Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT

Abstract
:1. Introduction
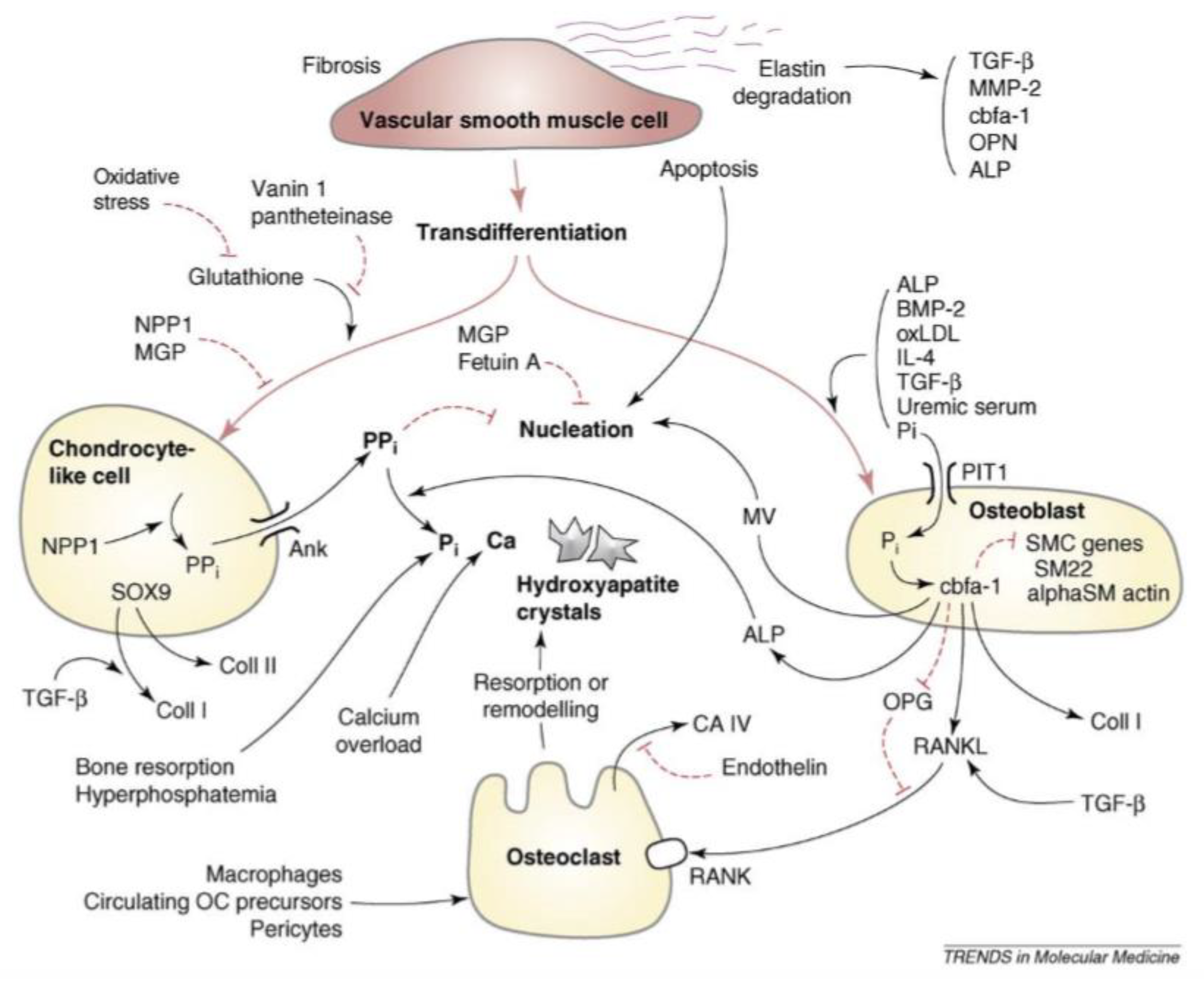
2. Phenotypic Switch
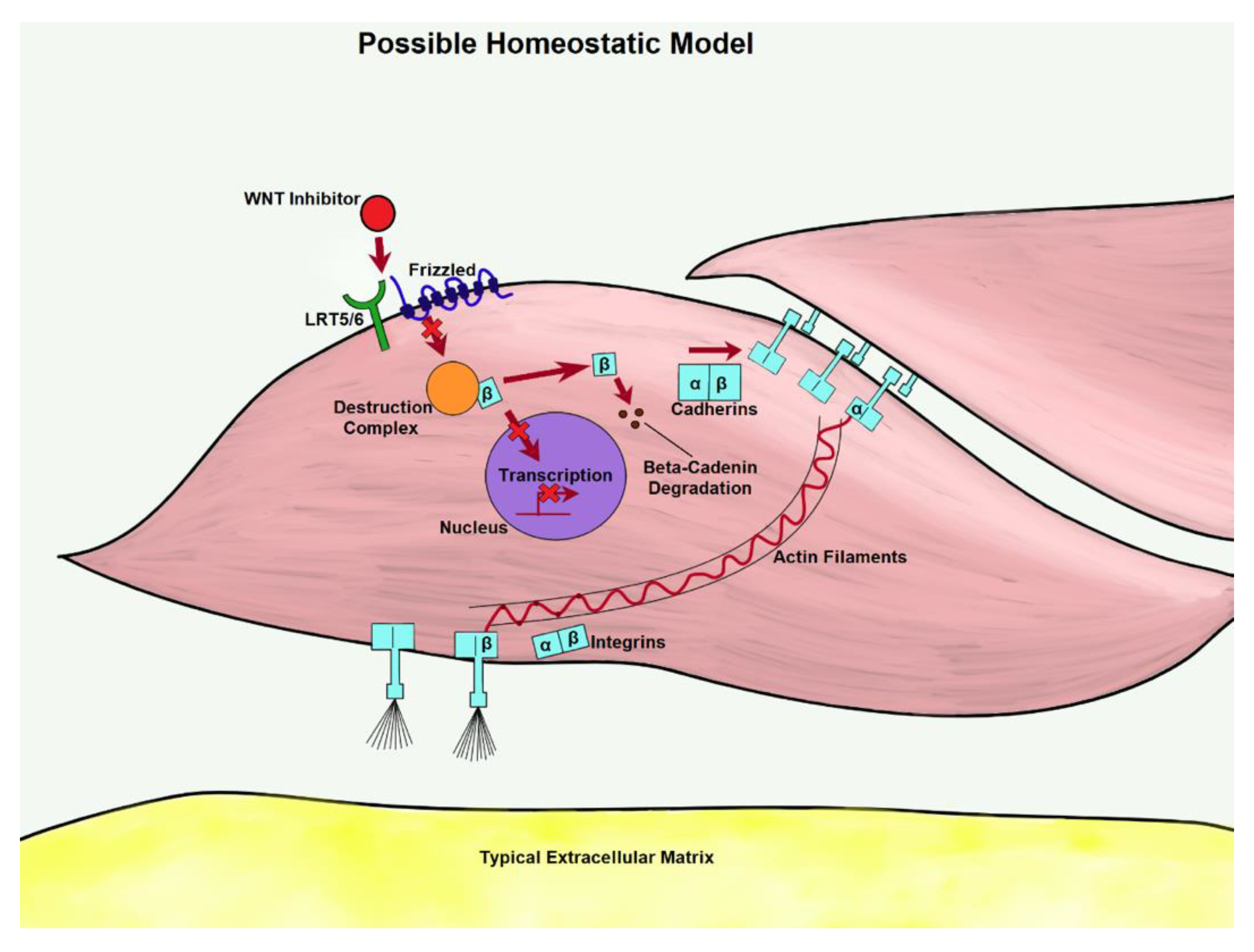
3. Canonical WNT Cascade
4. Relevant WNT-Targeted Genes
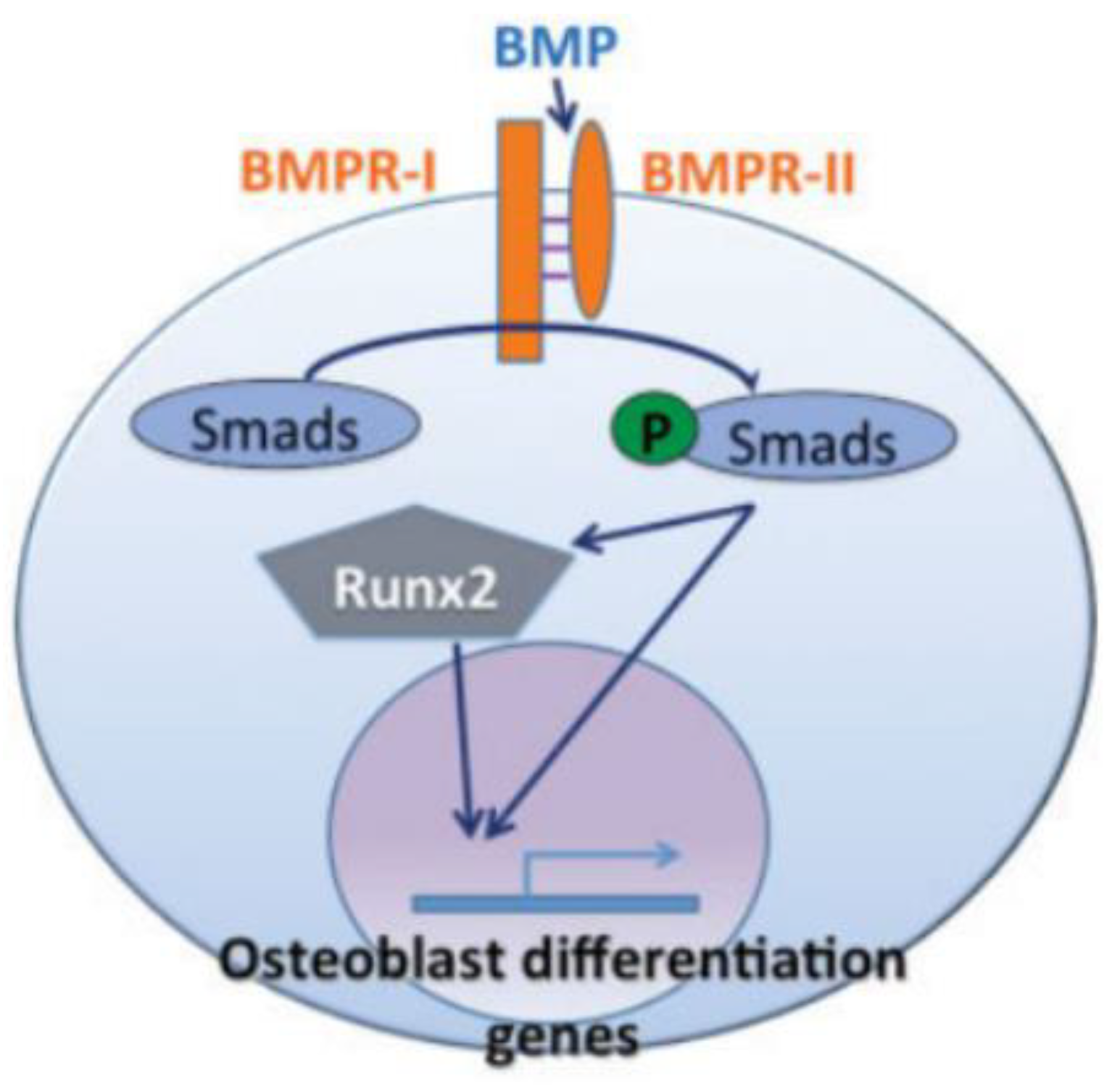
5. Bone Morphogenetic Proteins
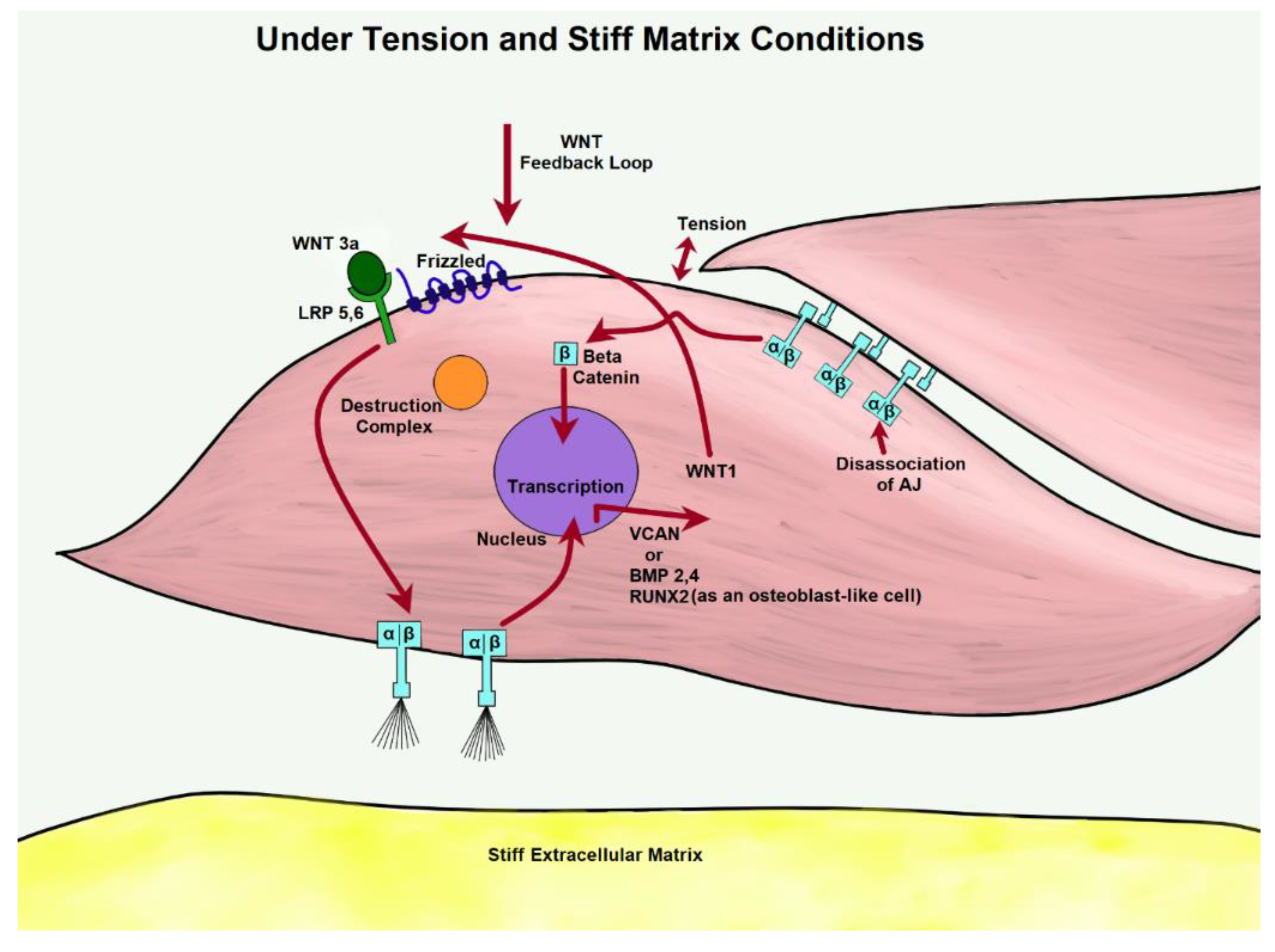
6. BMPs and Mechanical Stressors
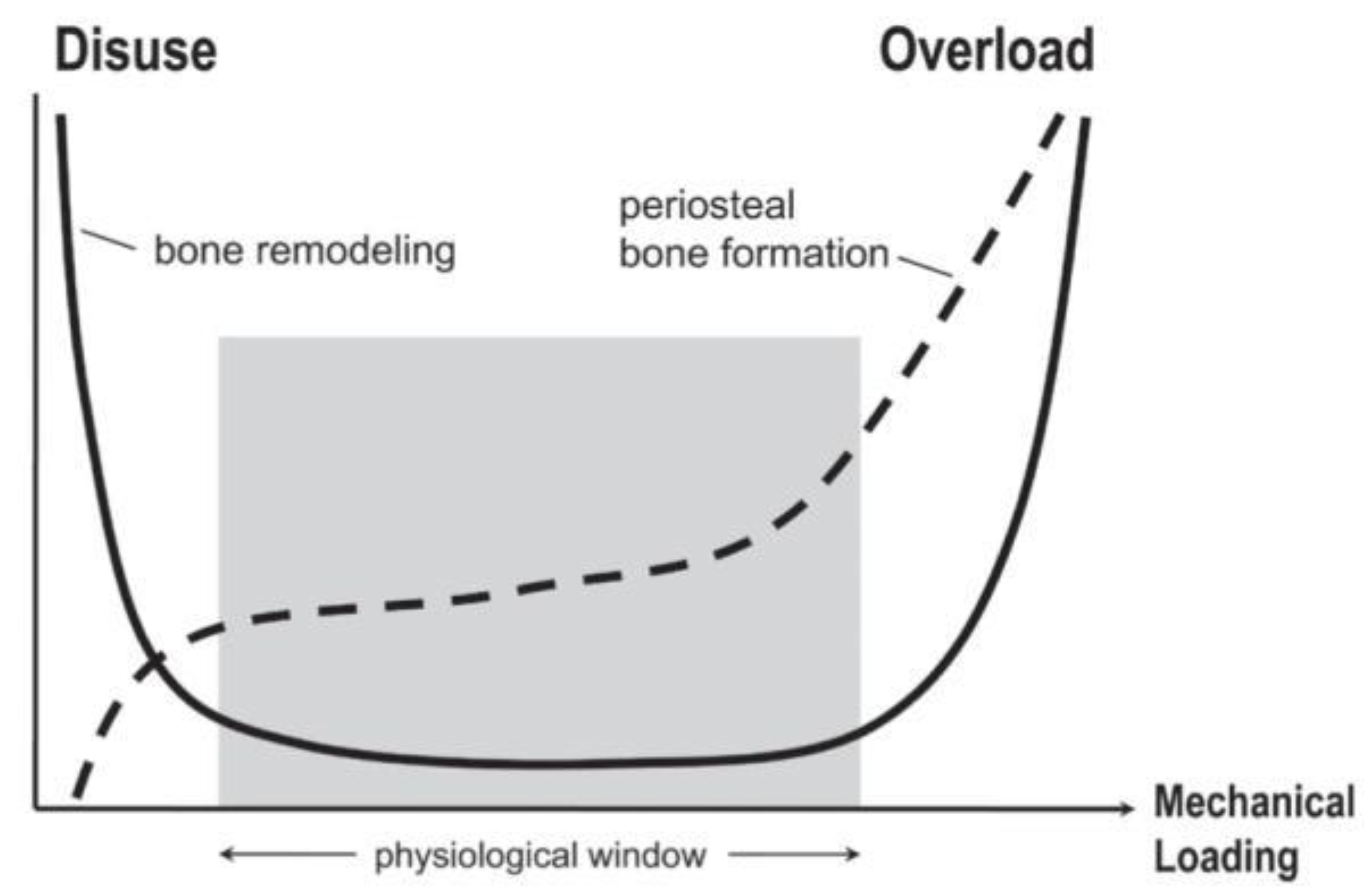
7. WNT Bone Remodeling and Mechanical Strain
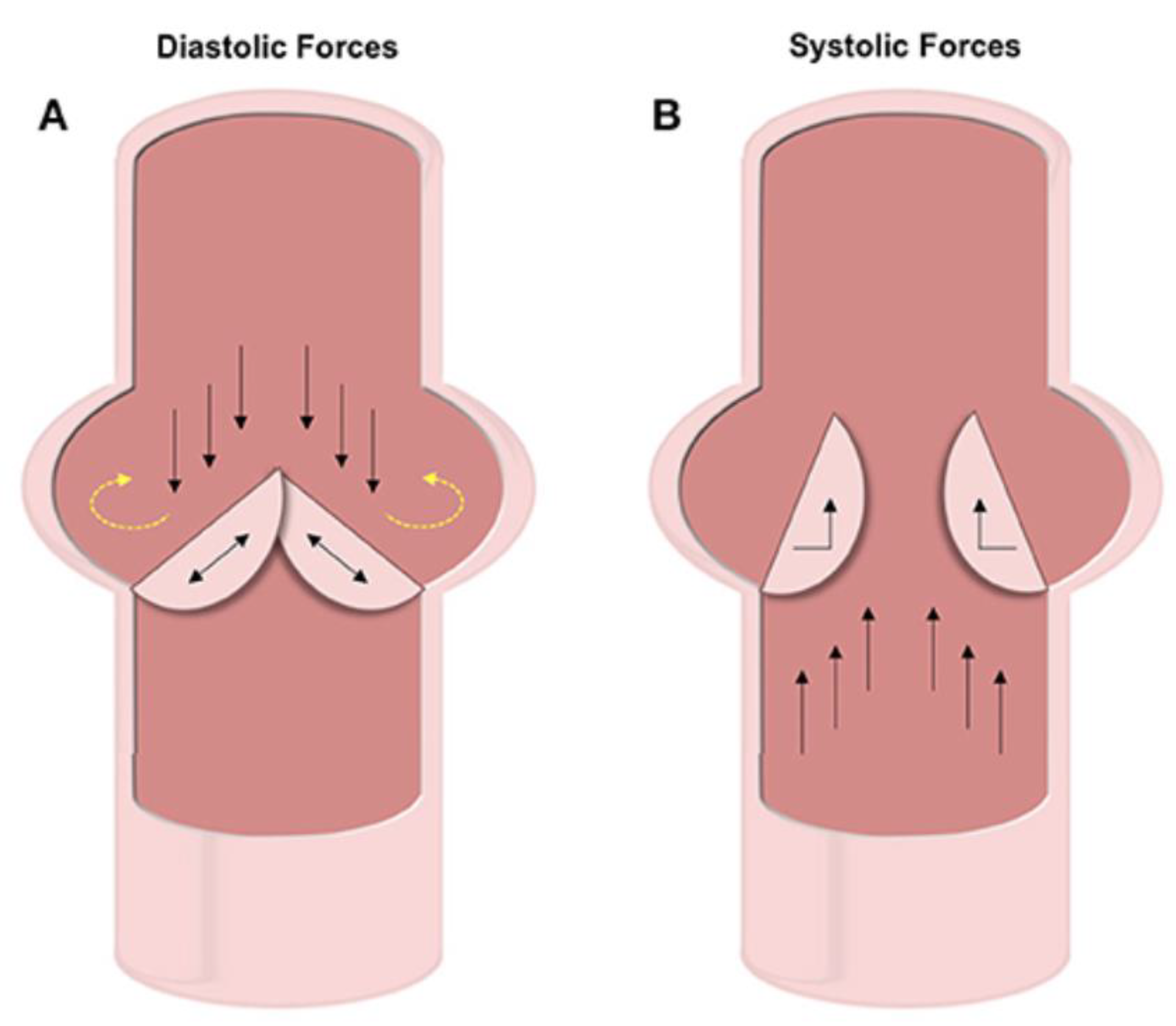
8. Mechanical Influence on Arterial Tissues under Pathological Conditions
9. Physiological Mechanical Forces
10. Arterial Matrix Stiffness under Pathological Conditions
11. Collagen Influence
12. Integrins and Relevant Functions
13. Integrins and Mechanical Stressors
14. Cadherins and Relevant Functions
15. Cadherins and Mechanical Stressors
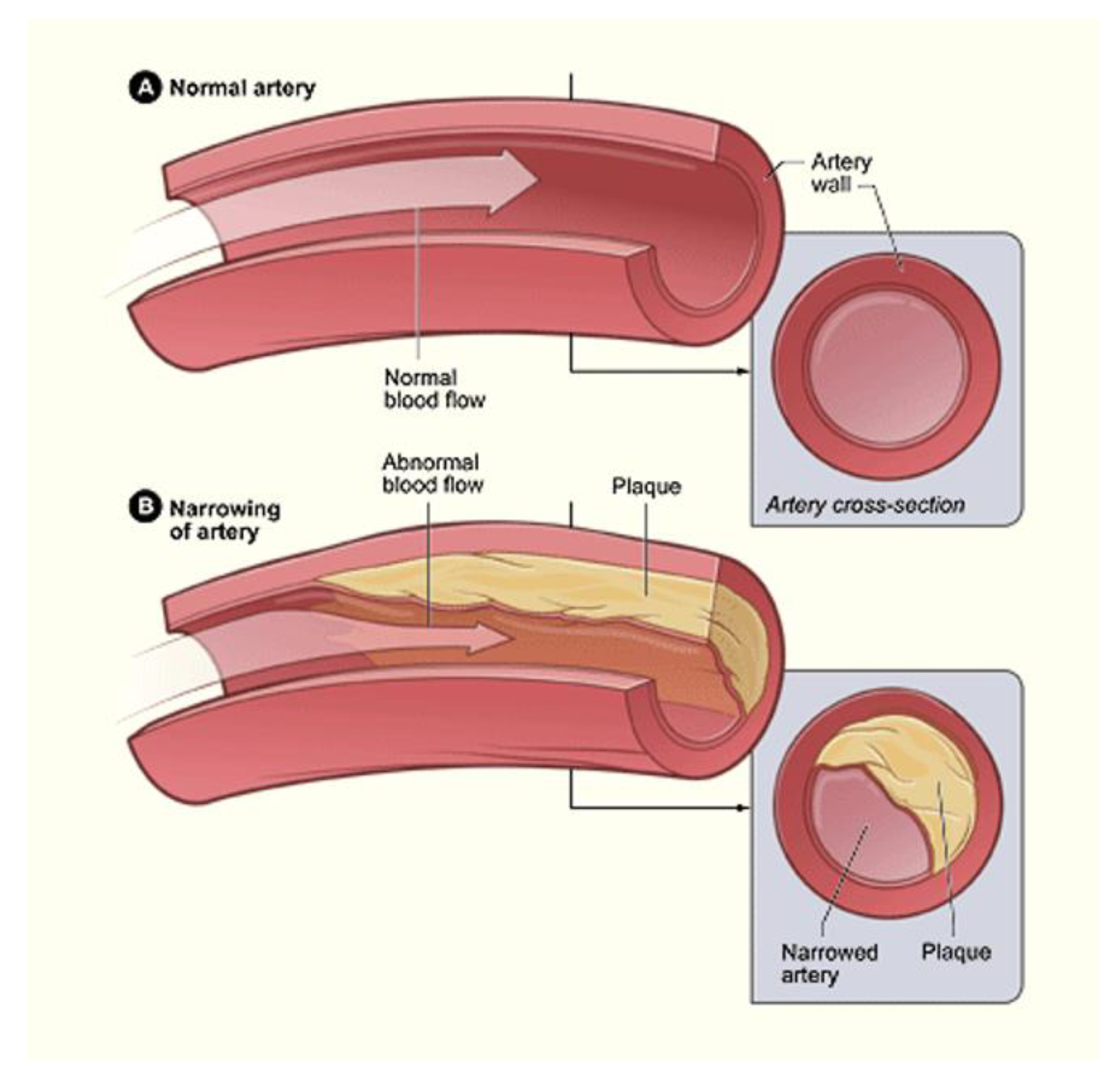
16. Seeding through Thrombus Release and Other Health Risks
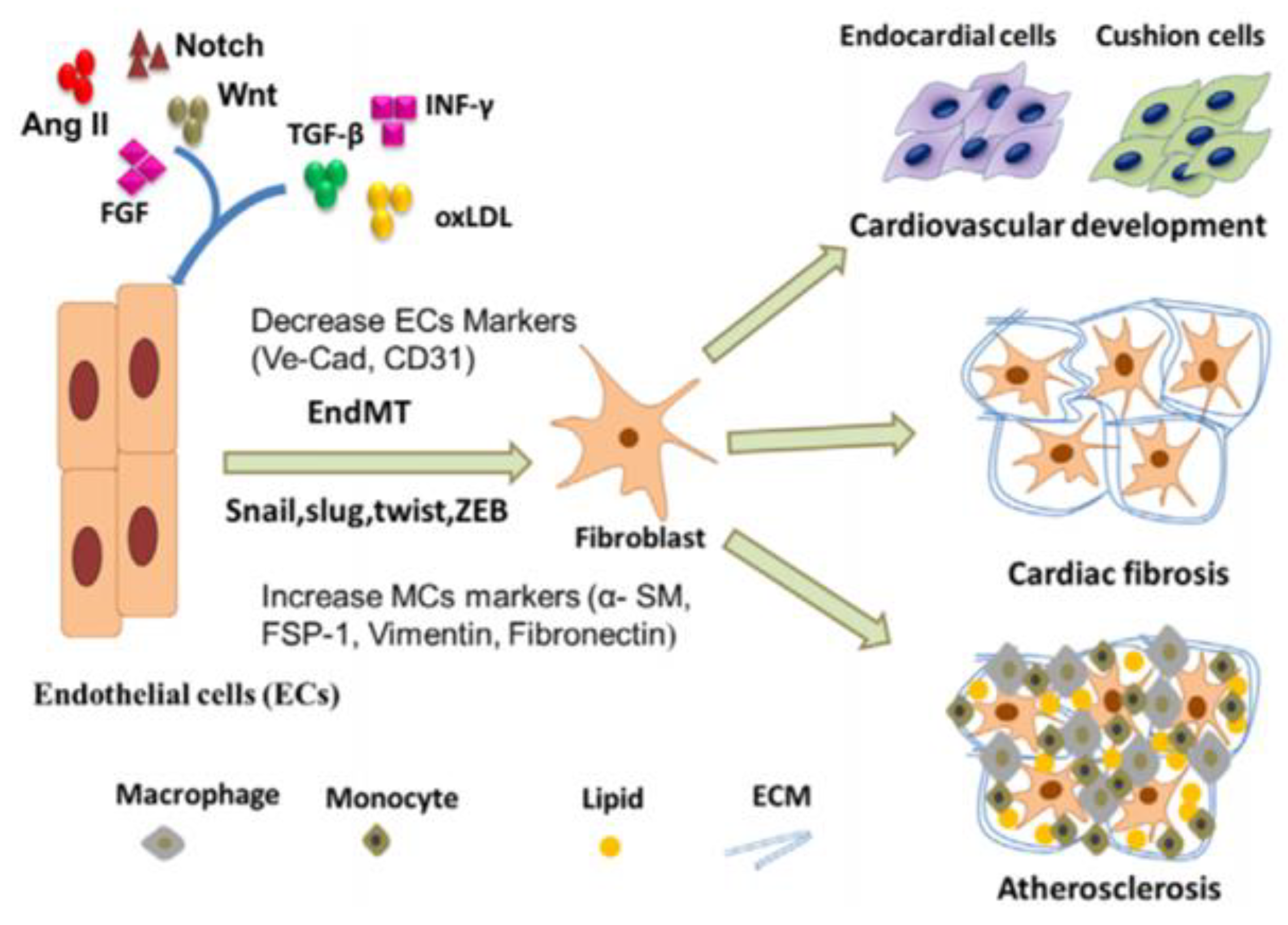
17. Endothelial to Mesenchymal Transition (EndMT)
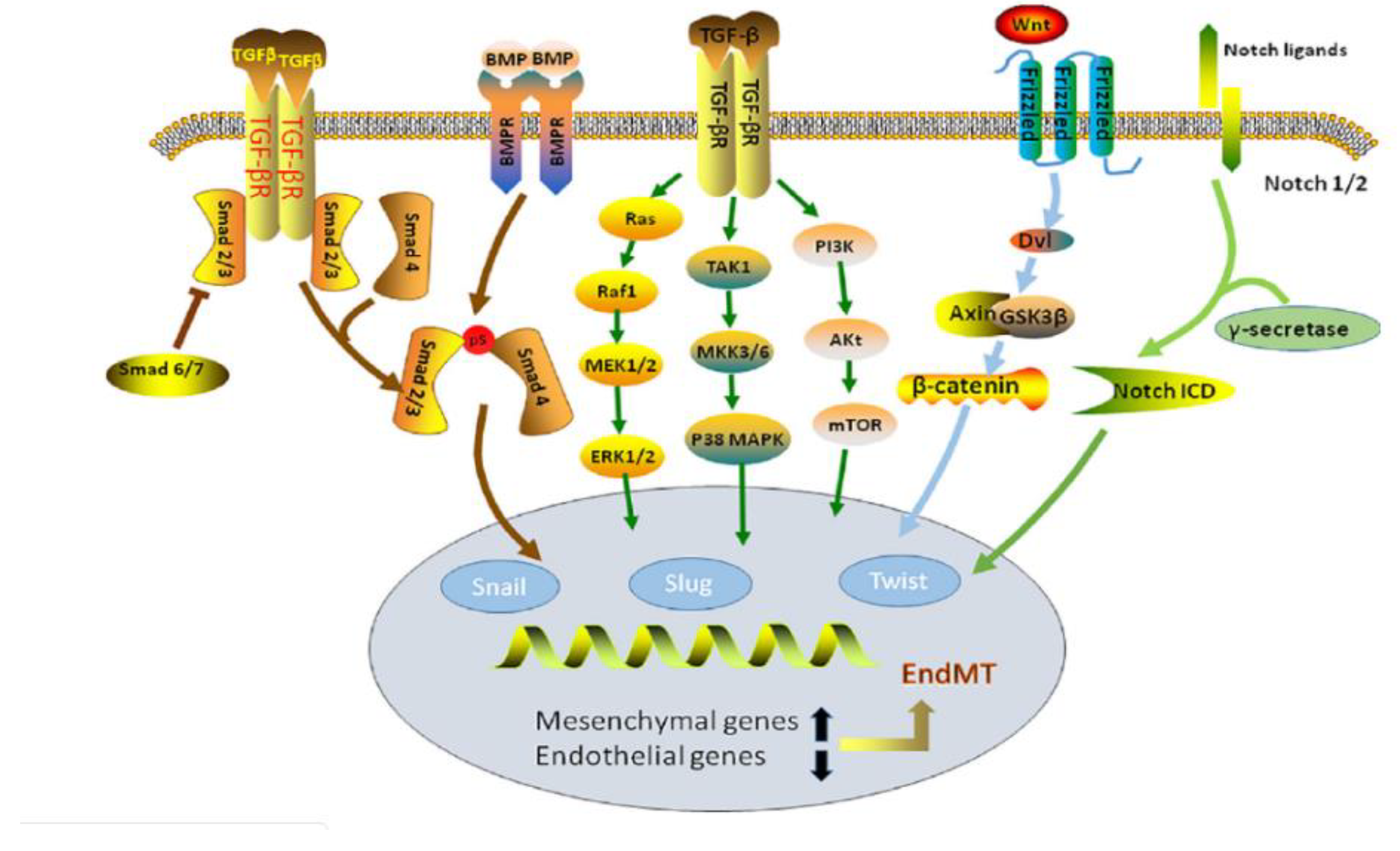
18. EndMT in TGF-β and BMP Signaling Pathways
19. EndMT in WNT Signaling Pathway
20. Calcification of Mechanically Active Vascular Devices
21. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Glossary
| Abbreviation | Definition |
| AJ | adherens junction |
| BMP | bone morphogenic protein |
| DLX3 | distal-less homeobox 3 |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EndMT | endothelial to mesenchymal transition |
| ERK | extracellular signal-regulated kinase |
| FAK | focal adhesion kinase |
| F-CPE | Full length Carboxypeptidase E |
| GSK-3 | glycogen synthase kinase-3 |
| LEF | lymphoid enhancer factor |
| LRP | lipoprotein receptor-related protein |
| MAPK | mitogen activated protein kinase |
| M-CSF | macrophage colony-stimulating factor |
| MGP | matrix gla protein |
| OPG | osteoprotegerin |
| RANKL | nuclear factor-κB ligand |
| RUNX2 | runt related transcription factor |
| SOST | sclerostin |
| TCF | T-cell factor |
| TGF-β | transforming growth factor-β |
| VCAN | versican |
| VEC | vascular endothelial cells |
| VEGF | vascular endothelial growth factor |
| VIC | vascular interstitial cells |
| VSMC | vascular smooth muscle cell |
| WNT | wingless-related integration site |
| ΔN-CPE | splice variant of carboxypeptidase E |
References
- Amin, S. Mechanical Factors and Bone Health: Effects of Weightlessness and Neurologic Injury. Curr. Rheumatol. Rep. 2010, 12, 170–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robling, A.G.; Turner, C.H. Mechanical Signaling for Bone Modeling and Remodeling. Crit. Rev. Eukaryot. Gene Expr. 2009, 19, 319–338. [Google Scholar] [CrossRef] [Green Version]
- Giachelli, C.M. Ectopic Calcification: Gathering Hard Facts about Soft Tissue Mineralization. Am. J. Pathol. 1999, 154, 671–674. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular Calcification Mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegemann, J.P.; Nerem, R.M. Altered Response of Vascular Smooth Muscle Cells to Exogenous Biochemical Stimulation in Two- and Three-Dimensional Culture. Exp. Cell Res. 2003, 283, 146–155. [Google Scholar] [CrossRef]
- Kohn, J.C.; Zhou, D.W.; Bordeleau, F.; Zhou, A.L.; Mason, B.N.; Mitchell, M.J.; King, M.R.; Reinhart-King, C.A. Cooperative Effects of Matrix Stiffness and Fluid Shear Stress on Endothelial Cell Behavior. Biophys. J. 2015, 108, 471–478. [Google Scholar] [CrossRef] [Green Version]
- Soares, C.P.; Midlej, V.; de Oliveira, M.E.W.; Benchimol, M.; Costa, M.L.; Mermelstein, C. 2D and 3D-Organized Cardiac Cells Shows Differences in Cellular Morphology, Adhesion Junctions, Presence of Myofibrils and Protein Expression. PLoS ONE 2012, 7, e38147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoghue, P.S.; Sun, T.; Gadegaard, N.; Riehle, M.O.; Barnett, S.C. Development of a Novel 3D Culture System for Screening Features of a Complex Implantable Device for CNS Repair. Mol. Pharm. 2014, 11, 2143–2150. [Google Scholar] [CrossRef] [Green Version]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Montezano, A.C.; Zimmerman, D.; Yusuf, H.; Burger, D.; Chignalia, A.Z.; Wadhera, V.; Van Leeuwen, F.N.; Touyz, R.M. Vascular Smooth Muscle Cell Differentiation to an Osteogenic Phenotype Involves TRPM7 Modulation by Magnesium. Hypertension 2010, 56, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Demer, L.L.; Tintut, Y. Vascular Calcification: Pathobiology of a Multifaceted Disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef]
- Avogaro, A.; Fadini, G.P. Mechanisms of Ectopic Calcification: Implications for Diabetic Vasculopathy. Cardiovasc. Diagn. Ther. 2015, 5, 343–352. [Google Scholar] [CrossRef]
- Renna, N.F.; De Las Heras, N.; Miatello, R.M. Pathophysiology of Vascular Remodeling in Hypertension. Int. J. Hypertens. 2013. [Google Scholar] [CrossRef] [Green Version]
- Mantella, L.E.; Quan, A.; Verma, S. Variability in Vascular Smooth Muscle Cell Stretch-Induced Responses in 2D Culture. Vasc. Cell 2015, 7. [Google Scholar] [CrossRef] [Green Version]
- Feng, X. Chemical and Biochemical Basis of Cell-Bone Matrix Interaction in Health and Disease. Curr. Chem. Biol. 2009, 3, 189–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Liu, Y.; Xiao, Q.; Yao, Q.; Allen, M.; Wang, Y.; Gao, L.; Qi, Y.; Zhang, P. Pathological Cyclic Strain Promotes Proliferation of Vascular Smooth Muscle Cells via the ACTH/ERK/STAT3 Pathway. J. Cell. Biochem. 2018, 119, 8260–8270. [Google Scholar] [CrossRef] [PubMed]
- Schad, J.F.; Meltzer, K.R.; Hicks, M.R.; Beutler, D.S.; Cao, T.V.; Standley, P.R. Cyclic Strain Upregulates VEGF and Attenuates Proliferation of Vascular Smooth Muscle Cells. Vasc. Cell 2011, 3. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Oyajobi, B.O.; Harris, S.E.; Chen, D.; Tsao, C.; Deng, H.W.; Zhao, M. Wnt/β-Catenin Signaling Activates Bone Morphogenetic Protein 2 Expression in Osteoblasts. Bone 2013, 52, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Saidak, Z.; LeHenaff, C.; Azzi, S.; Marty, C.; Da Nascimento, S.; Sonnet, P.; Marie, P.J. Wnt/β-Catenin Signaling Mediates Osteoblast Differentiation Triggered by Peptide-Induced A5β1 Integrin Priming in Mesenchymal Skeletal Cells. J. Biol. Chem. 2015, 290, 6903–6912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.M.; Nakashima, A.; Nashimoto, M.; Yawaka, Y.; Tamura, M. Bone Morphogenetic Protein-2 Enhances Wnt/β-Catenin Signaling-Induced Osteoprotegerin Expression. Genes Cells 2009, 14, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.E.; Chen, T.; Leaf, E.M.; Speer, M.Y.; Giachelli, C.M. Runx2 Expression in Smooth Muscle Cells Is Required for Arterial Medial Calcification in Mice. Am. J. Pathol. 2015, 185, 1958–1969. [Google Scholar] [CrossRef]
- Lin, G.L.; Hankenson, K.D. Integration of BMP, Wnt, and Notch Signaling Pathways in Osteoblast Differentiation. J. Cell. Biochem. 2011, 112, 3491–3501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, G.F.; Bjerke, M.A.; DeSimone, D.W. Integrins and Cadherins Join Forces to Form Adhesive Networks. J. Cell Sci. 2011, 124, 1183–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Ochi, T.; Nakase, T.; Hirota, S.; Kitamura, Y.; Nomura, S.; Yasui, N. Mechanical Tension-Stress Induces Expression of Bone Morphogenetic Protein (BMP)-2 and BMP-4, but Not BMP-6, BMP-7, and GDF-5 MRNA, During Distraction Osteogenesis. J. Bone Miner. Res. 1999, 14, 1084–1095. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zu, Y.; Li, J.; Du, S.; Xu, Y.; Zhang, L.; Jiang, L.; Wang, Z.; Chien, S.; Yang, C. Extracellular Matrix Stiffness Dictates Wnt Expression through Integrin Pathway. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
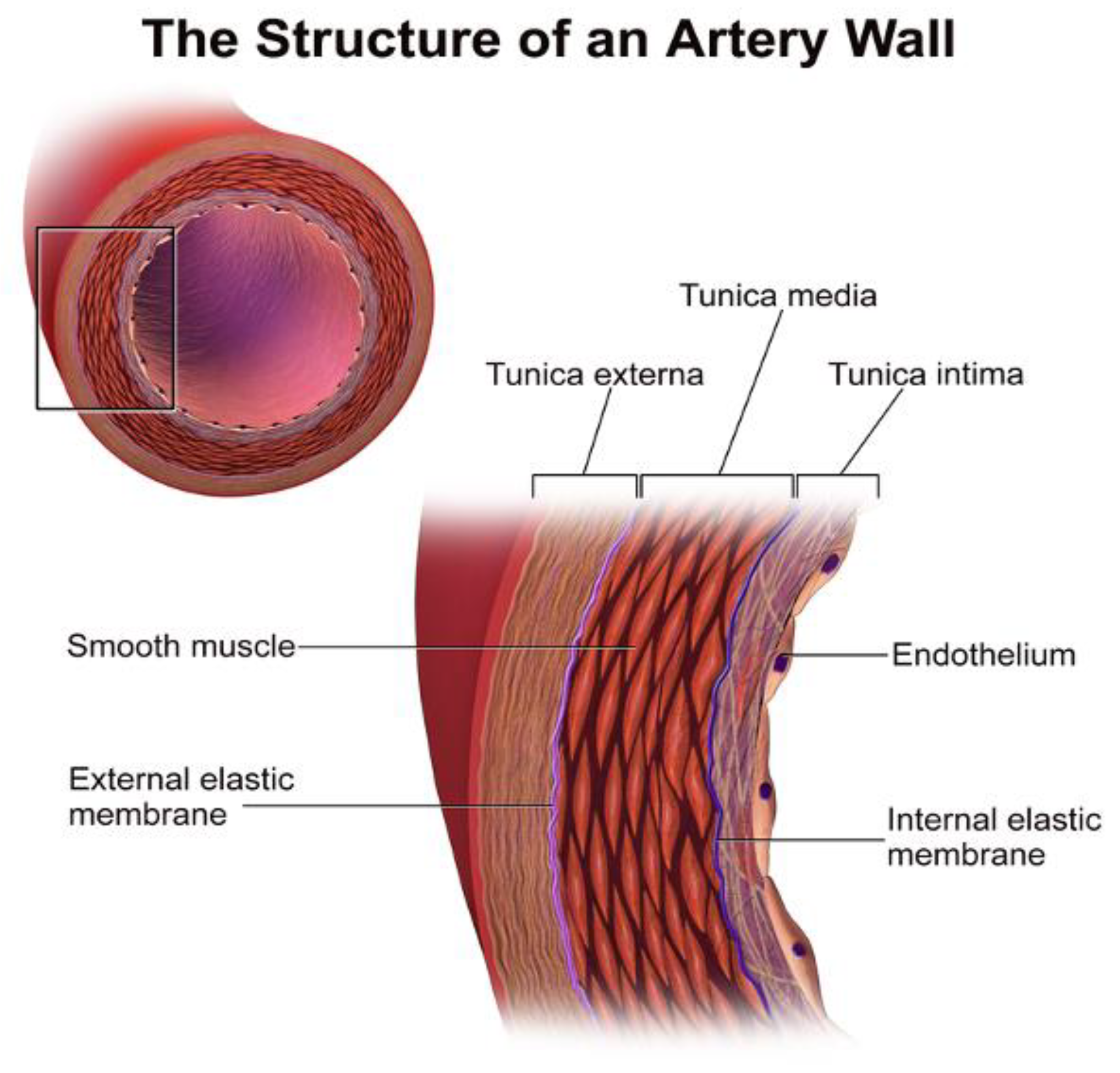
- Blood Vessel Structure and Function. Available online: https://courses.lumenlearning.com/boundless-ap/chapter/blood-vessel-structure-and-function/ (accessed on 26 June 2020).
- Essalihi, R.; Ouellette, V.; Dao, H.H.; McKee, M.D.; Moreau, P. Phenotypic Modulation of Vascular Smooth Muscle Cells during Medial Arterial Calcification: A Role for Endothelin? J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. 1), S147–S150. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.J.; Bourne, L.E.; Davies, B.K.; Arnett, T.R.; MacRae, V.E.; Wheeler-Jones, C.P.; Orriss, I.R. Differing Calcification Processes in Cultured Vascular Smooth Muscle Cells and Osteoblasts. Exp. Cell Res. 2019, 380, 100–113. [Google Scholar] [CrossRef]
- Giachelli, C.M. Vascular Calcification: In Vitro Evidence for the Role of Inorganic Phosphate. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 4), S300–S304. [Google Scholar] [CrossRef] [Green Version]
- Persy, V.; D’Haese, P. Vascular Calcification and Bone Disease: The Calcification Paradox. Trends Mol. Med. 2009, 15, 405–416. [Google Scholar] [CrossRef]
- Giachelli, C.M. The Emerging Role of Phosphate in Vascular Calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Schwarz-Romond, T.; Metcalfe, C.; Bienz, M. Dynamic Recruitment of Axin by Dishevelled Protein Assemblies. J. Cell Sci. 2007, 120, 2402–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamos, J.L.; Weis, W.I. The β-Catenin Destruction Complex. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, T.; Lengner, C.J.; Hovhannisyan, H.; Bhat, R.A.; Bodine, P.V.N.; Komm, B.S.; Javed, A.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; et al. Canonical WNT Signaling Promotes Osteogenesis by Directly Stimulating Runx2 Gene Expression. J. Biol. Chem. 2005, 280, 33132–33140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skalka, N.; Caspi, M.; Caspi, E.; Loh, Y.P.; Rosin-Arbesfeld, R. Carboxypeptidase E: A Negative Regulator of the Canonical Wnt Signaling Pathway. Oncogene 2013, 32, 2836–2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Y.; Zhang, Y. The Biomechanical Function of Arterial Elastin in Solutes. J. Biomech. Eng. 2012, 134, 071002. [Google Scholar] [CrossRef]
- Hampson, G.; Edwards, S.; Conroy, S.; Blake, G.M.; Fogelman, I.; Frost, M.L. The Relationship between Inhibitors of the Wnt Signalling Pathway (Dickkopf-1(DKK1) and Sclerostin), Bone Mineral Density, Vascular Calcification and Arterial Stiffness in Post-Menopausal Women. Bone 2013, 56, 42–47. [Google Scholar] [CrossRef]
- Mcarthur, K.M.; Kay, A.M.; Mosier, J.A.; Grant, J.N.; Stewart, J.A.; Simpson, C.L. Manipulating the Plasticity of Smooth Muscle Cells to Regulate Vascular Calcification. AIMS Cell Tissue Eng. 2017, 1, 165–179. [Google Scholar] [CrossRef]
- Rahmani, M.; Read, J.T.; Carthy, J.M.; McDonald, P.C.; Wong, B.W.; Esfandiarei, M.; Si, X.; Luo, Z.; Luo, H.; Rennie, P.S.; et al. Regulation of the Versican Promoter by the β-Catenin-T-Cell Factor Complex in Vascular Smooth Muscle Cells. J. Biol. Chem. 2005, 280, 13019–13028. [Google Scholar] [CrossRef] [Green Version]
- Wight, T.N.; Merrilees, M.J. Proteoglycans in Atherosclerosis and Restenosis: Key Roles for Versican. Circ. Res. 2004, 94, 1158–1167. [Google Scholar] [CrossRef] [Green Version]
- Sotoodehnejadnematalahi, F.; Burke, B. Structure, Function and Regulation of Versican: The Most Abundant Type of Proteoglycan in the Extracellular Matrix. Acta Med. Iran. 2013, 51, 740–750. [Google Scholar] [PubMed]
- Spencer, G.J.; Utting, J.C.; Etheridge, S.L.; Arnett, T.R.; Genever, P.G. Wnt Signalling in Osteoblasts Regulates Expression of the Receptor Activator of NFkappaB Ligand and Inhibits Osteoclastogenesis in Vitro. J. Cell Sci. 2006, 119, 1283–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradl, D.; Kuhl, M.; Wedlich, D. The Wnt/Wg Signal Transducer Beta-Catenin Controls Fibronectin Expression. Mol. Cell. Biol. 1999, 19, 5576–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, K.E.; Parhami, F.; Shin, V.; Demer, L.L. Fibronectin and Collagen I Matrixes Promote Calcification of Vascular Cells in Vitro, Whereas Collagen IV Matrix Is Inhibitory. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1964–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan, M.Q.; Tare, R.S.; Suk, H.L.; Mandeville, M.; Morasso, M.I.; Javed, A.; Van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. BMP2 Commitment to the Osteogenic Lineage Involves Activation of Runx2 by DLX3 and a Homeodomain Transcriptional Network. J. Biol. Chem. 2006, 281, 40515–40526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhaylova, L.; Malmquist, J.; Nurminskaya, M. Regulation of in Vitro Vascular Calcification by BMP4, VEGF and Wnt3a. Calcif. Tissue Int. 2007, 81, 372–381. [Google Scholar] [CrossRef]
- Dai, J.; Keller, J.; Zhang, J.; Lu, Y.; Yao, Z.; Keller, E.T. Bone Morphogenetic Protein-6 Promotes Osteoblastic Prostate Cancer Bone Metastases through a Dual Mechanism. Cancer Res. 2005, 65, 8274–8285. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.T.; Kang, D.I.; Ha, Y.S.; Jung, Y.S.; Chung, J.; Min, K.; Kim, T.H.; Moon, K.H.; Chung, J.M.; Lee, D.H.; et al. Prostate Cancer Bone Metastases Acquire Resistance to Androgen Deprivation via WNT5A-Mediated BMP-6 Induction. Br. J. Cancer 2014, 110. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.R.; Lund, R.J.; Hruska, K.A. BMP-7 Is an Efficacious Treatment of Vascular Calcification in a Murine Model of Atherosclerosis and Chronic Renal Failure. J. Am. Soc. Nephrol. 2003, 14, 1559–1567. [Google Scholar] [CrossRef] [Green Version]
- Godin, R.E.; Takaesu, N.T.; Robertson, E.J.; Dudley, A.T. Regulation of BMP7 Expression during Kidney Development. Development 1998, 125, 3473–3482. [Google Scholar]
- Morrell, N.W.; Bloch, D.B.; Ten Dijke, P.; Goumans, M.J.T.; Hata, A.; Smith, J.; Paul, B.Y.; Bloch, K.D. Targeting BMP Signalling in Cardiovascular Disease and Anaemia. Nat. Rev. Cardiol. 2016, 13, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Sakoda, S.; Shin, H.; Yamaji, K.; Takasaki, I.; Furuzono, T.; Kishida, A.; Akashi, M.; Kubo, T.; Nagaoka, E.; Maruyama, I.; et al. Mechanical Stretching of Human Osteoblast-like Cells Stimulates Bone Morphogenic Proteins and Macrophage Colony-Stimulating Factor Productions. Pathophysiology 1999, 6, 63–69. [Google Scholar] [CrossRef]
- Rui, Y.F.; Lui, P.P.Y.; Ni, M.; Chan, L.S.; Lee, Y.W.; Chan, K.M. Mechanical Loading Increased BMP-2 Expression Which Promoted Osteogenic Differentiation of Tendon-Derived Stem Cells. J. Orthop. Res. 2011, 29, 390–396. [Google Scholar] [CrossRef]
- Balachandran, K.; Sucosky, P.; Jo, H.; Yoganathan, A.P. Elevated Cyclic Stretch Induces Aortic Valve Calcification in a Bone Morphogenic Protein-Dependent Manner. Am. J. Pathol. 2010, 177, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Florencio-silva, R.; Sasso, G.; Sasso-cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viguet-Carrin, S.; Garnero, P.; Delmas, P.D. The Role of Collagen in Bone Strength. Osteoporos. Int. 2006, 17, 319–336. [Google Scholar] [CrossRef]
- Zhong, Z.; Zeng, X.L.; Ni, J.H.; Huang, X.F. Comparison of the Biological Response of Osteoblasts after Tension and Compression. Eur. J. Orthod. 2013, 35, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Ménard, A.L.; Grimard, G.; Londono, I.; Beaudry, F.; Vachon, P.; Moldovan, F.; Villemure, I. Bone Growth Resumption Following in Vivo Static and Dynamic Compression Removals on Rats. Bone 2015, 81, 662–668. [Google Scholar] [CrossRef]
- De Souza, R.L.; Matsuura, M.; Eckstein, F.; Rawlinson, S.C.F.; Lanyon, L.E.; Pitsillides, A.A. Non-Invasive Axial Loading of Mouse Tibiae Increases Cortical Bone Formation and Modifies Trabecular Organization: A New Model to Study Cortical and Cancellous Compartments in a Single Loaded Element. Bone 2005, 37, 810–818. [Google Scholar] [CrossRef]
- Takahashi, N.; Maeda, K.; Ishihara, A.; Uehara, S.; Kobayashi, Y. Regulatory Mechanism of Osteoclastogenesis by RANKL and Wnt Signals. Front. Biosci. 2011, 16, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Maeda, K.; Takahashi, N. Roles of Wnt Signaling in Bone Formation and Resorption. Jpn. Dent. Sci. Rev. 2008, 44, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Boyce, B.F.; Xing, L.; Chen, D. Osteoprotegerin, the Bone Protector, Is a Surprising Target for β-Catenin Signaling. Cell Metab. 2005, 2, 344–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spatz, J.M.; Wein, M.N.; Gooi, J.H.; Qu, Y.; Garr, J.L.; Liu, S.; Barry, K.J.; Uda, Y.; Lai, F.; Dedic, C.; et al. The Wnt Inhibitor Sclerostin Is Up-Regulated by Mechanical Unloading in Osteocytes in Vitro. J. Biol. Chem. 2015, 290, 16744–16758. [Google Scholar] [CrossRef] [Green Version]
- Duan, P.; Bonewald, L.F. The Role of the WNT/β-Catenin Signaling Pathway in Formation and Maintenance of Bone and Teeth. Int. J. Biochem. Cell Biol. 2016, 77, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Ndip, A.; Williams, A.; Jude, E.B.; Serracino-Inglott, F.; Richardson, S.; Smyth, J.V.; Boulton, A.J.M.; Alexander, M.Y. The RANKL/RANK/OPG Signaling Pathway Mediates Medial Arterial Calcification in Diabetic Charcot Neuroarthropathy. Diabetes 2011, 60, 2187–2196. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.Y. RANKL Links Arterial Calcification with Osteolysis. Circ. Res. 2009, 104, 1032–1034. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Shie, M.Y.; Hung, C.J.; Liu, S.L.; Huang, T.H.; Kao, C.T. Osteoblasts Subjected to Tensile Force Induce Osteoclastic Differentiation of Murine Macrophages in a Coculture System. J. Dent. Sci. 2015, 10, 81–87. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Hua, L.; Gao, L.J. A Computational Model for Biomechanical Effects of Arterial Compliance Mismatch. Appl. Bionics Biomech. 2015, 2015. [Google Scholar] [CrossRef]
- Thondapu, V.; Bourantas, C.V.; Foin, N.; Jang, I.K.; Serruys, P.W.; Barlis, P. Basic Science for the Clinician: Biomechanical Stress in Coronary Atherosclerosis: Emerging Insights from Computational Modelling. Eur. Heart J. 2017, 38, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, D.; Landau, S.; Shandalov, Y.; Raindel, N.; Freiman, A.; Shor, E.; Blinder, Y.; Vandenburgh, H.H.; Mooney, D.J.; Levenberg, S. Morphogenesis of 3D Vascular Networks Is Regulated by Tensile Forces. Proc. Natl. Acad. Sci. USA 2016, 113, 3215–3220. [Google Scholar] [CrossRef] [Green Version]
- Sharrett, A.; Hubbard, L. Retinal Arteriolar Diameters and Elevated Blood Pressure. Am. J. Epidemiol. 1999, 150, 263–270. [Google Scholar] [CrossRef]
- Jankowski, P.; Czarnecka, D. Pulse Pressure, Blood Flow, and Atherosclerosis. Am. J. Hypertens. 2012, 25, 1040–1041. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.A.; Shalhoub, J.; Lim, C.S.; Gohel, M.S.; Davies, A.H. The Effect of Pressure-Induced Mechanical Stretch on Vascular Wall Differential Gene Expression. J. Vasc. Res. 2012, 49, 463–478. [Google Scholar] [CrossRef]
- Kalra, S.S.; Shanahan, C.M. Vascular Calcification and Hypertension: Cause and Effect. Ann. Med. 2012, 44 (Suppl. 1), S85–S92. [Google Scholar] [CrossRef]
- Humphrey, J.D. Vascular Adaptation and Mechanical Homeostasis at Tissue, Cellular, and Sub-Cellular Levels. Cell Biochem. Biophys. 2008, 50, 53–78. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Tuzun, E.; Quick, C.M. Aortic Pulse Pressure Homeostasis Emerges from Physiological Adaptation of Systemic Arteries to Local Mechanical Stresses. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R522–R531. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.D.; Tian, Y.; Morrisey, E.E. Wnt Signaling: An Essential Regulator of Cardiovascular Differentiation, Morphogenesis and Progenitor Self-Renewal. Development 2008, 135, 789–798. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Sun, D.; Duan, Y.; Wen, P.; Dai, C.; Yang, J.; He, W.; Cohen, E.D.; Tian, Y.; Morrisey, E.E.; et al. WNT/β-Catenin Signaling Promotes VSMCs to Osteogenic Transdifferentiation and Calcification through Directly Modulating Runx2 Gene Expression. Exp. Cell Res. 2016, 345, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Shu, W.; Lu, M.M.; Morrisey, E.E. Wnt7b Activates Canonical Signaling in Epithelial and Vascular Smooth Muscle Cells through Interactions with Fzd1, Fzd10, and LRP5. Mol. Cell. Biol. 2005, 25, 5022–5030. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Qu, M.-J.; Qin, K.-R.; Li, H.; Li, Z.-K.; Shen, B.-R.; Jiang, Z.-L. Role of Cyclic Strain Frequency in Regulating the Alignment of Vascular Smooth Muscle Cells In Vitro. Biophys. J. 2008, 94, 1497–1507. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Hitomi, H.; Hosomi, N.; Lei, B.; Pelisch, N.; Nakano, D.; Kiyomoto, H.; Ma, H.; Nishiyama, A. Mechanical Stretch Potentiates Angiotensin II-Induced Proliferation in Spontaneously Hypertensive Rat Vascular Smooth Muscle Cells. Hypertens. Res. 2010, 33, 1250–1257. [Google Scholar] [CrossRef] [Green Version]
- Herencia, C.; Rodríguez-Ortiz, M.E.; Muñoz-Castañeda, J.R.; Martinez-Moreno, J.M.; Canalejo, R.; Montes de Oca, A.; Díaz-Tocados, J.M.; Peralbo-Santaella, E.; Marín, C.; Canalejo, A.; et al. Angiotensin II Prevents Calcification in Vascular Smooth Muscle Cells by Enhancing Magnesium Influx. Eur. J. Clin. Investig. 2015, 45, 1129–1144. [Google Scholar] [CrossRef]
- McCain, M.L.; Sheehy, S.P.; Grosberg, A.; Goss, J.A.; Parker, K.K. Recapitulating Maladaptive, Multiscale Remodeling of Failing Myocardium on a Chip. Proc. Natl. Acad. Sci. USA 2013, 110, 9770–9775. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Malashicheva, A.; Sullivan, G.; Bogdanova, M.; Kostareva, A.; Stensløkken, K.-O.; Fiane, A.; Vaage, J. Valve Interstitial Cells: The Key to Understanding the Pathophysiology of Heart Valve Calcification. J. Am. Heart Assoc. 2017, 6, e006339. [Google Scholar] [CrossRef]
- Shapero, K.; Wylie-Sears, J.; Levine, R.A.; Mayer, J.E.; Bischoff, J. Reciprocal Interactions between Mitral Valve Endothelial and Interstitial Cells Reduce Endothelial-to-Mesenchymal Transition and Myofibroblastic Activation. J. Mol. Cell. Cardiol. 2015, 80, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Hjortnaes, J.; Shapero, K.; Goettsch, C.; Hutcheson, J.D.; Keegan, J.; Kluin, J.; Mayer, J.E.; Bischoff, J.; Aikawa, E. Valvular Interstitial Cells Suppress Calcification of Valvular Endothelial Cells. Atherosclerosis 2015, 242, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Gomel, M.A.; Lee, R.; Grande-Allen, K.J. Comparing the Role of Mechanical Forces in Vascular and Valvular Calcification Progression. Front. Cardiovasc. Med. 2018, 5, 197. [Google Scholar] [CrossRef]
- Weinberg, E.J.; Mack, P.J.; Schoen, F.J.; García-Cardeña, G.; Kaazempur Mofrad, M.R. Hemodynamic Environments from Opposing Sides of Human Aortic Valve Leaflets Evoke Distinct Endothelial Phenotypes in Vitro. Cardiovasc. Eng. 2010, 10, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Song, S.; Lee, J.; Yoon, J.; Park, J.; Choi, S.; Park, J.K.; Choi, K.; Choi, C. Phenotypic Modulation of Primary Vascular Smooth Muscle Cells by Short-Term Culture on Micropatterned Substrate. PLoS ONE 2014, 9, e88089. [Google Scholar] [CrossRef] [PubMed]
- Osol, G. Mechanotransduction by Vascular Smooth Muscle. J. Vasc. Res. 1995, 32, 275–292. [Google Scholar] [CrossRef]
- Handorf, A.M.; Zhou, Y.; Halanski, M.A.; Li, W.J. Tissue Stiffness Dictates Development, Homeostasis, and Disease Progression. Organogenesis 2015, 11, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilhan, F. Atherosclerosis and the Role of Immune Cells. World J. Clin. Cases 2015, 3, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Steffens, S.; Mach, F. The Immune Response Is Involved in Atherosclerotic Plaque Calcification: Could the RANKL/RANK/OPG System Be a Marker of Plaque Instability? Clin. Dev. Immunol. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Cheng, W.; Huang, T.; Yuan, J.; Wang, X.; Song, M. Vascular Adventitia Calcification and Its Underlying Mechanism. PLoS ONE 2015, 10, e0132506. [Google Scholar] [CrossRef]
- Xu, J.; Shi, G.-P. Vascular Wall Extracellular Matrix Proteins and Vascular Diseases. Biochim. Biophys. Acta 2014, 1842, 2106–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moe, S.M.; O’Neill, K.D.; Duan, D.; Ahmed, S.; Chen, N.X.; Leapman, S.B.; Fineberg, N.; Kopecky, K. Medial Artery Calcification in ESRD Patients Is Associated with Deposition of Bone Matrix Proteins. Kidney Int. 2002, 61, 638–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ma, K.L.; Gao, M.; Wang, C.X.; Ni, J.; Zhang, Y.; Zhang, X.L.; Liu, H.; Wang, Y.L.; Liu, B.C. Inflammation Disrupts the LDL Receptor Pathway and Accelerates the Progression of Vascular Calcification in ESRD Patients. PLoS ONE 2012, 7, e47217. [Google Scholar] [CrossRef] [Green Version]
- Rekhter, M.D. Collagen Synthesis in Atherosclerosis: Too Much and Not Enough. Cardiovasc. Res. 1999, 41, 376–384. [Google Scholar] [CrossRef]
- Kuzan, A.; Chwiłkowska, A.; Pezowicz, C.; Witkiewicz, W.; Gamian, A.; Maksymowicz, K.; Kobielarz, M. The Content of Collagen Type II in Human Arteries Is Correlated with the Stage of Atherosclerosis and Calcification Foci. Cardiovasc. Pathol. 2017, 28, 21–27. [Google Scholar] [CrossRef]
- Huttenlocher, A.; Horwitz, A.R. Integrins in Cell Migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef]
- Crampton, S.P.; Wu, B.; Park, E.J.; Kim, J.H.; Solomon, C.; Waterman, M.L.; Hughes, C.C.W. Integration of the β-Catenin-Dependent Wnt Pathway with Integrin Signaling through the Adaptor Molecule Grb2. PLoS ONE 2009, 4, e7841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Wang, J.; Jiang, H.; Hu, Q.; Chen, J.; Zhang, J.; Zhu, R.; Liu, W.; Li, B. Wnt3a Activates Β1-Integrin and Regulates Migration and Adhesion of Vascular Smooth Muscle Cells. Mol. Med. Rep. 2014, 9, 1159–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puklin-Faucher, E.; Sheetz, M.P. The Mechanical Integrin Cycle. J. Cell Sci. 2009, 122, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Matthews, B.D. Cellular Adaptation to Mechanical Stress: Role of Integrins, Rho, Cytoskeletal Tension and Mechanosensitive Ion Channels. J. Cell Sci. 2006, 119, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.; Sudhir, K.; Ives, H.E. Mechanical Strain of Rat Vascular Smooth Muscle Cells Is Sensed by Specific Extracellular Matrix/Integrin Interactions. J. Clin. Investig. 1995, 96, 2364–2372. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, D.; Versaevel, M.; Bruyère, C.; Alaimo, L.; Luciano, M.; Vercruysse, E.; Procès, A.; Gabriele, S. Innovative Tools for Mechanobiology: Unraveling Outside-In and Inside-Out Mechanotransduction. Front. Bioeng. Biotechnol. 2019, 7, 162. [Google Scholar] [CrossRef]
- Tsai, J.; Kam, L. Rigidity-Dependent Cross Talk between Integrin and Cadherin Signaling. Biophys. J. 2009, 96, L39–L41. [Google Scholar] [CrossRef] [Green Version]
- Maître, J.L.; Heisenberg, C.P. Three Functions of Cadherins in Cell Adhesion. Curr. Biol. 2013, 23, R626–R633. [Google Scholar] [CrossRef] [Green Version]
- Hartsock, A.; Nelson, W.J. Adherens and Tight Junctions: Structure, Function and Connections to the Actin Cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Kam, Y.; Quaranta, V. Cadherin-Bound β-Catenin Feeds into the Wnt Pathway upon Adherens Junctions Dissociation: Evidence for an Intersection between β-Catenin Pools. PLoS ONE 2009, 4, e4580. [Google Scholar] [CrossRef]
- Leckband, D.E.; de Rooij, J. Cadherin Adhesion and Mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef]
- Mui, K.L.; Chen, C.S.; Assoian, R.K. The Mechanical Regulation of Integrin-Cadherin Crosstalk Organizes Cells, Signaling and Forces. J. Cell Sci. 2016, 129, 1093–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marie, P.J.; Haÿ, E.; Saidak, Z. Integrin and Cadherin Signaling in Bone: Role and Potential Therapeutic Targets. Trends Endocrinol. Metab. 2014, 25, 567–575. [Google Scholar] [CrossRef]
- Sawala, A.; Scarcia, M.; Sutcliffe, C.; Wilcockson, S.G.; Ashe, H.L. Peak BMP Responses in the Drosophila Embryo Are Dependent on the Activation of Integrin Signaling. Cell Rep. 2015, 12, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.U.L.I.; Lecanda, F.; Davidson, M.K.; Warlow, P.M.; Zhang, S.F.; Zhang, L.; Suzuki, S.; John, T.S.T.; Civitelli, R. Human Osteoblasts Express a Repertoire of Cadherins, Which Are Critical for BMP-2-Induced Osteogenic Differentiation. J. Bone Miner. Res. 1998, 13, 633–644. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis Formation on Atherosclerotic Lesions and Plaque Rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Calcifying Matrix Vesicles and Atherosclerosis. BioMed Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef] [PubMed]
- Snell-Bergeon, J.K.; Budoff, M.J.; Hokanson, J.E. Vascular Calcification in Diabetes: Mechanisms and Implications. Curr. Diab. Rep. 2013, 13, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X. Pathophysiology of Vascular Calcification in Chronic Kidney Disease. Circ. Res. 2004, 95, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Regents of the University of California. A Vascular & Endovascular Surgery-Atherosclerosis. Available online: https://vascularsurgery.ucsf.edu/conditions--procedures/atherosclerosis.aspx (accessed on 26 June 2020).
- Yao, Y.; Jumabay, M.; Ly, A.; Radparvar, M.; Cubberly, M.R.; Boström, K.I. A Role for the Endothelium in Vascular Calcification. Circ. Res. 2013, 113, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.O.; Zhang, J.; Jiang, Z.; Yin, K. Endothelial-to-Mesenchymal Transition: A Novel Therapeutic Target for Cardiovascular Diseases. Trends Cardiovasc. Med. 2017, 27, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Boström, K.I.; Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Yao, Y. Endothelial-Mesenchymal Transition in Atherosclerotic Lesion Calcification. Atherosclerosis 2016, 253, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Gong, H.; Lyu, X.; Wang, Q.; Hu, M.; Zhang, X. Endothelial to Mesenchymal Transition in the Cardiovascular System. Life Sci. 2017, 184, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Peng, W.; Xia, X.; Li, R.; Wang, Y.; Wei, D. Endothelial-to-Mesenchymal Transition: A Potential Mechanism for Atherosclerosis Plaque Progression and Destabilization. DNA Cell Biol. 2017, 36, 883–891. [Google Scholar] [CrossRef]
- Mosier, J.; Nguyen, N.; Parker, K.; Simpson, C. Calcification of Biomaterials and Diseased States; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Kiousis, D.E.; Gasser, T.C.; Holzapfel, G.A. A Numerical Model to Study the Interaction of Vascular Stents with Human Atherosclerotic Lesions. Ann. Biomed. Eng. 2007, 35, 1857–1869. [Google Scholar] [CrossRef]
- Yang, J.H.; Briggs, W.H.; Libby, P.; Lee, R.T. Small Mechanical Strains Selectively Suppress Matrix Metalloproteinase-1 Expression by Human Vascular Smooth Muscle Cells. J. Biol. Chem. 1998, 273, 6550–6555. [Google Scholar] [CrossRef] [Green Version]
- Wight, T.N.; Kinsella, M.G.; Evanko, S.P.; Potter-Perigo, S.; Merrilees, M.J. Versican and the Regulation of Cell Phenotype in Disease. Biochim. Biophys. Acta 2014, 1840, 2441–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulari, M.T.K.; Qu, Q.; Härkönen, P.L.; Väänänen, H.K. Osteoblast-like Cells Complete Osteoclastic Bone Resorption and Form New Mineralized Bone Matrix in Vitro. Calcif. Tissue Int. 2004, 75, 253–261. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inducers | Inhibitors |
|---|---|
| N-carboxypeptidase E | F-Carboxypeptidase E |
| Hyperphosphatemia | Sclerostin |
| Hypercalcemia | MGP |
| BMP-2 and BMP-4 | OPG |
| RUNX2 | Collagen type IV |
| Injury and stress |
| Gene | Effect |
|---|---|
| RUNX2 | osteogenic differentiation |
| RANKL | Recruitment of osteoblast-like cell precursors |
| OPG | regulates bone turnover, blocks RANKL |
| VCAN | cell proliferation and migration |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tyson, J.; Bundy, K.; Roach, C.; Douglas, H.; Ventura, V.; Segars, M.F.; Schwartz, O.; Simpson, C.L. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering 2020, 7, 88. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7030088
Tyson J, Bundy K, Roach C, Douglas H, Ventura V, Segars MF, Schwartz O, Simpson CL. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering. 2020; 7(3):88. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7030088
Chicago/Turabian StyleTyson, John, Kaylee Bundy, Cameron Roach, Hannah Douglas, Valerie Ventura, Mary Frances Segars, Olivia Schwartz, and C. LaShan Simpson. 2020. "Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT" Bioengineering 7, no. 3: 88. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering7030088