Stabilization of Poly (β-Amino Ester) Nanoparticles for the Efficient Intracellular Delivery of PiggyBac Transposon





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PEG-PDHA
2.3. PEG-PDHA Characterization
2.4. Plasmid Encapsulation with PEG-PDHA
2.5. Plasmid Encapsulation with Layer-by-Layer
2.6. Plasmid Encapsulation and Crosslinking
2.7. Plasmid Release Kinetics
2.8. PEG-PDHA NPs Cellular Uptake
2.9. In Vitro Transfection Efficiency
2.10. PEG-PDHA NP Toxicity
3. Results
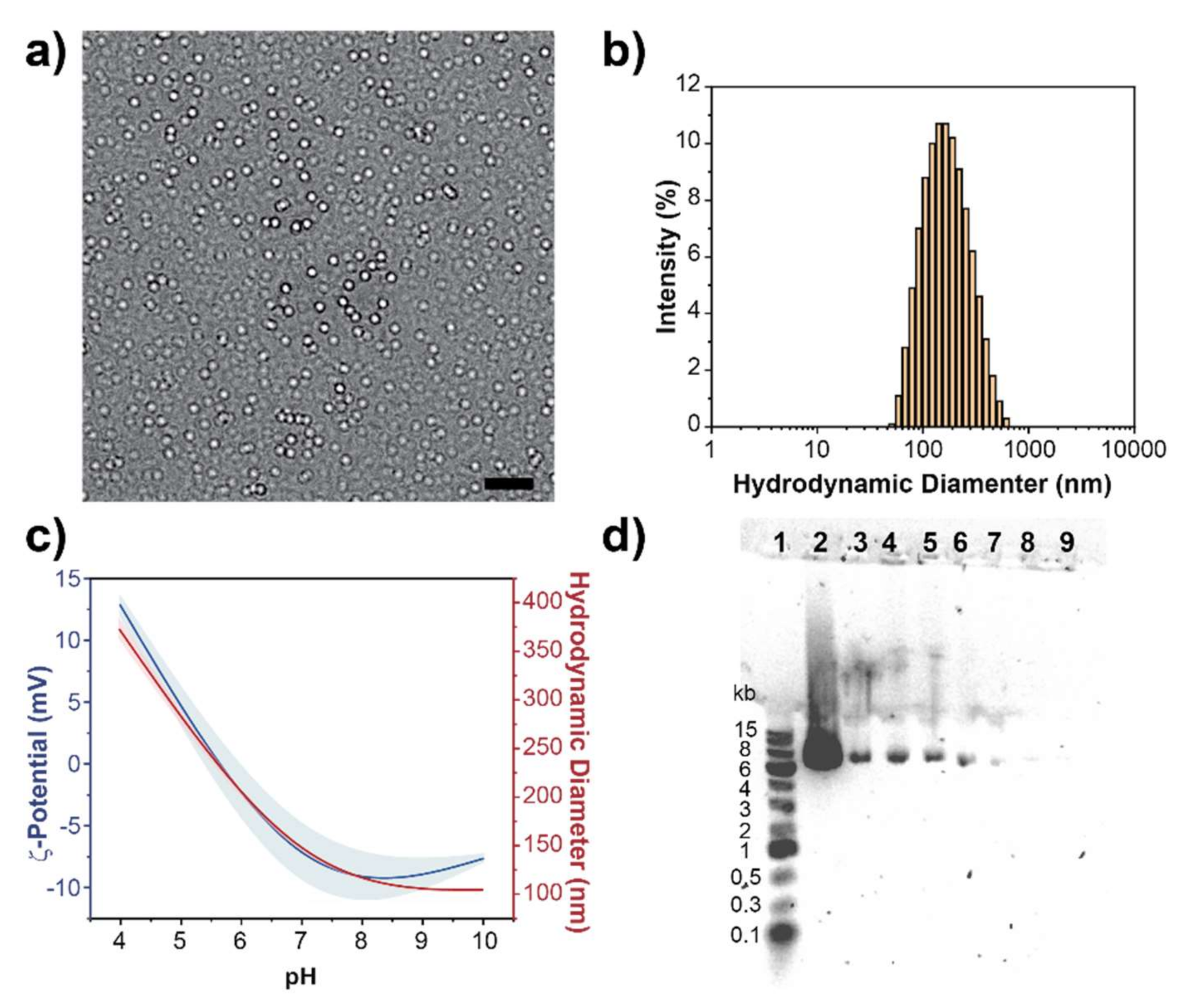
3.1. Synthesis and Characterization of PEG-PDHA
3.2. Fabrication of PEG-PDHA NPs
3.3. Plasmid Encapsulation into PEG-PDHA NPs
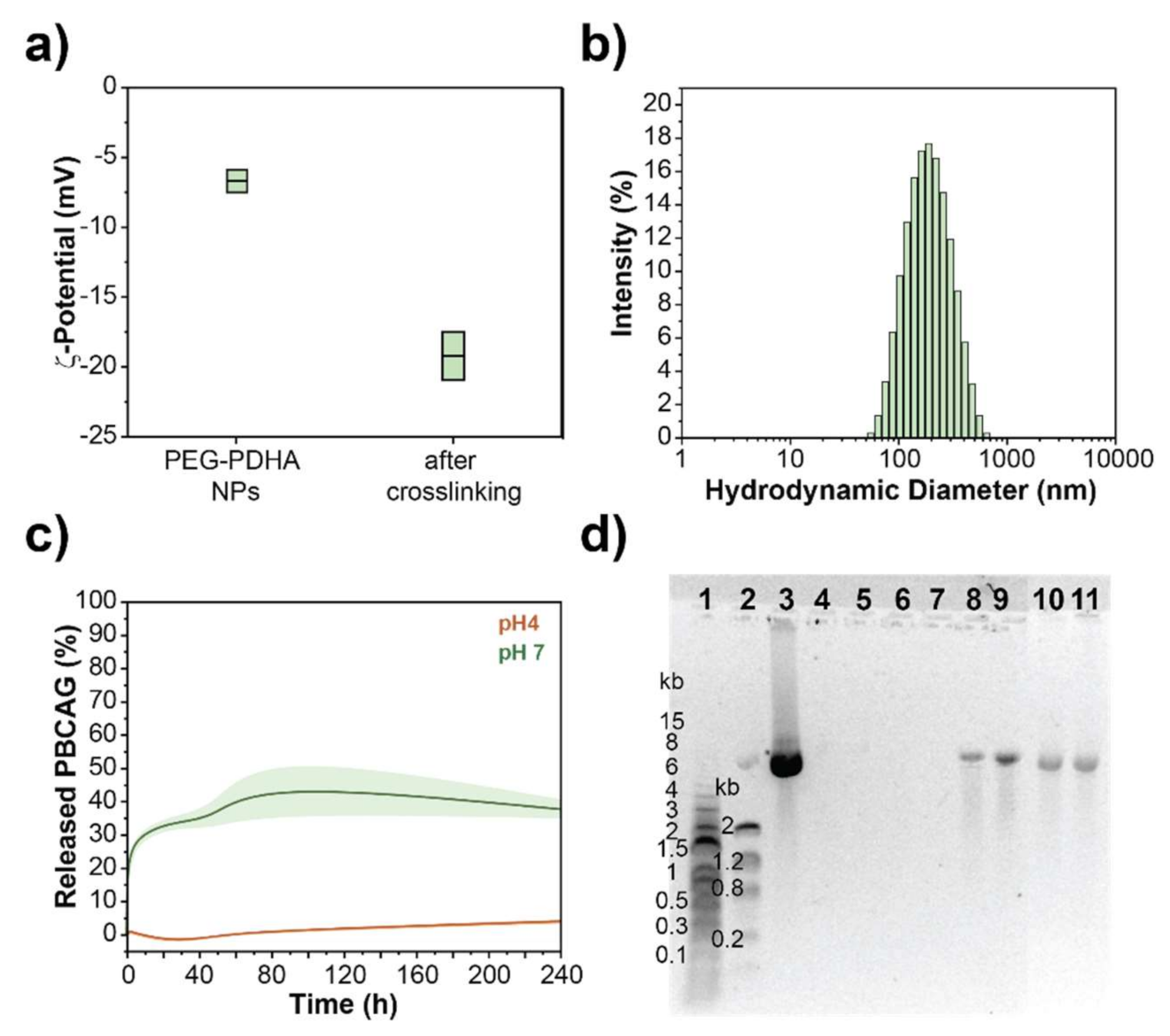
3.4. PBCAG Release Kinetics from PEG-PDHA NPs
3.4.1. PEG-PDHA NPs Stabilization by Layer-by-Layer Surface Engineering
3.4.2. PEG-PDHA NPs Stabilization by Crosslinking
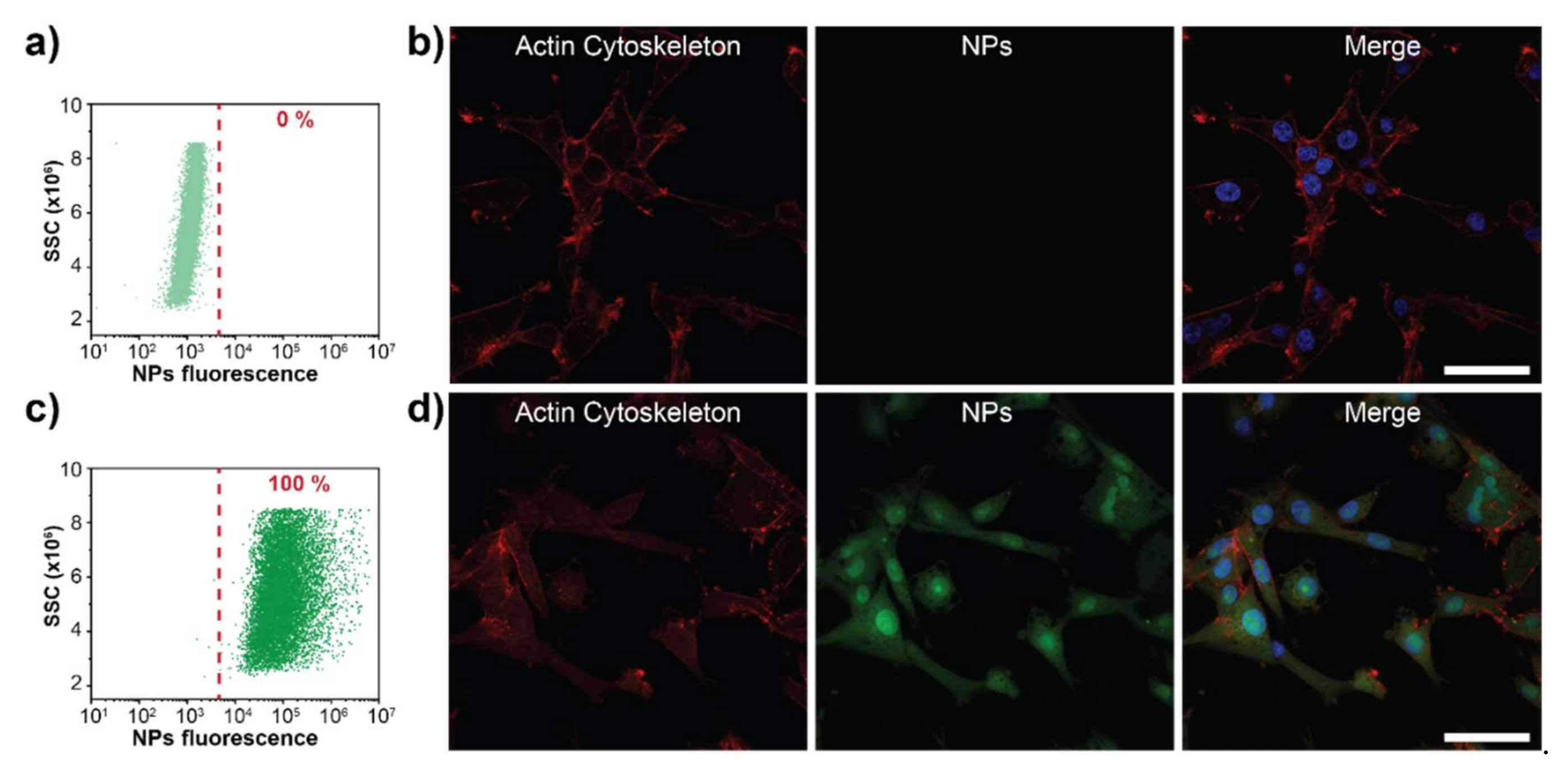
3.5. PEG-PDHA NPs Cellular Uptake
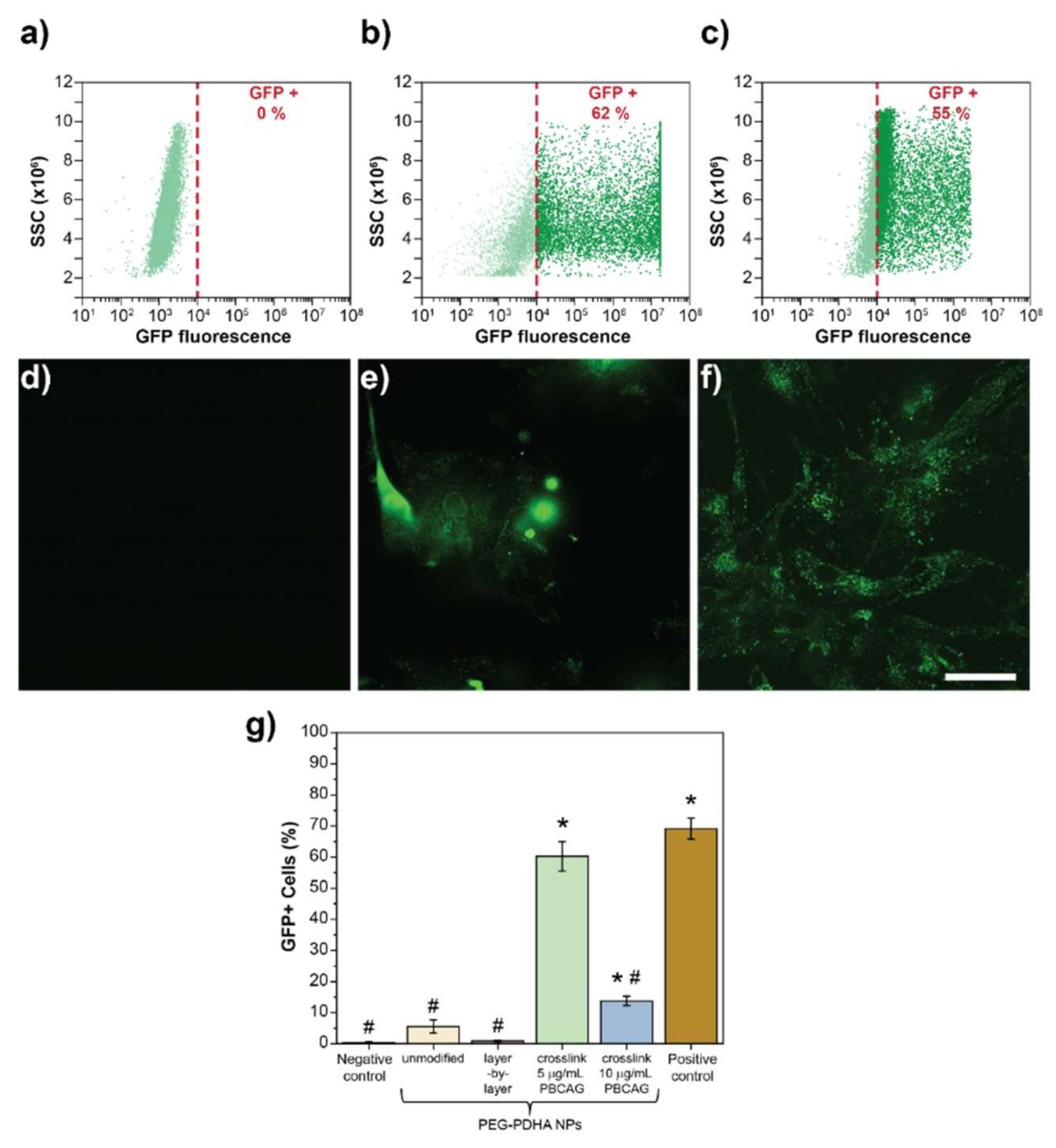
3.6. In Vitro Transfection Efficiency of PEG-PDHA NPs
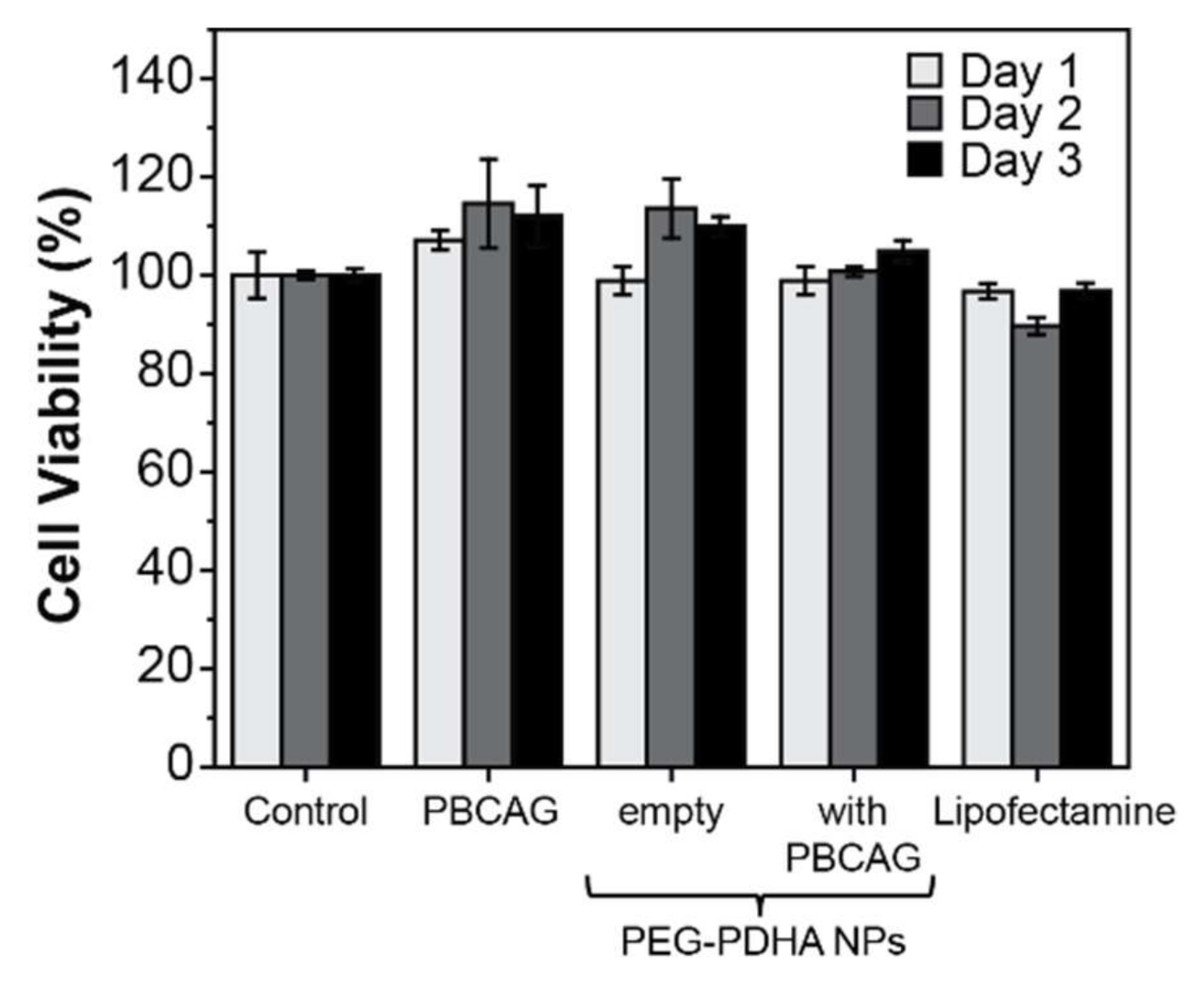
3.7. PEG-PDHA NPs Cytotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Wu, S.-S.; Li, Q.-C.; Yin, C.-Q.; Xue, W.; Song, C.-Q. Advances in CRISPR/Cas-based gene therapy in human genetic diseases. Theranostics 2020, 10, 4374–4382. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Hu, W.; Khalili, K. The CRISPR/Cas9 genome editing methodology as a weapon against human viruses. Discov. Med. 2015, 19, 255–262. [Google Scholar] [PubMed]
- Aubert, M.; Strongin, D.E.; Roychoudhury, P.; Loprieno, M.A.; Haick, A.K.; Klouser, L.M.; Stensland, L.; Huang, M.-L.; Makhsous, N.; Tait, A.; et al. Gene editing and elimination of latent herpes simplex virus in vivo. Nat. Commun. 2020, 11, 4148. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Troike, K.; Lathia, J.D. Optimising gene editing for cancer therapy. Nat. Cell Biol. 2020, 22, 259–261. [Google Scholar] [CrossRef]
- Miri, S.M.; Tafsiri, E.; Cho, W.C.S.; Ghaemi, A. CRISPR-Cas, a robust gene-editing technology in the era of modern cancer immunotherapy. Cancer Cell Int. 2020, 20, 456. [Google Scholar] [CrossRef]
- Azangou-Khyavy, M.; Ghasemi, M.; Khanali, J.; Boroomand-Saboor, M.; Jamalkhah, M.; Soleimani, M.; Kiani, J. CRISPR/Cas: From Tumor Gene Editing to T Cell-Based Immunotherapy of Cancer. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Wittrup, A.; Lieberman, J. Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet. 2015, 16, 543–552. [Google Scholar] [CrossRef]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef]
- Paschon, D.E.; Lussier, S.; Wangzor, T.; Xia, D.F.; Li, P.W.; Hinkley, S.J.; Scarlott, N.A.; Lam, S.C.; Waite, A.J.; Truong, L.N.; et al. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun. 2019, 10, 1133. [Google Scholar] [CrossRef] [Green Version]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug Ii, R.G.; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.H.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, R.; Berges, B.K.; Solis-Leal, A.; Igbinedion, O.; Strong, C.L.; Schiller, M.R. TALEN gene editing takes aim on HIV. Hum. Genet. 2016, 135, 1059–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlotorynski, E. CRISPR–Cas in its prime. Nat. Rev. Mol. Cell Biol. 2019, 20, 718–719. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Burnight, E.R.; Cooney, A.L.; Malani, N.; Brady, T.; Sander, J.D.; Staber, J.; Wheelan, S.J.; Joung, J.K.; McCray, P.B.; et al. piggyBac transposase tools for genome engineering. Proc. Natl. Acad. Sci. USA 2013, 110, E2279–E2287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Bus, T.; Traeger, A.; Schubert, U.S. The great escape: How cationic polyplexes overcome the endosomal barrier. J. Mater. Chem. B 2018, 6, 6904–6918. [Google Scholar] [CrossRef]
- Park, T.G.; Jeong, J.H.; Kim, S.W. Current status of polymeric gene delivery systems. Adv. Drug Deliv. Rev. 2006, 58, 467–486. [Google Scholar] [CrossRef]
- Pouton, C.W.; Seymour, L.W. Key issues in non-viral gene delivery1PII of original article: S0169-409X(98)00048-9. The article was originally published in Advanced Drug Delivery Reviews 34 (1998) 3–19.1. Adv. Drug Deliv. Rev. 2001, 46, 187–203. [Google Scholar] [CrossRef]
- Caffery, B.; Lee, J.S.; Alexander-Bryant, A.A. Vectors for glioblastoma gene therapy: Viral & non-viral delivery strategies. Nanomaterials 2019, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.D.; Ghivizzani, S.C. Viral vectors for gene therapy. Pharmacol. Ther. 1998, 80, 35–47. [Google Scholar] [CrossRef]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Loo, J.C.M.; Wright, J.F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 2016, 25, R42–R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Liu, F.; Chen, Y.; Liu, J.; Wang, X.; Chen, A.T.; Deng, G.; Zhang, H.; Liu, J.; Hong, Z.; et al. Targeted delivery of CRISPR/Cas9-mediated cancer gene therapy via liposome-templated hydrogel nanoparticles. Adv. Funct. Mater. 2017, 27, 1703036. [Google Scholar] [CrossRef]
- Zylberberg, C.; Gaskill, K.; Pasley, S.; Matosevic, S. Engineering liposomal nanoparticles for targeted gene therapy. Gene Ther. 2017, 24, 441. [Google Scholar] [CrossRef]
- Rohovie, M.J.; Nagasawa, M.; Swartz, J.R. Virus-like particles: Next-generation nanoparticles for targeted therapeutic delivery. Bioeng. Transl. Med. 2017, 2, 43–57. [Google Scholar] [CrossRef]
- Xiang, Y.; Oo, N.N.L.; Lee, J.P.; Li, Z.; Loh, X.J. Recent development of synthetic nonviral systems for sustained gene delivery. Drug Discov. Today 2017, 22, 1318–1335. [Google Scholar] [CrossRef] [PubMed]
- Romero, G.; Ochoteco, O.; Sanz, D.J.; Estrela-Lopis, I.; Donath, E.; Moya, S.E. Poly (Lactide-co-Glycolide) nanoparticles, layer by layer engineered for the sustainable delivery of AntiTNF-α. Macromol. Biosci. 2013, 13, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Ghoroghchian, P.P.; ROMERO, U.G.; Ostertag, E. Poly(histidine)-Based Micelles For Complexation And Delivery of Proteins and Nucleic Acids. U.S. Patent WO2017190091 A1, 8 March 2017. [Google Scholar]
- Shim, G.; Kim, D.; Le, Q.-V.; Park, G.T.; Kwon, T.; Oh, Y.-K. Nonviral delivery systems for cancer gene therapy: Strategies and challenges. Curr. Gene Ther. 2018, 18, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Dowaidar, M.; Abdelhamid, H.N.; Hällbrink, M.; Freimann, K.; Kurrikoff, K.; Zou, X.; Langel, Ü. Magnetic nanoparticle assisted self-assembly of cell penetrating peptides-oligonucleotides complexes for gene delivery. Sci. Rep. 2017, 7, 9159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina-Kauwe, L.K.; Xie, J.; Hamm-Alvarez, S. Intracellular trafficking of nonviral vectors. Gene Ther. 2005, 12, 1734–1751. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Maeder, M.L.; Joung, J.K.; Chen, Z.-Y.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.W.; Kim, J.D.; Park, K. Polycation gene delivery systems: Escape from endosomes to cytosol. J. Pharm. Pharmacol. 2003, 55, 721–734. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Yin, Q.; Zhang, Z.; Gu, W.; Chen, L.; Yu, H.; Huang, Y.; Chen, X.; Xu, M.; Li, Y. Co-delivery of doxorubicin and RNA using pH-sensitive poly (beta-amino ester) nanoparticles for reversal of multidrug resistance of breast cancer. Biomaterials 2014, 35, 6047–6059. [Google Scholar] [CrossRef]
- Green, J.J.; Langer, R.; Anderson, D.G. A combinatorial polymer library approach yields insight into nonviral gene delivery. Acc. Chem. Res. 2008, 41, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Y.; Keskin, D.; Shi, L. Poly(β-Amino Esters): Synthesis, formulations, and their biomedical applications. Adv. Healthc. Mater. 2019, 8, 1801359. [Google Scholar] [CrossRef]
- Swamy, T.; Raviteja, P.; Subba Reddy, B.V.; Ravinder, V. Efficient method for the synthesis of benzamides from benzoic acids and aryl isothiocyanates using K2HPO4. ChemistrySelect 2017, 2, 7612–7614. [Google Scholar] [CrossRef]
- Tang, S.; Meng, Q.; Sun, H.; Su, J.; Yin, Q.; Zhang, Z.; Yu, H.; Chen, L.; Chen, Y.; Gu, W.; et al. Tumor-microenvironment-adaptive nanoparticles codeliver paclitaxel and siRNA to inhibit growth and lung metastasis of breast cancer. Adv. Funct. Mater. 2016, 26, 6033–6046. [Google Scholar] [CrossRef]
- Tang, S.; Yin, Q.; Su, J.; Sun, H.; Meng, Q.; Chen, Y.; Chen, L.; Huang, Y.; Gu, W.; Xu, M.; et al. Inhibition of metastasis and growth of breast cancer by pH-sensitive poly (β-amino ester) nanoparticles co-delivering two siRNA and paclitaxel. Biomaterials 2015, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shamul, J.G.; Kang, Y.; Kim, J.; Green, J.J. Poly (beta-amino ester)-poly (ethylene glycol) micelles for intracellular therapeutic drug delivery. Front. Bioeng. Biotech. 2016. [Google Scholar] [CrossRef]
- Li, W.; Sun, J.; Zhang, X.; Jia, L.; Qiao, M.; Zhao, X.; Hu, H.; Chen, D.; Wang, Y. Synthesis and characterization of pH-responsive PEG-Poly(β-Amino Ester) block copolymer micelles as drug carriers to eliminate cancer stem cells. Pharmaceutics 2020, 12, 111. [Google Scholar] [CrossRef] [Green Version]
- San Juan, A.M.T.; Rodgers, T.; Bedolla, C.; Noriega, F.; Romero, G. Layer by layer surface engineering of poly(lactide-co-glycolide) nanoparticles for plasmid DNA delivery. J. Appl. Polym. Sci. 2020, 137, 49377. [Google Scholar] [CrossRef]
- Mangraviti, A.; Tzeng, S.Y.; Kozielski, K.L.; Wang, Y.; Jin, Y.; Gullotti, D.; Pedone, M.; Buaron, N.; Liu, A.; Wilson, D.R.; et al. Polymeric nanoparticles for nonviral gene therapy extend brain tumor survival in vivo. ACS Nano 2015, 9, 1236–1249. [Google Scholar] [CrossRef] [Green Version]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Morille, M.; Passirani, C.; Vonarbourg, A.; Clavreul, A.; Benoit, J.-P. Progress in developing cationic vectors for non-viral systemic gene therapy against cancer. Biomaterials 2008, 29, 3477–3496. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, K.; Yen, S.K.; Dou, Q.; Padmanabhan, P.; Sudhaharan, T.; Ahmed, S.; Ying, J.Y.; Selvan, S.T. Mimicking cellular transport mechanism in stem cells through endosomal escape of new peptide-coated quantum dots. Sci. Rep. 2013, 3, 2184. [Google Scholar] [CrossRef] [Green Version]
- Mislick, K.A.; Baldeschwieler, J.D. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc. Natl. Acad. Sci. USA 1996, 93, 12349–12354. [Google Scholar] [CrossRef] [Green Version]
- Nayerossadat, N.; Maedeh, T.; Ali, P.A. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodgers, T.; Muzzio, N.; Watson, C.; Romero, G. Stabilization of Poly (β-Amino Ester) Nanoparticles for the Efficient Intracellular Delivery of PiggyBac Transposon. Bioengineering 2021, 8, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8020016
Rodgers T, Muzzio N, Watson C, Romero G. Stabilization of Poly (β-Amino Ester) Nanoparticles for the Efficient Intracellular Delivery of PiggyBac Transposon. Bioengineering. 2021; 8(2):16. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8020016
Chicago/Turabian StyleRodgers, Tina, Nicolas Muzzio, Caleb Watson, and Gabriela Romero. 2021. "Stabilization of Poly (β-Amino Ester) Nanoparticles for the Efficient Intracellular Delivery of PiggyBac Transposon" Bioengineering 8, no. 2: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8020016