
A Unique Anti-Cancer 3-Styrylchromone Suppresses Inflammatory Response via HMGB1-RAGE Signaling


 ,
,
Abstract
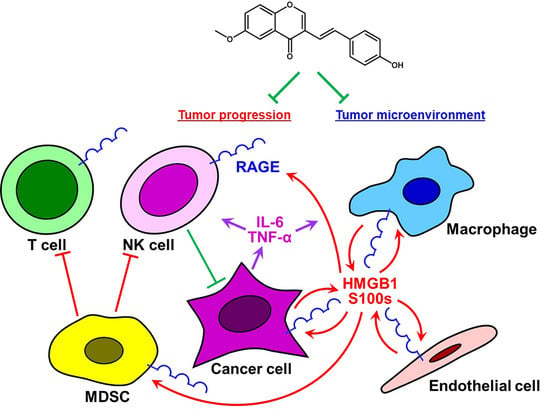
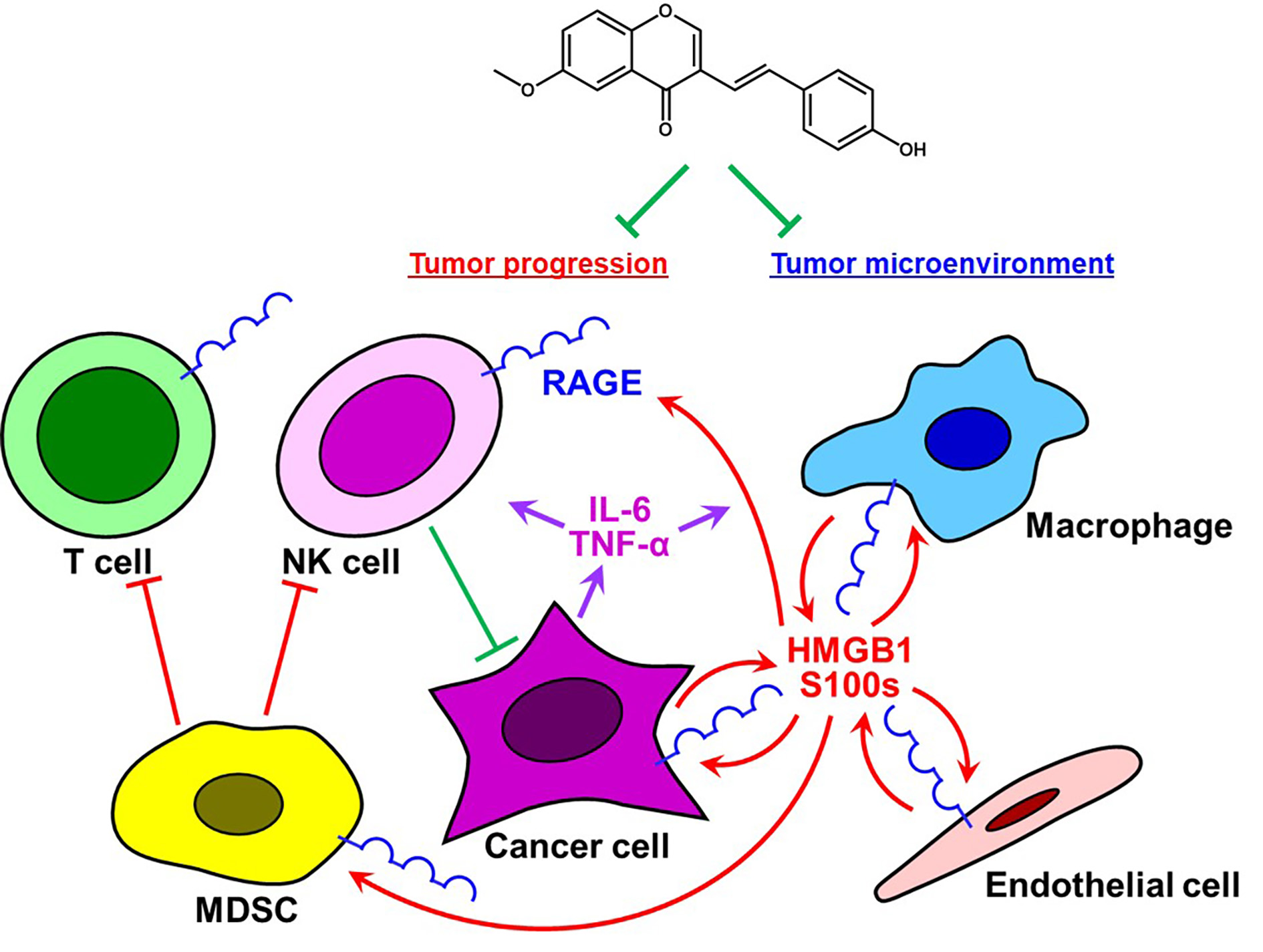
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Synthesis of Test Compounds
2.3. Cells and Cell Culture
2.4. ELISA Assay for IL-6
2.5. Assay for Cell Viability
2.6. Western Blot Analysis
2.7. In Silico 3D Pharmacophore Analysis
2.8. Statistical Analysis
3. Results
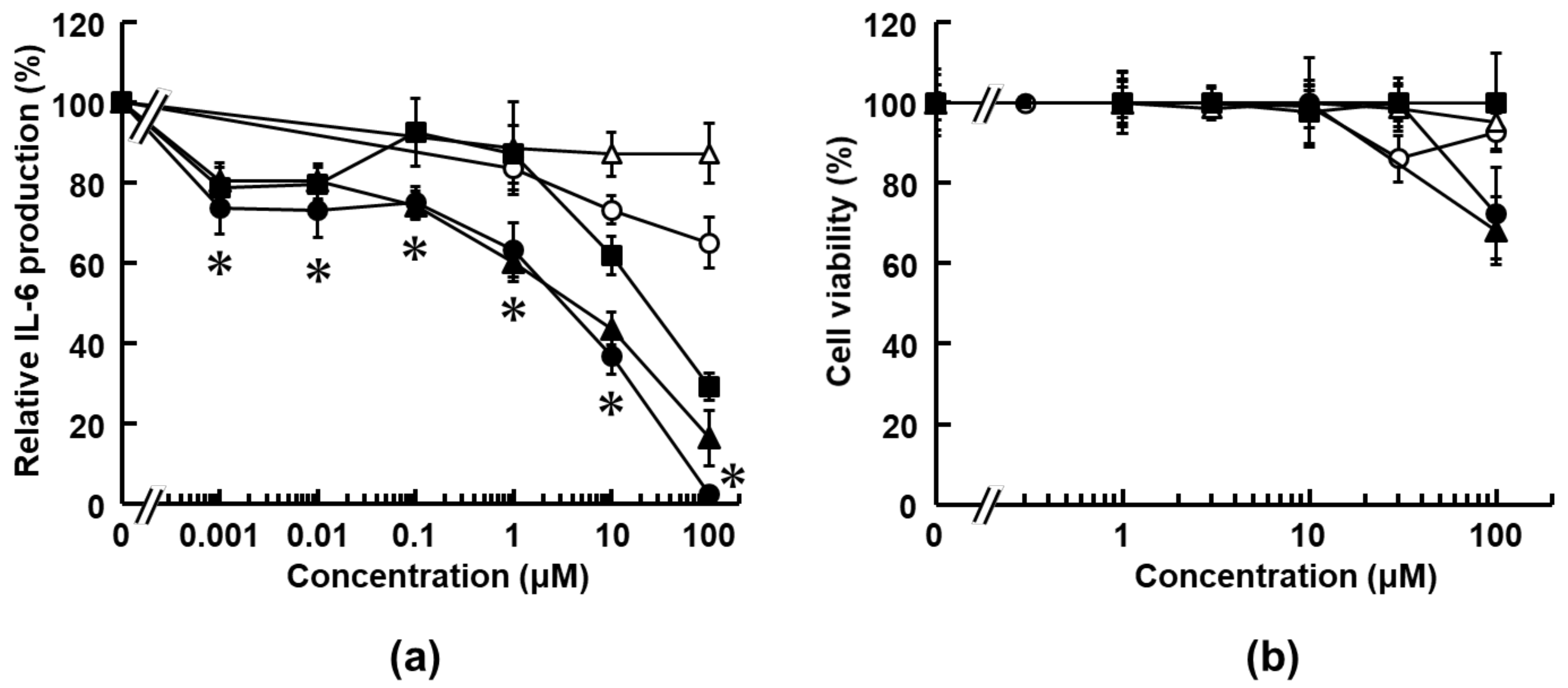
3.1. Anti-Inflammatory Activities of 6M3SC Derivatives Assessed by Suppression of IL-6 Production in HMGB1-Stimulated RAW264.7 Cells
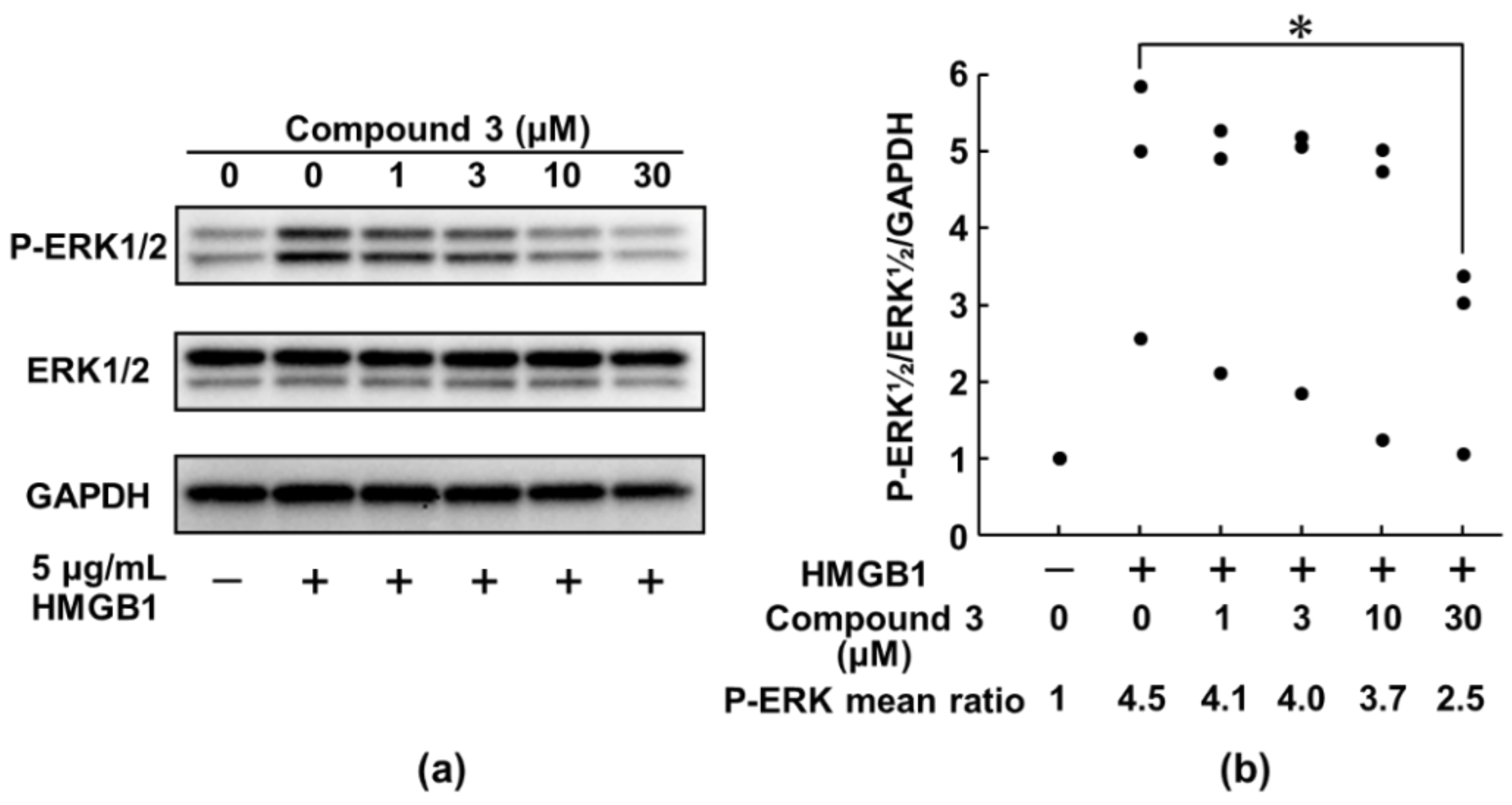
3.2. Compound 3 Suppresses HMGB1-RAGE Signaling by Inhibiting the Activation of ERK 1/2
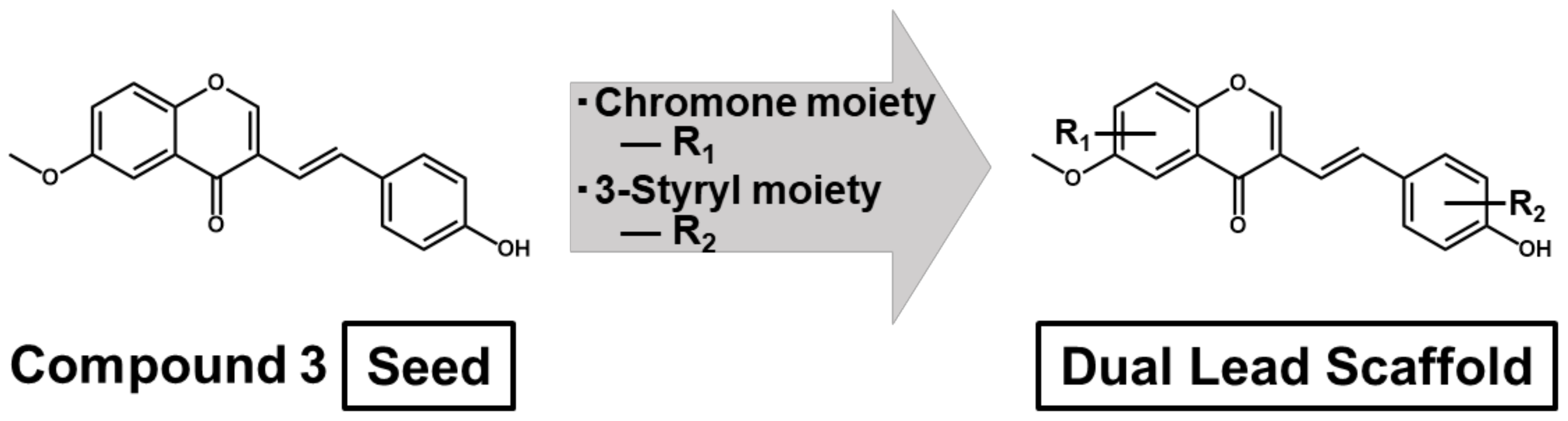
3.3. Structural Insights into 6M3SC Derivatives for Designing Novel Dual Anti-Inflammatory and Anti-Cancer Agents
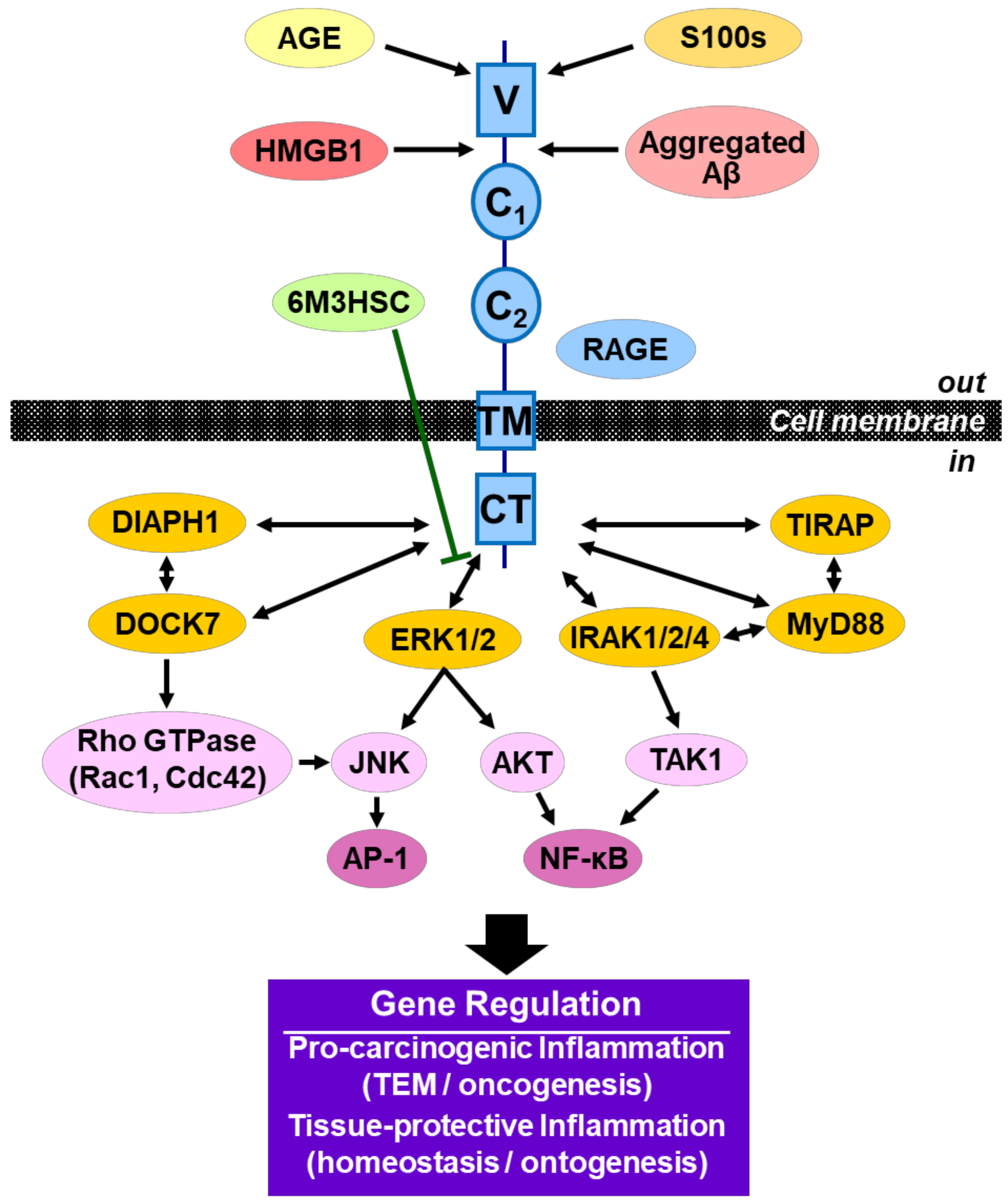
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kuper, H.; Adami, H.O.; Trichopoulos, D. Infections as a major preventable cause of human cancer. J. Intern. Med. 2000, 248, 171–183. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F.R. Cancer-related inflammation. Nat. Cell Biol. 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Nisticò, P.; Ciliberto, G. Biological mechanisms linked to inflammation in cancer: Discovery of tumor microenvironment-related biomarkers and their clinical application in solid tumors. Int. J. Biol. Markers 2020, 35 (Suppl. 1), 8–11. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A.; Höckel, M. Tumor Hypoxia and Malignant Progression. Methods Enzymol. 2004, 381, 335–354. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: Linking Inflammation and Immunity to Cancer Development and Progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-kappaB and the Link between Inflammation and Cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.; Mallon, E.A.; Horgan, P.G.; Paul, A.; McMillan, D.C.; Edwards, J. The relationship between members of the canonical NF-κB pathway, components of tumour microenvironment and survival in patients with invasive ductal breast cancer. Oncotarget 2017, 8, 33002–33013. [Google Scholar] [CrossRef]
- Mortezaee, K.; Najafi, M.; Farhood, B.; Ahmadi, A.; Shabeeb, D.; Musa, A.E. NF-kappaB targeting for Overcoming Tumor Resistance and Normal Tissues Toxicity. J. Cell Physiol. 2019, 234, 17187–17204. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Dai, L.; Ma, Y.; Wang, J.; Liu, Z. Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Lin, W.-W.; Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Investig. 2007, 117, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Allavena, P.; Mantovani, A. Cytokines as a key component of cancer-related inflammation. Cytokine 2008, 43, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M. Interleukin-6: From an inflammatory marker to a target for inflammatory diseases. Trends Immunol. 2012, 33, 571–577. [Google Scholar] [CrossRef]
- Van Schie, K.A.; Hart, M.H.; de Groot, E.R.; Kruithof, S.; Aarden, L.A.; Wolbink, G.J.; Rispens, T. The antibody response against human and chimeric anti-TNF therapeutic antibodies primarily targets the TNF binding region. Ann. Rheum. Dis. 2015, 74, 311–314. [Google Scholar] [CrossRef]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.-S. Role of high mobility group box 1 in inflammatory disease: Focus on sepsis. Arch. Pharmacal Res. 2012, 35, 1511–1523. [Google Scholar] [CrossRef] [PubMed]
- González, H.; Elgueta, D.; Montoya, A.; Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 2014, 274, 1–13. [Google Scholar] [CrossRef]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef]
- Tamada, K.; Nakajima, S.; Ogawa, N.; Inada, M.; Shibasaki, H.; Sato, A.; Takasawa, R.; Yoshimori, A.; Suzuki, Y.; Watanabe, N.; et al. Papaverine identified as an inhibitor of high mobility group box 1/receptor for advanced glycation end-products interaction suppresses high mobility group box 1-mediated inflammatory responses. Biochem. Biophys. Res. Commun. 2019, 511, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Ogawa, N.; Yokoue, N.; Tachibana, H.; Tamada, K.; Okazawa, M.; Sato, A.; Oyama, T.; Abe, H.; Kamiya, T.; et al. Trimebutine attenuates high mobility group box 1–receptor for advanced glycation end-products inflammatory signaling pathways. Biochem. Biophys. Res. Commun. 2020, 533, 1155–1161. [Google Scholar] [CrossRef]
- Inada, M.; Shindo, M.; Kobayashi, K.; Sato, A.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.-I. Anticancer effects of a non-narcotic opium alkaloid medicine, papaverine, in human glioblastoma cells. PLoS ONE 2019, 14, e0216358. [Google Scholar] [CrossRef] [PubMed]
- Inada, M.; Sato, A.; Shindo, M.; Yamamoto, Y.; Akasaki, Y.; Ichimura, K.; Tanuma, S.-I. Anticancer Non-narcotic Opium Alkaloid Papaverine Suppresses Human Glioblastoma Cell Growth. Anticancer Res. 2019, 39, 6743–6750. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Du Yan, S.; Stern, D.M.; Yan, S.F. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Clynes, R.; Moser, B.; Yan, S.F.; Ramasamy, R.; Herold, K. Receptor for AGE (RAGE): Weaving Tangled Webs Within the Inflammatory Response. Curr. Mol. Med. 2007, 7, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Stern, D.M.; Arnold, B.; Nawroth, P.P. Advanced Glycation End Product Receptor-Mediated Cellular Dysfunction. Ann. N. Y. Acad. Sci. 2005, 1043, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Furstenberger, G.; Muller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE signaling sustains inflammation and promotes tumor development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Qamri, Z.; Deol, Y.S.; Ravi, J.; Powell, C.A.; Trikha, P.; Schwendener, R.A.; Bai, X.-F.; Shilo, K.; Zou, X.; et al. S100A7 Enhances Mammary Tumorigenesis through Upregulation of Inflammatory Pathways. Cancer Res. 2012, 72, 604–615. [Google Scholar] [CrossRef]
- Chen, Y.; Akirav, E.M.; Chen, W.; Henegariu, O.; Moser, B.; Desai, D.; Shen, J.M.; Webster, J.C.; Andrews, R.C.; Mjalli, A.M.; et al. RAGE Ligation Affects T Cell Activation and Controls T Cell Differentiation. J. Immunol. 2008, 181, 4272–4278. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef]
- Takao, K.; Ishikawa, R.; Sugita, Y. Synthesis and Biological Evaluation of 3-Styrylchromone Derivatives as Free Radical Scavengers and α-Glucosidase Inhibitors. Chem. Pharm. Bull. 2014, 62, 810–815. [Google Scholar] [CrossRef]
- Shimada, C.; Uesawa, Y.; Ishii-Nozawa, R.; Ishihara, M.; Kagaya, H.; Kanamoto, T.; Terakubo, S.; Nakashima, H.; Takao, K.; Sugita, Y.; et al. Quantitative structure-cytotoxicity relationship of 3-styrylchromones. Anticancer Res. 2014, 34, 5405–5411. [Google Scholar]
- Sakagami, H.; Shimada, C.; Kanda, Y.; Amano, O.; Sugimoto, M.; Ota, S.; Soga, T.; Tomita, M.; Sato, A.; Tanuma, S.-I.; et al. Effects of 3-styrylchromones on metabolic profiles and cell death in oral squamous cell carcinoma cells. Toxicol. Rep. 2015, 2, 1281–1290. [Google Scholar] [CrossRef]
- Takao, K.; Hoshi, K.; Sakagami, H.; Shi, H.; Bandow, K.; Nagai, J.; Uesawa, Y.; Tomomura, A.; Tomomura, M.; Sugita, Y. Further Quantitative Structure–Cytotoxicity Relationship Analysis of 3-Styrylchromones. Anticancer Res. 2019, 40, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D Pharmacophores Derived from Protein-Bound Ligands and Their Use as Virtual Screening Filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Shibui, Y.; Oyama, T.; Okazawa, M.; Yoshimori, A.; Abe, H.; Uchiumi, F.; Tanuma, S.-I. Structural insights into the active site of poly(ADP-ribose) glycohydrolase using docking modes of 6-hydroxy-3H-xanthen-3-one derivative inhibitors. Bioorg. Med. Chem. 2020, 28, 115249. [Google Scholar] [CrossRef]
- Tanuma, S.-I.; Katsuragi, K.; Oyama, T.; Yoshimori, A.; Shibasaki, Y.; Asawa, Y.; Yamazaki, H.; Makino, K.; Okazawa, M.; Ogino, Y.; et al. Structural Basis of Beneficial Design for Effective Nicotinamide Phosphoribosyltransferase Inhibitors. Molecules 2020, 25, 3633. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.-C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (Hmg-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Riehl, A.; Németh, J.; Angel, P.; Hess, J. The receptor RAGE: Bridging inflammation and cancer. Cell Commun. Signal. 2009, 7, 12. [Google Scholar] [CrossRef]
- Rojas, A.; Figueroa, H.; Morales, E. Fueling inflammation at tumor microenvironment: The role of multiligand/rage axis. Carcinogenesis 2009, 31, 334–341. [Google Scholar] [CrossRef]
- He, S.-J.; Cheng, J.; Feng, X.; Yu, Y.; Tian, L.; Huang, Q. The dual role and therapeutic potential of high-mobility group box 1 in cancer. Oncotarget 2017, 8, 64534–64550. [Google Scholar] [CrossRef]
- Wu, L.; Yang, L. The function and mechanism of HMGB1 in lung cancer and its potential therapeutic implications (Review). Oncol. Lett. 2018, 15, 6799–6805. [Google Scholar] [CrossRef]
- El-Far, A.; Munesue, S.; Harashima, A.; Sato, A.; Shindo, M.; Nakajima, S.; Inada, M.; Tanaka, M.; Takeuchi, A.; Tsuchiya, H.; et al. In vitro anticancer effects of a RAGE inhibitor discovered using a structure-based drug design system. Oncol. Lett. 2018, 15, 4627–4634. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Liu, Z.; Xu, Z.; Liu, J.; Zhang, J. High mobility group box 1 (HMGB1): A pivotal regulator of hematopoietic malignancies. J. Hematol. Oncol. 2020, 13, 91. [Google Scholar] [CrossRef]
- Waetzig, V.; Herdegen, T. Context-specific inhibition of JNKs: Overcoming the dilemma of protection and damage. Trends Pharmacol. Sci. 2005, 26, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Kalea, A.Z.; Arriero, M.D.M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE Cytoplasmic Domain with Diaphanous-1 is Required for Ligand-Stimulated Cellular Migration through Activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hisamoto, N.; Nix, P.; Kanao, S.; Mizuno, T.; Bastiani, M.J.; Matsumoto, K. The growth factor SVH-1 regulates axon regeneration in C. elegans via the JNK MAPK cascade. Nat. Neurosci. 2012, 15, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Wani, N.A.; Ahirwar, D.K.; Powell, C.A.; Ravi, J.; Elbaz, M.; Zhao, H.; Padilla, L.; Zhang, X.; Shilo, K.; et al. RAGE mediates S100A7-induced breast cancer growth and metastasis by modulating the tumor microenvironment. Cancer Res. 2015, 75, 974–985. [Google Scholar] [CrossRef]
- Bierhaus, A.; Stern, D.M.; Nawroth, P.P. RAGE in Inflammation: A New Therapeutic Target? Curr. Opin. Investig. Drugs 2006, 7, 985–991. [Google Scholar]
- El-Far, A.H.; Sroga, G.; Al Jaouni, S.K.; Mousa, S.A. Role and Mechanisms of RAGE-Ligand Complexes and RAGE-Inhibitors in Cancer Progression. Int. J. Mol. Sci. 2020, 21, 3613. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Bianchi, M.E. High mobility group box 1 protein, a cue for stem cell recruitment. Biochem. Pharmacol. 2004, 68, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Sampaolesi, M.; De Marchis, F.; Tonlorenzi, R.; Colombetti, S.; Mondino, A.; Cossu, G.; Bianchi, M.E. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J. Cell Biol. 2004, 164, 441–449. [Google Scholar] [CrossRef]
- Chavakis, E.; Hain, A.; Vinci, M.; Carmona, G.; Bianchi, M.E.; Vajkoczy, P.; Zeiher, A.M.; Chavakis, T.; Dimmeler, S. High-Mobility Group Box 1 Activates Integrin-Dependent Homing of Endothelial Progenitor Cells. Circ. Res. 2007, 100, 204–212. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
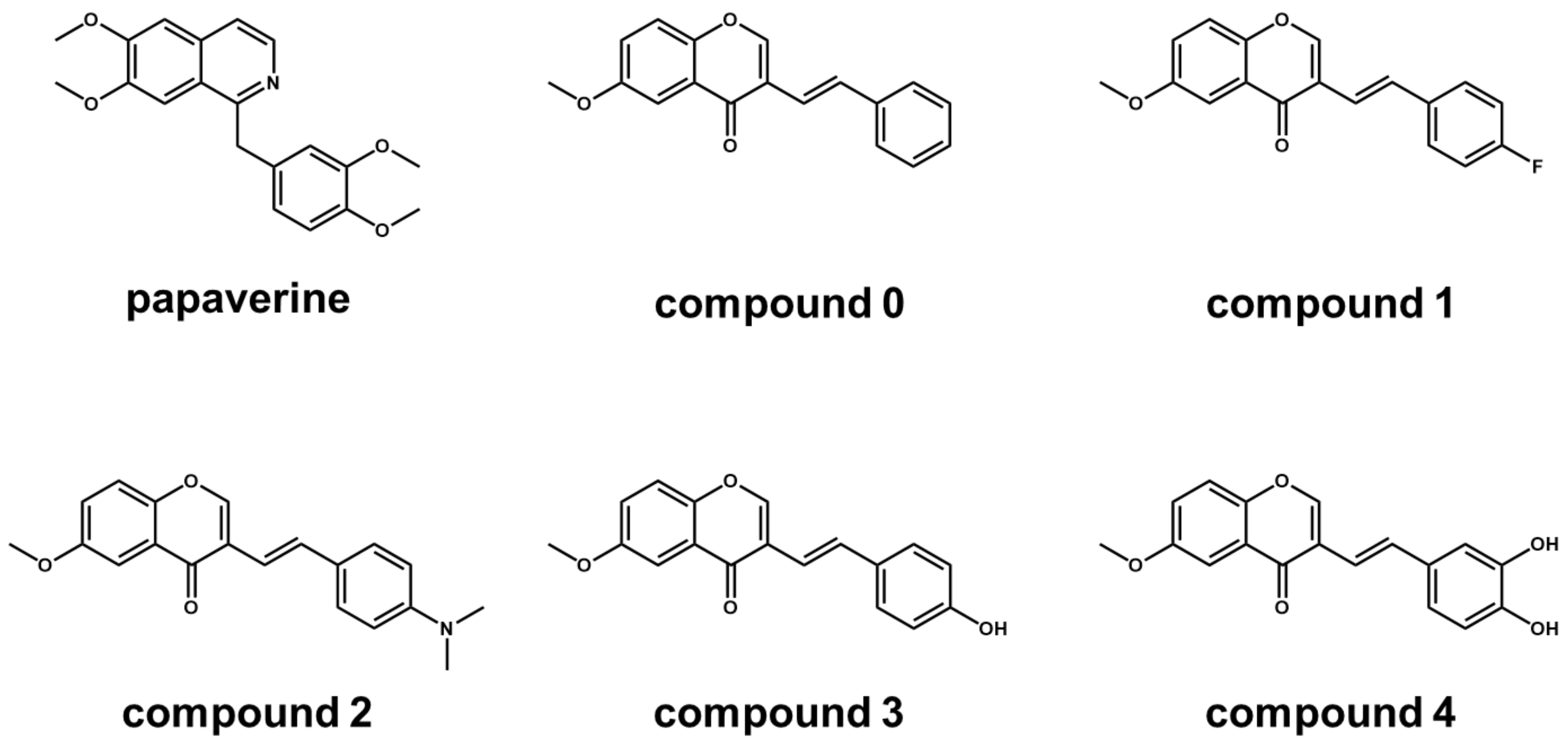
| Compound | Anti-Cancer Activity CC50 (μM) a | 3D Pharmacophore Similarity 3DPFS b |
|---|---|---|
| papaverine | 40.0 | 1 |
| 0 | >50 | 0.51 |
| 1 | 49.0 | 0.51 |
| 2 | 6.5 | 0.42 |
| 3 | 2.0 | 0.42 |
| 4 | 16.0 | 0.42 |
| Compound | Number | |||
|---|---|---|---|---|
| AR a | H b | HBA c | HBD d | |
| 1 | 3 | 4 | 3 | 0 |
| 2 | 3 | 4 | 2 | 0 |
| 3 | 3 | 3 | 3 | 1 |
| 4 | 3 | 3 | 4 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, H.; Okazawa, M.; Oyama, T.; Yamazaki, H.; Yoshimori, A.; Kamiya, T.; Tsukimoto, M.; Takao, K.; Sugita, Y.; Sakagami, H.; et al. A Unique Anti-Cancer 3-Styrylchromone Suppresses Inflammatory Response via HMGB1-RAGE Signaling. Medicines 2021, 8, 17. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8040017
Abe H, Okazawa M, Oyama T, Yamazaki H, Yoshimori A, Kamiya T, Tsukimoto M, Takao K, Sugita Y, Sakagami H, et al. A Unique Anti-Cancer 3-Styrylchromone Suppresses Inflammatory Response via HMGB1-RAGE Signaling. Medicines. 2021; 8(4):17. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8040017
Chicago/Turabian StyleAbe, Hideaki, Miwa Okazawa, Takahiro Oyama, Hiroaki Yamazaki, Atsushi Yoshimori, Takanori Kamiya, Mitsutoshi Tsukimoto, Koichi Takao, Yoshiaki Sugita, Hiroshi Sakagami, and et al. 2021. "A Unique Anti-Cancer 3-Styrylchromone Suppresses Inflammatory Response via HMGB1-RAGE Signaling" Medicines 8, no. 4: 17. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8040017