
Gut Microbiota Regulates the Interaction between Diet and Genetics to Influence Glucose Tolerance †


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
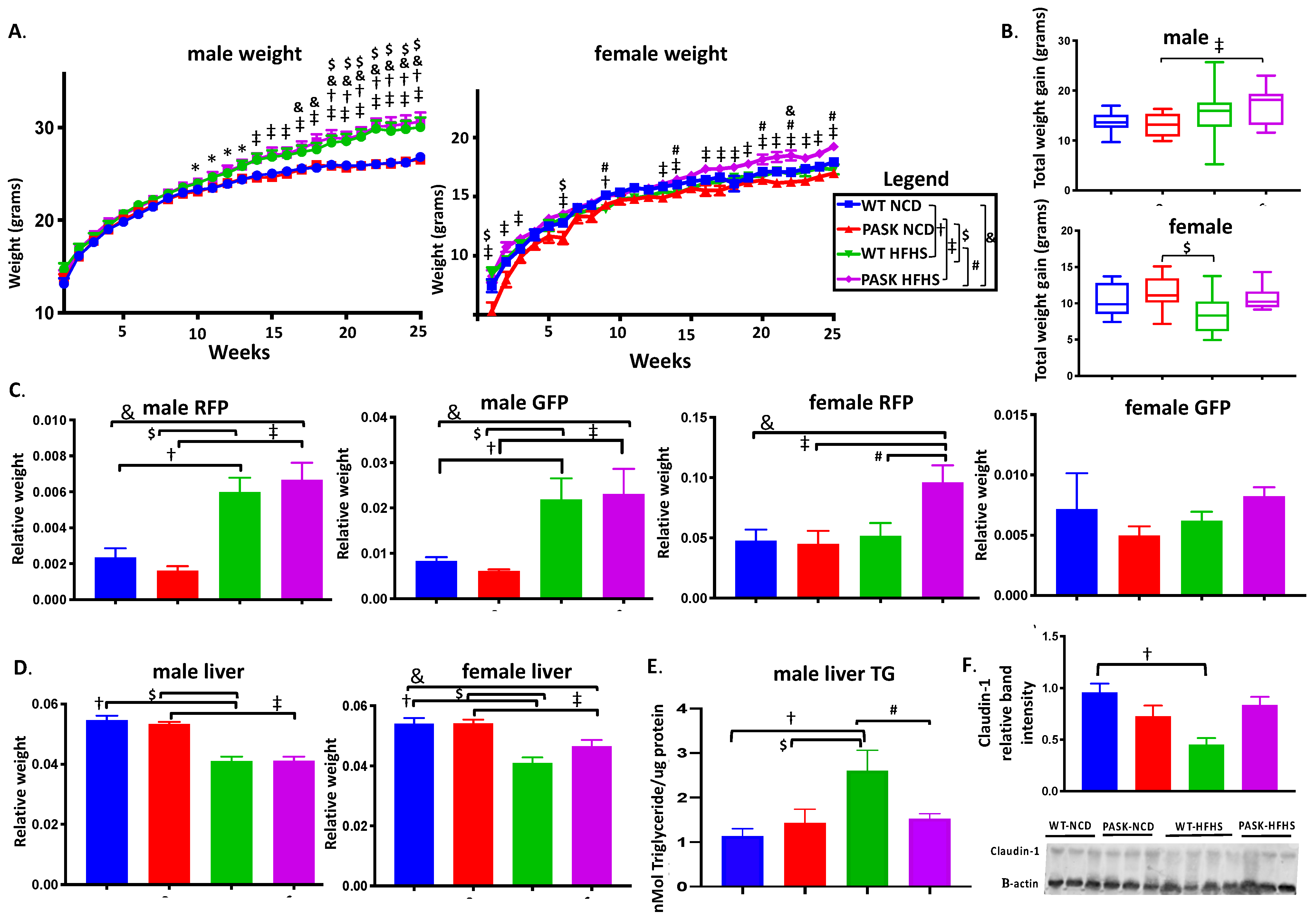
3.1. PASK−/− Deletion Does Not Protect against Weight Gain on an HFHS Diet, but Does Protect against Hepatic Triglyceride Accumulation and Claudin-1 Decrease in Male Mice While Causing Increased RFP in Female HFHS Mice
3.2. PASK Deletion Does Not Significantly Alter Blood Glucose Levels
3.3. Gut Microbiota Composition Is Determined by an Interaction between Diet and Genotype
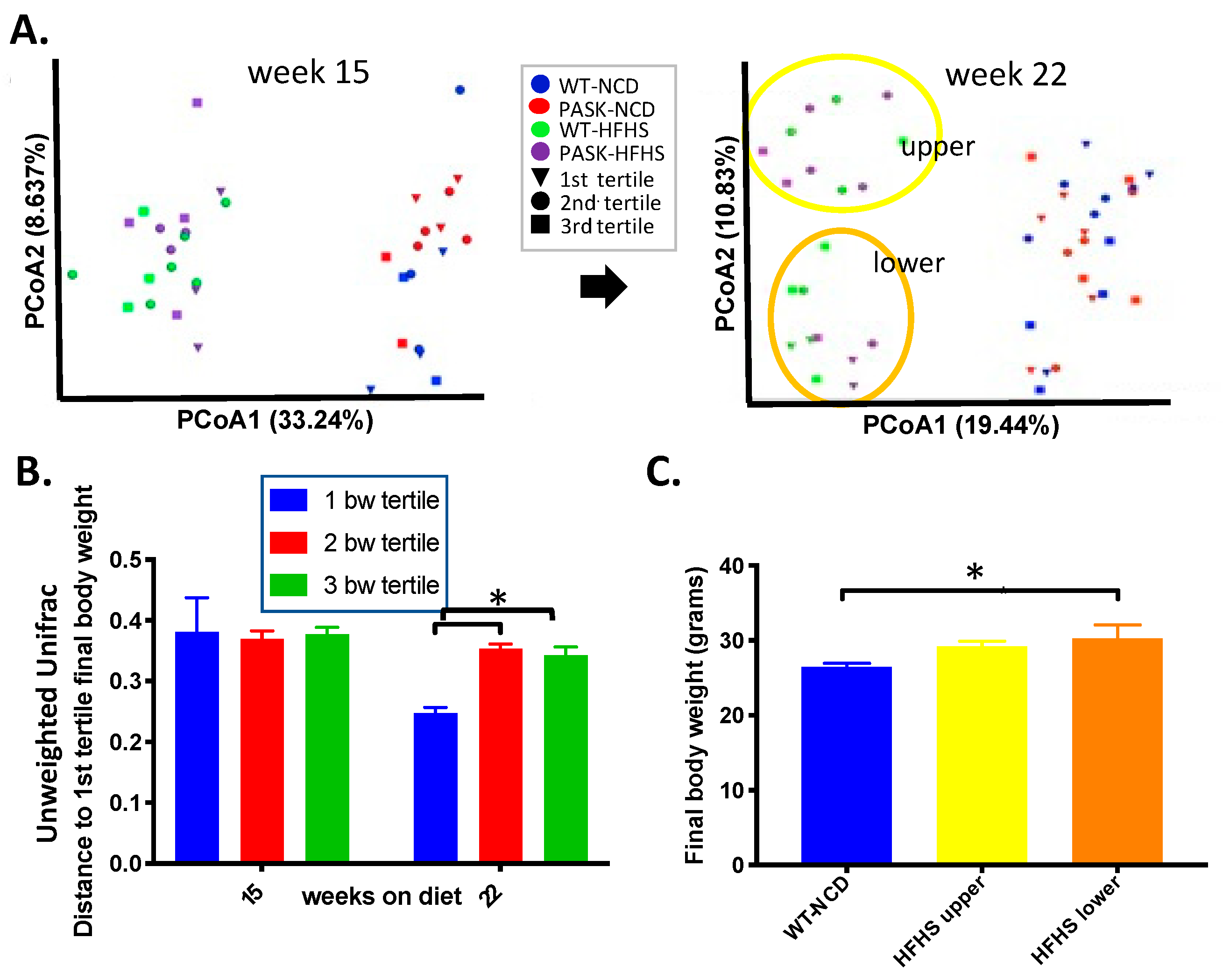
3.4. Bacterial Composition Is Associated with Weight Gain and Undergoes a Population Fork at Week 22 on the HFHS Diet—A Reanalysis of the Data by Microbiome Composition
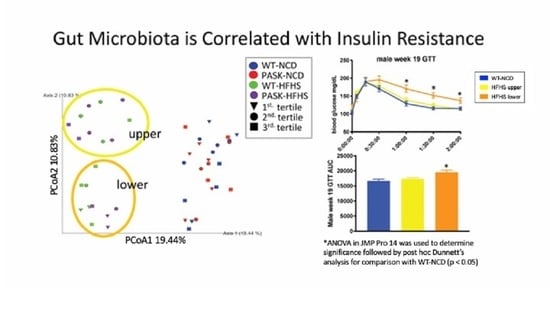
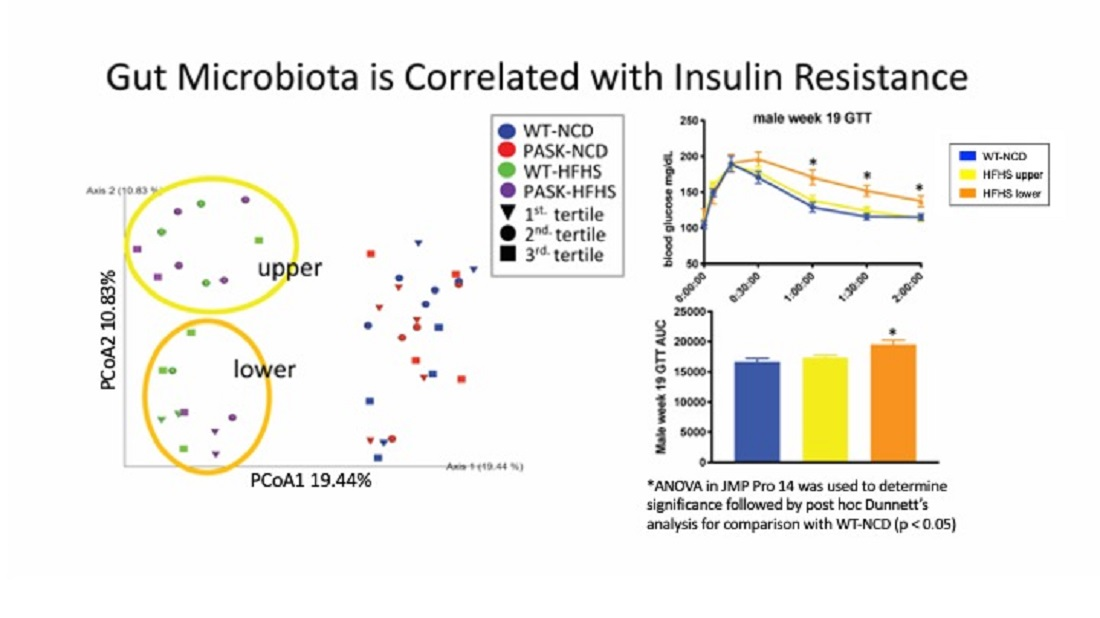
3.5. Microbiome Composition Is Associated with Glucose Tolerance in Mice on an HFHS Diet
3.6. Genotype Influences Bacterial Composition, and Their Interplay Is Associated with Glucose Response in HFHS Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kasuga, M.; Karlsson, F.A.; Kahn, C.R. Insulin stimulates the phosphorylation of the 95,000-dalton subunit of its own receptor. Science 1982, 215, 185–187. [Google Scholar] [CrossRef]
- Bryant, N.J.; Govers, R.; James, D. Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell Biol. 2002, 3, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of Obesity, Diabetes, and Obesity-Related Health Risk Factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef]
- Mendola, N.D.; Chen, T.C.; Gu, Q.; Eberhardt, M.S.; Saydah, S. Prevalen, of Total, Diagnosed, and Undiagnosed Diabetes among Adults: United States, 2013–2016; National Center for Health Statistics: Hyattsville, MD, USA, 2018.
- Centers for Disease Control and Prevention. National Diabetes Statistics Report, 2017; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2017.
- Fryar, C.D.; Hughes, J.P.; Herrick, K.A.; Ahluwalia, N. Fast food consumption among adults in the United States, 2013–2016. In NCHS Data Brief; National Center for Health Statistics: Hyattsville, MD, USA, 2018. [Google Scholar]
- Yang, Q.; Zhang, Z.; Gregg, E.W.; Flanders, W.D.; Merritt, R.; Hu, F.B. Added Sugar Intake and Cardiovascular Diseases Mortality among US Adults. JAMA Intern. Med. 2014, 174, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanhope, K.L. Sugar consumption, metabolic disease and obesity: The state of the controversy. Crit. Rev. Clin. Lab. Sci. 2016, 53, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.A.; Rodríguez, G.; Fleta, J.; Lozano, M.B.; Lázaro, A.; Bueno, G. Trends of Dietary Habits in Adolescents. Crit. Rev. Food Sci. Nutr. 2010, 50, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Sommer, F.; Bäckhed, F. Know your neighbor: Microbiota and host epithelial cells interact locally to control intestinal function and physiology. BioEssays 2016, 38, 455–464. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Macfarlane, S. Fermentation in the Human Large Intestine. J. Clin. Gastroenterol. 2011, 45, S120–S127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egrenham, S.; Clarke, G.; Cryan, J.F.; Dinan, T.G. Brain-Gut-Microbe Communication in Health and Disease. Front. Physiol. 2011, 2, 94. [Google Scholar] [CrossRef] [Green Version]
- Godon, J.J.; Zumstein, E.; Dabert, P.; Habouzit, F.; Moletta, R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997, 63, 2802–2813. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Indias, I.; Sánchez-Alcoholado, L.; García-Fuentes, E.; Cardona, F.; Queipo-Ortuño, M.I.; Tinahones, F.J. Insulin resistance is associated with specific gut microbiota in appendix samples from morbidly obese patients. Am. J. Transl. Res. 2016, 8, 5672–5684. [Google Scholar] [PubMed]
- Cani, P.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.; Delzenne, N.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet-Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Medina, M.; Denizot, J.; Dreux, N.; Robin, F.; Billard, E.; Bonnet, R.; Darfeuille-Michaud, A.; Barnich, N. Western diet induces dysbiosis with increasedE coliin CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 2014, 63, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, N.; Zhao, L. An opportunistic pathogen isolated from the gut of an obese human causes obesity in germfree mice. ISME J. 2012, 7, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nat. Cell Biol. 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.-R.; Lee, J.-C.; Lee, H.-Y.; Kim, M.-S.; Whon, T.W.; Lee, M.-S.; Bae, J.-W. An increase in theAkkermansiaspp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [Green Version]
- Everard, A.; Belzer, C.; Geurts, L.; Ouwerkerk, J.P.; Druart, C.; Bindels, L.B.; Guiot, Y.; Derrien, M.; Muccioli, G.G.; Delzenne, N.M.; et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 2013, 110, 9066–9071. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, C.L.; Önnerfält, J.; Xu, J.; Molin, G.; Ahrné, S.; Thorngren-Jerneck, K. The Microbiota of the Gut in Preschool Children with Normal and Excessive Body Weight. Obesity 2012, 20, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.-M.; Jeong, J.-J.; Woo, K.H.; Han, M.J.; Kim, D.-H. Lactobacillus sakei OK67 ameliorates high-fat diet–induced blood glucose intolerance and obesity in mice by inhibiting gut microbiota lipopolysaccharide production and inducing colon tight junction protein expression. Nutr. Res. 2016, 36, 337–348. [Google Scholar] [CrossRef]
- Ashida, H.; Ogawa, M.; Kim, M.; Mimuro, H.; Sasakawa, C. Bacteria and host interactions in the gut epithelial barrier. Nat. Chem. Biol. 2011, 8, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.-X.; Cardon, C.M.; Swiatek, W.; Cooksey, R.C.; Smith, T.L.; Wilde, J.; Boudina, S.; Abel, E.D.; McClain, D.; Rutter, J. PAS kinase is required for normal cellular energy balance. Proc. Natl. Acad. Sci. USA 2007, 104, 15466–15471. [Google Scholar] [CrossRef] [Green Version]
- García, A.P.; Dongil, P.; Hurtado-Carneiro, V.; Blázquez, E.; Sanz, C.; Álvarez, E. High-fat diet alters PAS kinase regulation by fasting and feeding in liver. J. Nutr. Biochem. 2018, 57, 14–25. [Google Scholar] [CrossRef]
- García, A.P.; Dongil, P.; Hurtado-Carneiro, V.; Blazquez, E.; Sanz, C.; Alvarez, E. PAS Kinase deficiency alters the glucokinase function and hepatic metabolism. Sci. Rep. 2018, 8, 11091. [Google Scholar] [CrossRef] [PubMed]
- Pape, J.A.; Newey, C.R.; Burrell, H.R.; Workman, A.; Perry, K.; Bikman, B.T.; Bridgewater, L.C.; Grose, J.H. Per-Arnt-Sim Kinase (PASK) Deficiency Increases Cellular Respiration on a Standard Diet and Decreases Liver Triglyceride Accumulation on a Western High-Fat High-Sugar Diet. Nutrients 2018, 10, 1990. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Romero, D.; Swiatek, W.I.; Dorweiler, I.; Kikani, C.; Sabic, H.; Zweifel, B.S.; McKearn, J.; Blitzer, J.T.; Nickols, G.A.; et al. PAS Kinase Drives Lipogenesis through SREBP-1 Maturation. Cell Rep. 2014, 8, 242–255. [Google Scholar] [CrossRef] [Green Version]
- DeMille, D.; Pape, J.A.; Bikman, B.T.; Ghassemian, M.; Grose, J.H. The Regulation of Cbf1 by PAS Kinase Is a Pivotal Control Point for Lipogenesis vs. Respiration in Saccharomyces cerevisiae. G3 2019, 9, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-D.; Zhang, J.-G.; Wang, Y.-Z.; Liu, Y.; Liu, G.-L.; Li, X.-Y. Per-Arnt-Sim Kinase (PASK): An Emerging Regulator of Mammalian Glucose and Lipid Metabolism. Nutrients 2015, 7, 7437–7450. [Google Scholar] [CrossRef] [Green Version]
- DeMille, D.; Grose, J.H. PAS kinase: A nutrient sensing regulator of glucose homeostasis. IUBMB Life 2013, 65, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, R.; Xavier, G.D.S.; Hao, H.-X.; Semplici, F.; Rutter, J.; Rutter, G. Regulation by Per-Arnt-Sim (PAS) kinase of pancreatic duodenal homeobox-1 nuclear import in pancreatic β-cells. Biochem. Soc. Trans. 2006, 34, 791–793. [Google Scholar] [CrossRef]
- Borter, E.; Niessen, M.; Zuellig, R.; Spinas, G.A.; Spielmann, P.; Camenisch, G.; Wenger, R.H. Glucose-Stimulated Insulin Production in Mice Deficient for the PAS Kinase PASKIN. Diabetes 2007, 56, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Xavier, G.D.S.; Farhan, H.; Kim, H.; Caxaria, S.; Johnson, P.; Hughes, S.; Bugliani, M.; Marselli, L.; Marchetti, P.; Birzele, F.; et al. Per-arnt-sim (PAS) domain-containing protein kinase is downregulated in human islets in type 2 diabetes and regulates glucagon secretion. Diabetologia 2011, 54, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Fontés, G.; Semache, M.; Hagman, D.K.; Tremblay, C.; Shah, R.; Rhodes, C.J.; Rutter, J.; Poitout, V. Involvement of Per-Arnt-Sim Kinase and Extracellular-Regulated Kinases-1/2 in Palmitate Inhibition of Insulin Gene Expression in Pancreatic β-Cells. Diabetes 2009, 58, 2048–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semache, M.; Zarrouki, B.; Fontés, G.; Fogarty, S.; Kikani, C.; Chawki, M.B.; Rutter, J.; Poitout, V. Per-Arnt-Sim Kinase Regulates Pancreatic Duodenal Homeobox-1 Protein Stability via Phosphorylation of Glycogen Synthase Kinase 3β in Pancreatic β-Cells. J. Biol. Chem. 2013, 288, 24825–24833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semplici, F.; Mondragon, A.; MacIntyre, B.; Madeyski-Bengston, K.; Persson-Kry, A.; Barr, S.; Ramne, A.; Marley, A.; McGinty, J.; French, P.; et al. Cell type-specific deletion in mice reveals roles for PAS kinase in insulin and glucagon production. Diabetologia 2016, 59, 1938–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semplici, F.; Vaxillaire, M.; Fogarty, S.; Semache, M.; Bonnefond, A.; Fontés, G.; Philippe, J.; Meur, G.; Diraison, F.; Sessions, R.B.; et al. Human Mutation within Per-Arnt-Sim (PAS) Domain-containing Protein Kinase (PASK) Causes Basal Insulin Hypersecretion. J. Biol. Chem. 2011, 286, 44005–44014. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME Allows Analysis of High-Throughput Community Sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Gregory Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Vázquez-Baeza, Y.; Gonzalez, A.; Smarr, L.; McDonald, D.; Morton, J.T.; Navas-Molina, J.A.; Knight, R. Bringing the Dynamic Microbiome to Life with Animations. Cell Host Microbe 2017, 21, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. GigaScience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Kruskal, W.H.; Wallis, W.A. Use of ranks in one-criterion variance analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Faith, D.P. Conservation evaluation and phylogenetic diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Schroeder, A.B.; Dobson, E.T.A.; Rueden, C.T.; Tomancak, P.; Jug, F.; Eliceiri, K.W. The ImageJ ecosystem: Open-source software for image visualization, processing, and analysis. Protein Sci. 2021, 30, 234–249. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiatek, W.; Parnell, K.M.; Nickols, G.A.; Scharschmidt, B.F.; Rutter, J. Validation of PAS Kinase, a Regulator of Hepatic Fatty Acid and Triglyceride Synthesis, as a Therapeutic Target for Nonalcoholic Steatohepatitis. Hepatol. Commun. 2020, 4, 696–707. [Google Scholar] [CrossRef] [PubMed]
- DeMille, D.; Bikman, B.T.; Mathis, A.D.; Prince, J.T.; Mackay, J.T.; Sowa, S.W.; Hall, T.D.; Grose, J.H. A comprehensive protein–protein interactome for yeast PAS kinase 1 reveals direct inhibition of respiration through the phosphorylation of Cbf1. Mol. Biol. Cell 2014, 25, 2199–2215. [Google Scholar] [CrossRef] [Green Version]
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ji, Y.; Wu, G.; Sun, K.; Sun, Y.; Li, W.; Wang, B.; He, B.; Zhang, Q.; Dai, Z.; et al. L-Tryptophan Activates Mammalian Target of Rapamycin and Enhances Expression of Tight Junction Proteins in Intestinal Porcine Epithelial Cells. J. Nutr. 2015, 145, 1156–1162. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Ajuwon, K.M. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS ONE 2017, 12, e0179586. [Google Scholar] [CrossRef]
- Bridgewater, L.C.; Zhang, C.; Wu, Y.; Hu, W.; Zhang, Q.; Wang, J.; Li, S.; Zhao, L. Gender-based differences in host behavior and gut microbiota composition in response to high fat diet and stress in a mouse model. Sci. Rep. 2017, 7, 10776. [Google Scholar] [CrossRef] [Green Version]
- Demaude, J.; Salvador-Cartier, C.; Fioramonti, J.; Ferrier, L.; Bueno, L. Phenotypic changes in colonocytes following acute stress or activation of mast cells in mice: Implications for delayed epithelial barrier dysfunction. Gut 2006, 55, 655–661. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franson, J.J.; Grose, J.H.; Larson, K.W.; Bridgewater, L.C. Gut Microbiota Regulates the Interaction between Diet and Genetics to Influence Glucose Tolerance. Medicines 2021, 8, 34. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8070034
Franson JJ, Grose JH, Larson KW, Bridgewater LC. Gut Microbiota Regulates the Interaction between Diet and Genetics to Influence Glucose Tolerance. Medicines. 2021; 8(7):34. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8070034
Chicago/Turabian StyleFranson, Jeralyn J., Julianne H. Grose, Kaitlyn W. Larson, and Laura C. Bridgewater. 2021. "Gut Microbiota Regulates the Interaction between Diet and Genetics to Influence Glucose Tolerance" Medicines 8, no. 7: 34. https://0-doi-org.brum.beds.ac.uk/10.3390/medicines8070034