y.; Wada-Isoe, K.; Nakashima, K.; Akatsu, H.; Furukawa, K.; et al. Sugar chains of cerebrospinal fluid transferrin as a new biological marker of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2008, 26, 117–122. [Google Scholar] [CrossRef]
- van Rensburg, S.J.; Berman, P.; Potocnik, F.; MacGregor, P.; Hon, D.; de Villiers, N. 5- and 6-glycosylation of transferrin in patients with Alzheimer’s disease. Metab. Brain Dis. 2004, 19, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Sherman, S.; Norman, K.A.; Kirchhoff, B.A.; Nicolas, M.M.; Greicius, M.D.; Cramer, S.C.; Breiter, H.C.; Hasselmo, M.E.; Stern, C.E. Blockade of central cholinergic receptors impairs new learning and increases proactive interference in a word paired-associate memory task. Behav. Neurosci. 2004, 118, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Kihara, T.; Shimohama, S. Alzheimer’s disease and acetylcholine receptors. Acta Neurobiologiae Experimentalis 2004, 64, 99–105. [Google Scholar] [PubMed]
- Sáez-Valero, J.; de Ceballos, M.A.L.; Small, D.H.; de Felipe, C. Changes in molecular isoform distribution of acetylcholinesterase in rat cortex and cerebrospinal fluid after intracerebroventricular administration of amyloid β-peptide. Neurosci. Lett. 2002, 325, 199–202. [Google Scholar] [CrossRef]
- Fodero, L.R.; Saez-Valero, J.; McLean, C.A.; Martins, R.N.; Beyreuther, K.; Masters, C.L.; Robertson, T.A.; Small, D.H. Altered glycosylation of acetylcholinesterase in APP (SW) Tg2576 transgenic mice occurs prior to amyloid plaque deposition. J. Neurochem. 2002, 81, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Saez-Valero, J.; Fodero, L.R.; Sjögren, M.; Andreasen, N.; Amici, S.; Gallai, V.; Vanderstichele, H.; Vanmechelen, E.; Parnetti, L.; Blennow, K.; et al. Glycosylation of acetylcholinesterase and butyrylcholinesterase changes as a function of the duration of Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Durakoglugil, M.S.; Chen, Y.; White, C.L.; Kavalali, E.T.; Herz, J. Reelin signaling antagonizes β-amyloid at the synapse. Proc. Natl. Acad. Sci. USA 2009, 106, 15938–15943. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Ballif, B.A.; Cooper, J.A. Regulation of Protein Tyrosine Kinase Signaling by Substrate Degradation during Brain Development. Mol. Cell. Biol. 2003, 23, 9293–9302. [Google Scholar] [CrossRef] [Green Version]
- Qiu, S.; Zhao, L.F.; Korwek, K.M.; Weeber, E.J. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J. Neurosci. 2006, 26, 12943–12955. [Google Scholar] [CrossRef] [PubMed]
- Botella-López, A.; Burgaya, F.; Gavín, R.; García-Ayllón, M.S.; Gómez-Tortosa, E.; Peña-Casanova, J.; Ureña, J.M.; Del Río, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, D.; Wang, Y.; Huang, H.; Zhao, Y.; Zhou, H. Meta-analysis of expression and function of neprilysin in Alzheimer’s disease. Neurosci. Lett. 2017, 657, 69–76. [Google Scholar] [CrossRef]
- Lafrance, M.H.; Vézina, C.; Wang, Q.; Boileau, G.; Crine, P.; Lemay, G. Role of glycosylation in transport and enzymic activity of neutral endopeptidase-24.11. Biochem. J. 1994, 302, 451. [Google Scholar] [CrossRef] [PubMed]
- Sato, B.; Katagiri, Y.U.; Iijima, K.; Yamada, H.; Ito, S.; Kawasaki, N.; Okita, H.; Fujimoto, J.; Kiyokawa, N. The human CD10 lacking an N-glycan at Asn (628) is deficient in surface expression and neutral endopeptidase activity. Biochim. Biophys. Acta 2012, 1820, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Tan, L. TREM2 in Alzheimer’s disease. Mol. Neurobiol. 2013, 48, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Ji, I.J.; Kim, D.H.; An, H.J.; Yoon, S.Y. The Alzheimer’s Disease-Associated R47H Variant of TREM2 Has an Altered Glycosylation Pattern and Protein Stability. Front. Neurosci. 2016, 10, 618. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, S.L.; Yang, H.; Lyutvinskiy, Y.; Rutishauser, D.; Herukka, S.K.; Soininen, H.; Zubarev, R.A. Blood plasma IgG Fc glycans are significantly altered in Alzheimer’s disease and progressive mild cognitive impairment. J. Alzheimer’s Dis. 2014, 38, 567–579. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef]
- Leeman, M.; Choi, J.; Hansson, S.; Storm, M.U.; Nilsson, L. Proteins and antibodies in serum, plasma, and whole blood-size characterization using asymmetrical flow field-flow fractionation (AF4). Anal. Bioanal. Chem. 2018, 410, 4867–4873. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
N-glycan profiling of a glycoprotein mixture. N-linked glycans are cleaved from glycoproteins by PNGase F digestion and fluorescently labelled in preparation for exoglycosidase sequencing and subsequent ultra-high performance liquid chromatography (UPLC) analysis. Terminal monosaccharides are removed from the non-reducing end of a glycan structure during an exoglycosidase digestion. Black dotted lines indicate the points of enzymatic digestion in this example of an exoglycosidase digestion. ASN, Asparagine residue in a protein chain; UPLC, ultra high performance liquid chromatography; PNGase F, peptide-N4-(N-acetyl-β-glucosaminyl) asparagine amidase. Images were created using Adobe Photoshop.
Figure 1.
N-glycan profiling of a glycoprotein mixture. N-linked glycans are cleaved from glycoproteins by PNGase F digestion and fluorescently labelled in preparation for exoglycosidase sequencing and subsequent ultra-high performance liquid chromatography (UPLC) analysis. Terminal monosaccharides are removed from the non-reducing end of a glycan structure during an exoglycosidase digestion. Black dotted lines indicate the points of enzymatic digestion in this example of an exoglycosidase digestion. ASN, Asparagine residue in a protein chain; UPLC, ultra high performance liquid chromatography; PNGase F, peptide-N4-(N-acetyl-β-glucosaminyl) asparagine amidase. Images were created using Adobe Photoshop.

Figure 2.
Examples of glycan alterations in disease related glycoproteins in Alzheimer’s disease (AD). (A) Tau, a central protein in the AD pathology, is O-GlcNAcylated which acts as a defense against hyperphosphorylation. In AD, O-glycosylation is downregulated and tau becomes uncharacteristically N-glycosylated. Both normal and hyperphosphorylated tau contain N-glycans in AD. Truncated glycans are more abundant on paired helical filaments (PHF) which are present later in the disease pathology. (B) Bisecting GlcNAc on β-secretase β-site APP-cleaving enzyme 1 (BACE1) occurs prior to toxic Aβ formation in AD. In contrast, GlcNAc bisection on amyloid precursor protein (APP) stimulates α-secretase production and acts as a protective mechanism against amyloid beta (Aβ) peptide formation (not shown). (C) Decreased terminal sialic acids are present on AD cerebrospinal fluid (CSF) glycoproteins, the major glycoprotein being transferrin which is critical to the survival of neuronal cells. Additionally, BACE1 and presenilin, a subunit of γ-secretase, both have a direct role in the sialylation of AD relevant proteins. Grey dotted lines indicate location of changes on glycan structures. Alzheimer’s disease, AD; N-acetyl glucosamine, GlcNAc; amyloid beta, Aβ; amyloid precursor protein, APP; cerebrospinal fluid, CSF; β-site APP-cleaving enzyme 1, BACE1; SER, serine; THR, threonine; ASN, asparagine; pTau, hyperphosphorylated tau; PHF, paired helical filaments. Images were created using Adobe Photoshop.
Figure 2.
Examples of glycan alterations in disease related glycoproteins in Alzheimer’s disease (AD). (A) Tau, a central protein in the AD pathology, is O-GlcNAcylated which acts as a defense against hyperphosphorylation. In AD, O-glycosylation is downregulated and tau becomes uncharacteristically N-glycosylated. Both normal and hyperphosphorylated tau contain N-glycans in AD. Truncated glycans are more abundant on paired helical filaments (PHF) which are present later in the disease pathology. (B) Bisecting GlcNAc on β-secretase β-site APP-cleaving enzyme 1 (BACE1) occurs prior to toxic Aβ formation in AD. In contrast, GlcNAc bisection on amyloid precursor protein (APP) stimulates α-secretase production and acts as a protective mechanism against amyloid beta (Aβ) peptide formation (not shown). (C) Decreased terminal sialic acids are present on AD cerebrospinal fluid (CSF) glycoproteins, the major glycoprotein being transferrin which is critical to the survival of neuronal cells. Additionally, BACE1 and presenilin, a subunit of γ-secretase, both have a direct role in the sialylation of AD relevant proteins. Grey dotted lines indicate location of changes on glycan structures. Alzheimer’s disease, AD; N-acetyl glucosamine, GlcNAc; amyloid beta, Aβ; amyloid precursor protein, APP; cerebrospinal fluid, CSF; β-site APP-cleaving enzyme 1, BACE1; SER, serine; THR, threonine; ASN, asparagine; pTau, hyperphosphorylated tau; PHF, paired helical filaments. Images were created using Adobe Photoshop.


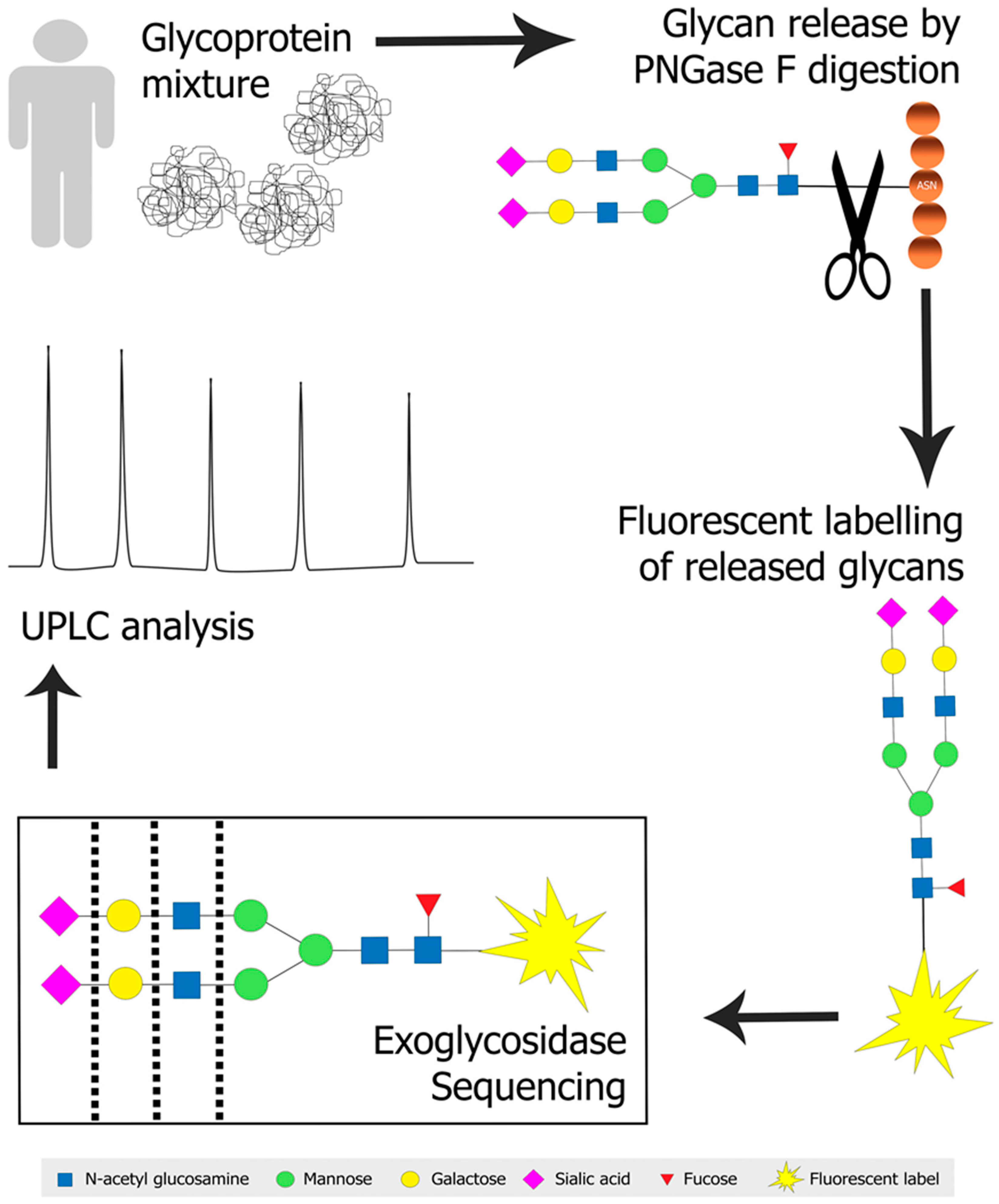{kind=link}
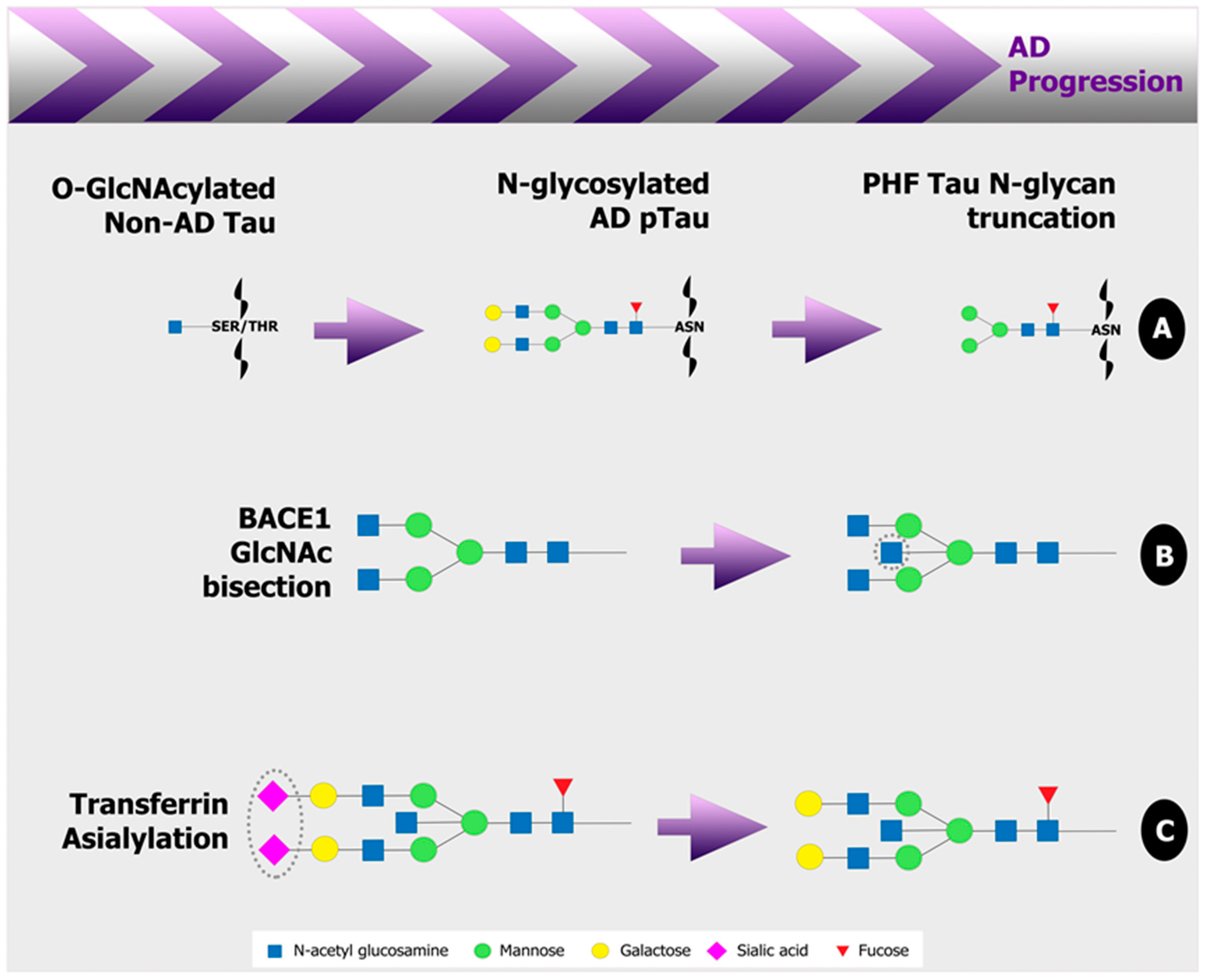{kind=link}
Table 1.
Candidate human biomarkers of glycosylation in AD blood and CSF.
| Location | Analysis Method | Biomarker | Cohorts | Additional Comments |
|---|---|---|---|---|
| Serum and CSF | Glyco-blotting and MS | Increased bisect type, core fucosylated, highly branched species [127]. | AD patients (n = 2–3) versus sex-matched non-AD controls (n = 2–3). | |
| Serum | Radio-enzymatic assay | Decreased sialyltransferase activity [129]. | AD patients (n = 12) versus age and sex matched non-AD controls (n = 12). | Although both moderate and severe AD cases were assessed, there was no correlation between serum sialyltransferase activity and degree of AD. Considerable variation in the control group was observed. |
| Serum | DSA-FACE | Decreased bi-galactosylated core fucosylated bi-antennary glycan [132]. | Population of primarily moderate/severe AD patients (n = 48) versus age and sex matched healthy (n = 149) and non-AD (n = 31) controls. | Desialylated serum assessed. Difference not observed between non-AD patients and age and sex matched controls. Discriminated AD patients (n = 48) from non-AD patients and healthy controls (n = 180) with a diagnostic accuracy of 85.7% ± 2.8%, 92% specificity and 70% sensitivity. |
| CSF | Matrix-assisted laser de-sorption/ionization-MS | Increased bisect type species and decreased sialylated species [124]. | Pre-dementia (n = 11) and sporadic AD (n = 24) cases versus age matched healthy controls (n = 21). | 40–50% of the diseased patients had this altered glycoprofile versus controls. All pre-dementia cases that converted to AD displayed an altered glycoprofile. |
| CSF | LC-MS/MS | Increased ratio of tyrosine linked O-glycosylated Aβ peptides to corresponding unglycosylated peptides [142]. | AD patients (n = 6) versus non-AD patients (n = 7). | Patients not cognitively assessed in detail. Diagnosis based on sensitive and specific CSF biomarker detection of pathological tau and Aβ levels. |
| Plasma | LC-MS/MS | Decreased N-glycosylation of clusterin [181]. | Mild/moderate AD patients with high hippocampal atrophy (n = 14) versus those with low hippocampal atrophy (n = 13). | N-glycans modified with mannose, galactose, sialic acid and GlcNAc. Determined that decreased glycans all present at a common N-glycosylation site on clusterin. |
| CSF | Lectin blotting, isoelectric focusing and MS | Decreased sialylation of transferrin [186]. | Diagnosed probable AD patients (n = 43) versus non-AD (n = 13) and non-demented (n = 32) controls. | Combined with phosphorylated tau detection, specificity and sensitivity was 88.4% and 92.3%, respectively. CSF transferrin levels did not differ between groups. |
| Serum | Isoelectric focusing and immuno-blotting | Increased penta- and hexa-sialylation of transferrin [187]. | AD patients (n = 11) versus non-demented, age-matched controls (n = 14). | |
| CSF | Lectin blotting | Increased mannosylated glycans on reelin [196]. | AD patients (n = 11) versus non-demented, age- and sex-matched controls (n = 9). | Combining two lectin stains increased discrimination of AD from controls. 10 of 11 AD cases were below an arbitrary cutoff point, and 7 of 9 controls were above this cutoff. |
| Plasma | LC-MS/MS | Decreased complex, galactosylated and sialylated glycans on IgG [204]. | AD patients (n = 31) versus non-demented controls (n = 26). | One such bi-antennary, complex, bi-galactosylated glycan decreased in females (n = 93) steadily prior to disease onset from earlier to later stage cases, but an inverse trend was true for males (n = 65). |
Mass spectrometry, MS; Alzheimer’s disease, AD; DNA sequencer-assisted, fluorophore-assisted carbohydrate electrophoresis, DSA-FACE; cerebrospinal fluid, CSF; liquid chromatography, LC; amyloid beta, Aβ; N-acetyl glucosamine, GlcNAc.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Regan, P.; McClean, P.L.; Smyth, T.; Doherty, M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines 2019, 6, 92. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6030092
AMA Style
Regan P, McClean PL, Smyth T, Doherty M. Early Stage Glycosylation Biomarkers in Alzheimer’s Disease. Medicines. 2019; 6(3):92. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6030092
Chicago/Turabian StyleRegan, Patricia, Paula L. McClean, Thomas Smyth, and Margaret Doherty. 2019. "Early Stage Glycosylation Biomarkers in Alzheimer’s Disease" Medicines 6, no. 3: 92. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines6030092
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.