
Kinetic Analysis of Lidocaine Elimination by Pig Liver Cells Cultured in 3D Multi-Compartment Hollow Fiber Membrane Network Perfusion Bioreactors

 , and
, and
Abstract
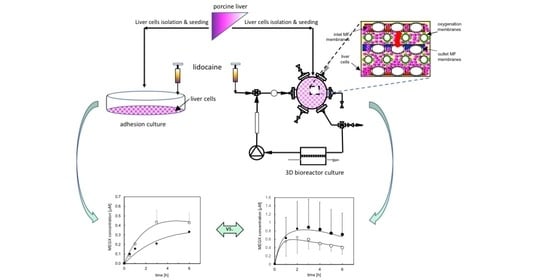
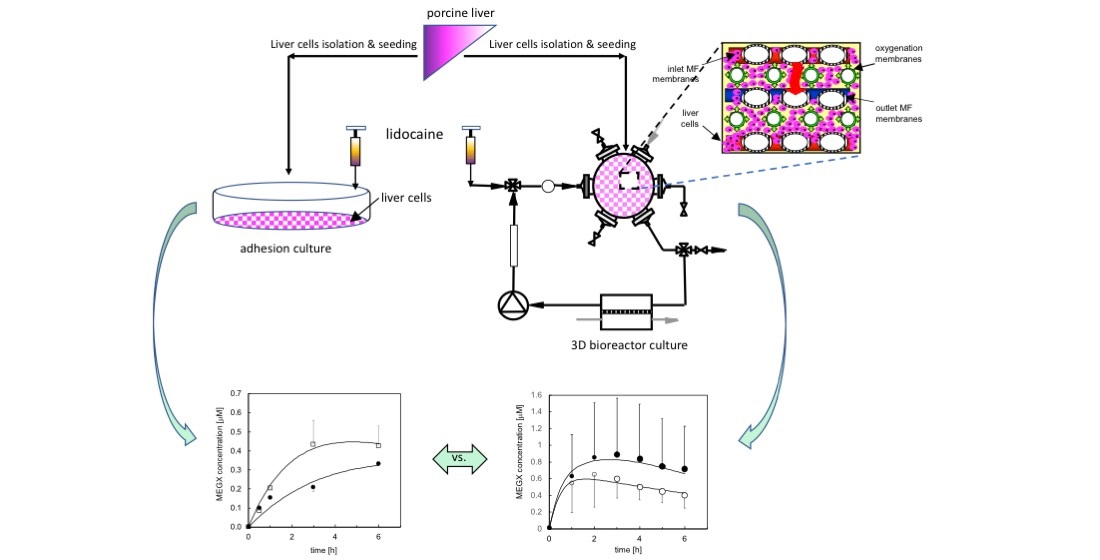
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Lidocaine Adsorption Tests
2.3. Cell Isolation and Culture
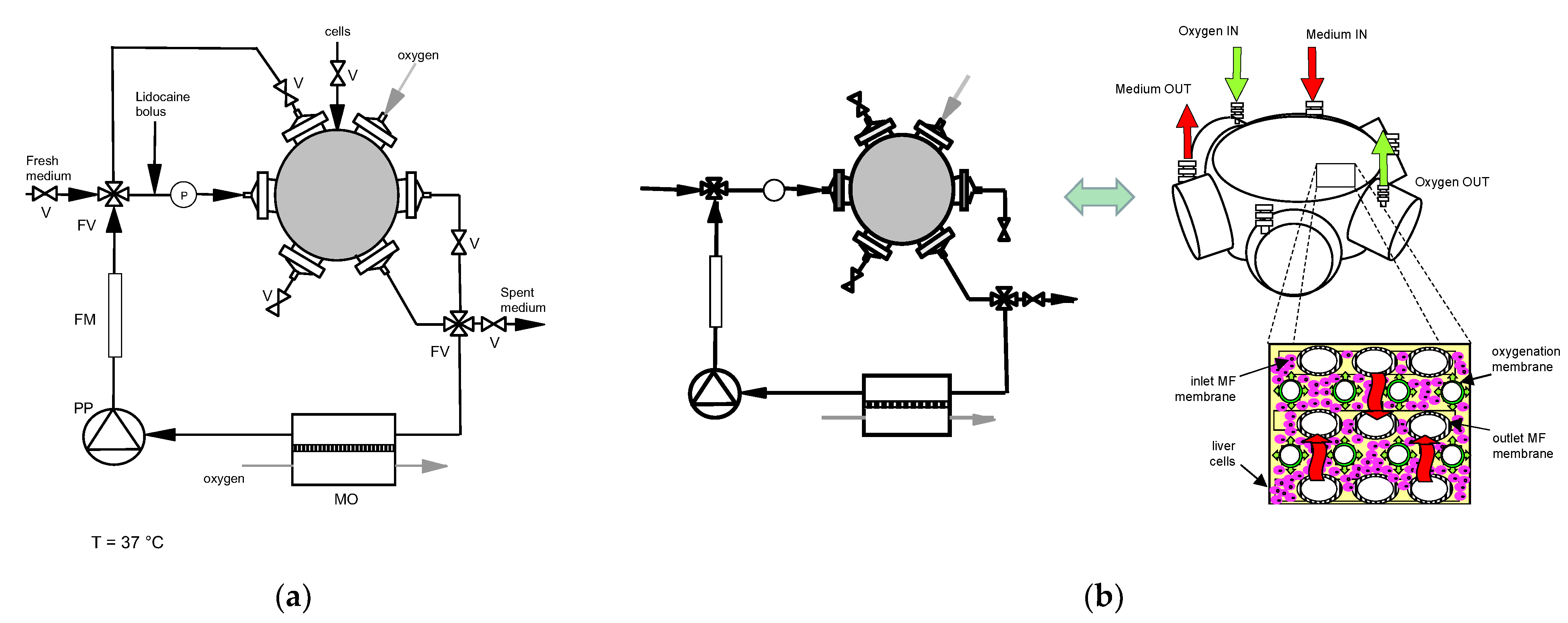
2.3.1. Adhesion Culture
2.3.2. Three-Dimensional Bioreactor Culture
2.4. Kinetic Tests
2.5. Analytical Methods
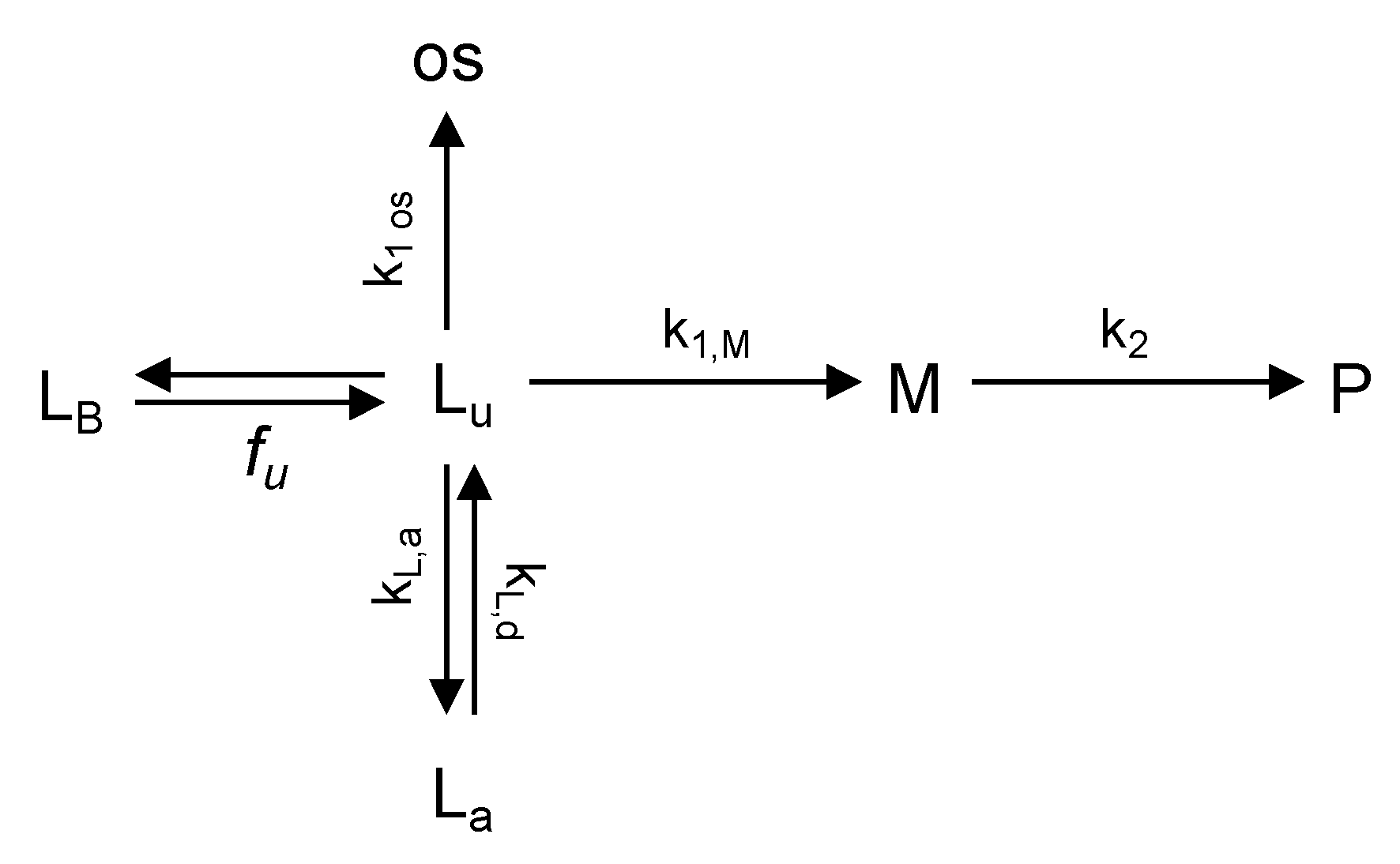
2.6. Data Analysis
3. Results
3.1. Lidocaine Adsorption in Cell-Free Bioreactors
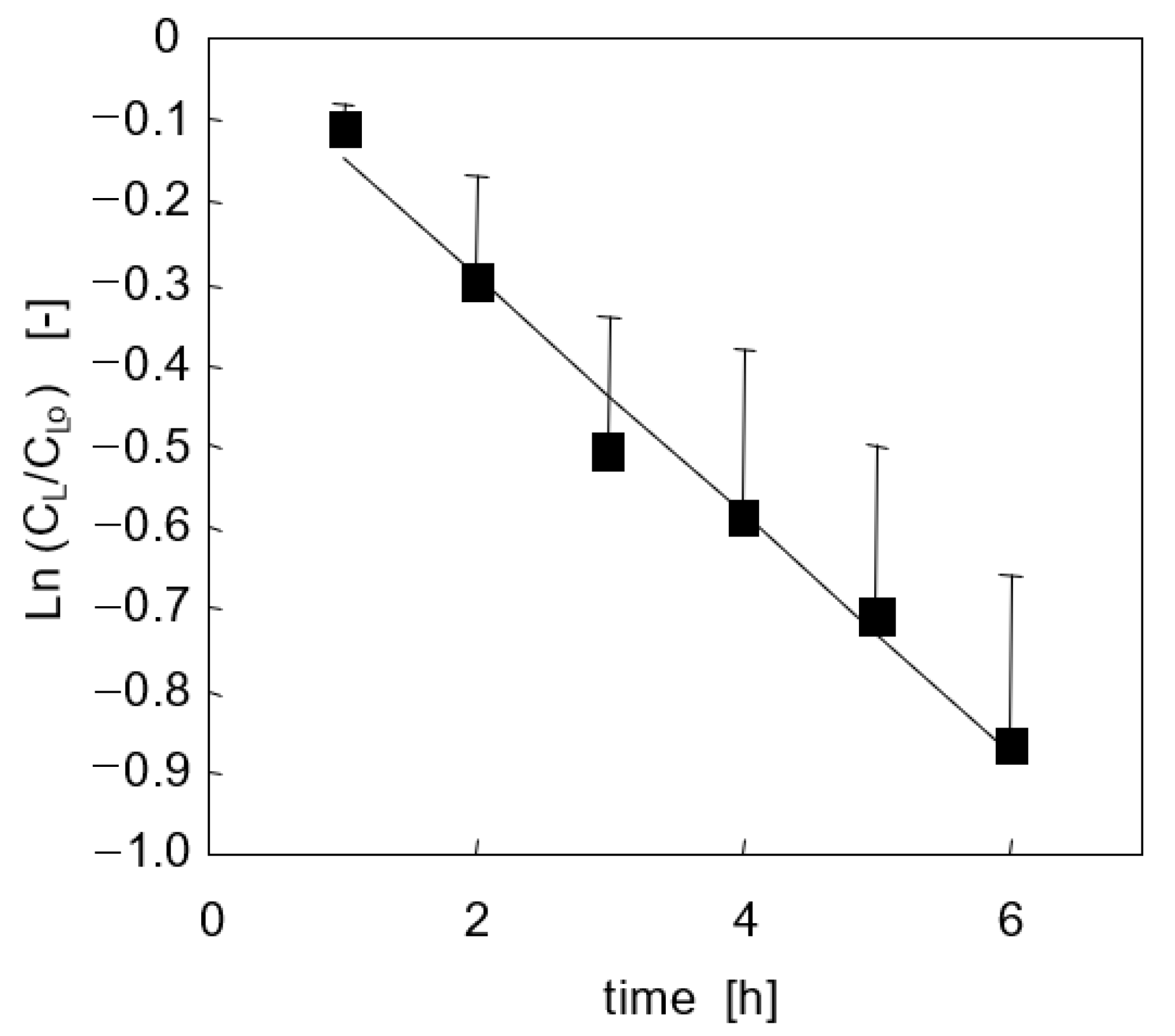
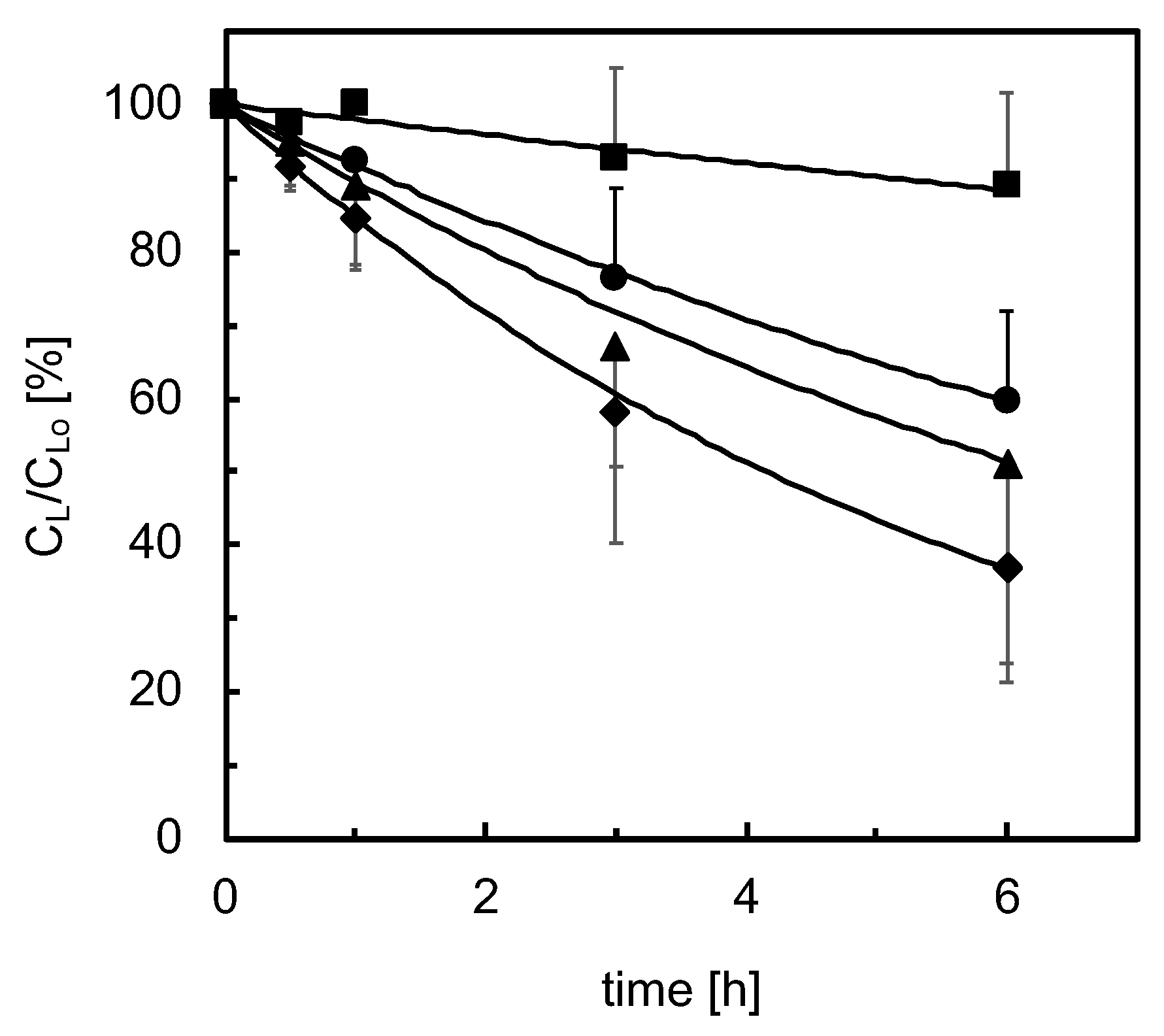
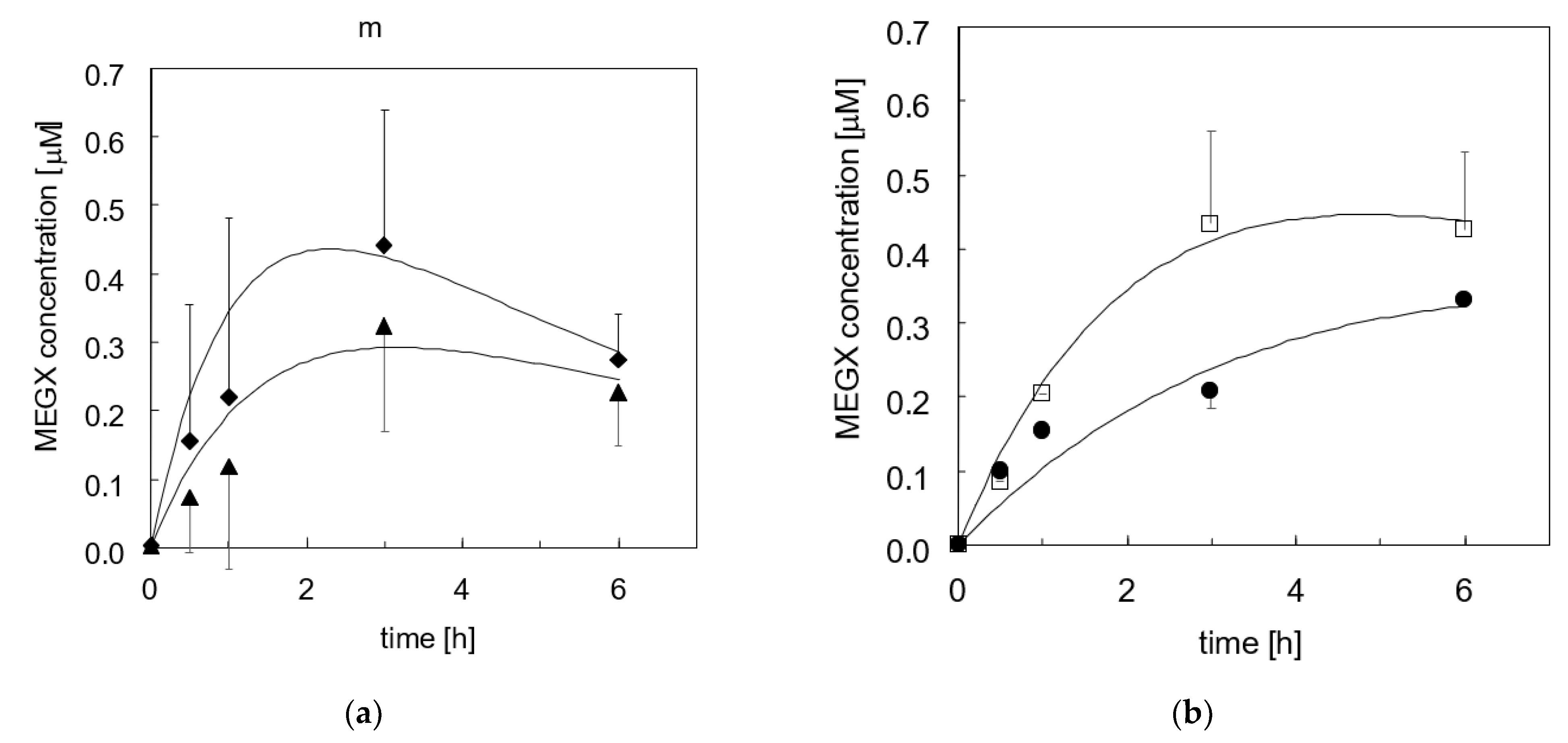
3.2. Lidocaine Disappearance in Cell-Seeded Bioreactors
3.2.1. Adhesion Culture
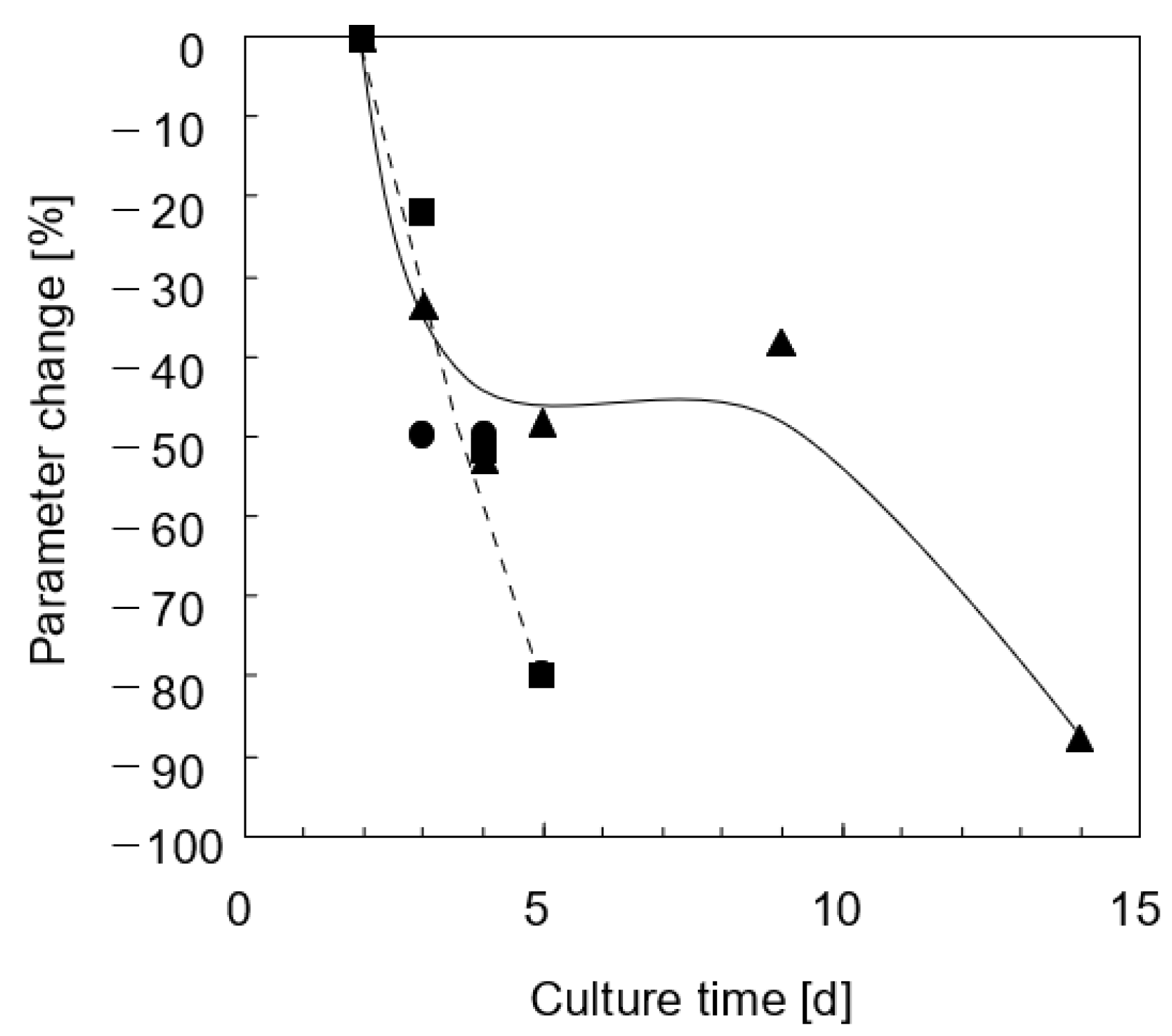
3.2.2. Three-Dimensional Bioreactor Culture
4. Discussion and Conclusions
4.1. Lidocaine Adsorption
4.2. Lidocaine Metabolic Elimination
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| C | solute concentration, M |
| Ccell | cell density, cell/cm2 |
| CL,a | lidocaine concentration in the adsorbed phase, moles/cm2 |
| CO2,eq | dissolved oxygen concentration in medium equilibrating the above gas, M |
| Damkoehler number, | |
| DO2 | oxygen diffusion coefficient in medium, cm2/s |
| fu | unbound lidocaine fraction, |
| h | medium thickness above cell surface in adhesion culture, cm |
| ki | kinetic constant for the i-th transformation, s−1 |
| ki’ = ki/Ccell | cell-specific kinetic constant for the i-th transformation, cm3/(s ncell) |
| KM | Michaelis constant, M |
| m | number of experiments, |
| n | number of data points, |
| p | number of model parameters, |
| -r | metabolite disappearance rate, M/s |
| Subscripts/Superscripts | |
| a | adsorption |
| d | desorption |
| exp | experimental |
| i | i-th metabolite |
| j | j-th data point |
| k | k-th experiment |
| mod | model-predicted |
| os | species other than MEGX produced by lidocaine metabolic transformation |
| A | adhesion culture |
| B | bioreactor culture |
| L | lidocaine |
| M | MEGX |
| O2 | dissolved oxygen |
| 0 | initial value |
| 1 | lidocaine metabolic transformation |
| 2 | MEGX further metabolic transformation |
| α | reaction rate order |
References
- Lautt, W. Hepatic Circulation: Physiology and Pathophysiology. In Colloquium Series on Integrated Systems Physiology: From Molecule to Function; Morgan & Claypool Life Sciences: Williston, VT, USA, 2009. [Google Scholar]
- La Moncloa. World Transplant Registry Reports 135,860 Transplants Performed Worldwide Last Year, up 7.2% (29 August 2018). Available online: https://www.lamoncloa.gob.es/lang/en/gobierno/news/Paginas/2018/20180829transplants.aspx (accessed on 10 June 2021).
- EMA, E.M.A. Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes text with EEA relevance. Off. J. Eur. Union 2010, 53, 33–79. [Google Scholar]
- Anzenbacher, P.; Soucek, P.; Anzenbacherova, E.; Gut, I.; Svoboda, Z.; Kvetina, J. Presence and and activity of cytochrome P450 isoforms in minipig liver microsomes. Drug Metab. Dispos. 1998, 26, 56–59. [Google Scholar]
- Soucek, P.; Zuber, R.; Anzenbacherova, E.; Anzenbach, P.; Guengerich, F.P. Minipig cytochrome P450 3A, 2A and 2C enzymes have similar properties to human analogs. BMC Pharmacol. 2001, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Selden, C.; Fuller, B. Role of bioreactor technology in tissue engineering for clinical use and therapeutic target design. Bioengineering 2018, 5, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimkhani, M.R.; Neiman, J.; Raredon, M.; Hughes, D.J.; Griffith, L. Bioreactor technologies to support liver function in vitro. Adv. Drug Deliv. Rev. 2014, 69, 132–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauer, I.M.; Kardassis, D.; Zeillinger, K.; Pascher, A.; Gruenwald, A.; Pless, G.; Irgang, M.; Kraemer, M.; Puhl, G.; Frank, J.; et al. Clinical extracorporeal hybrid liver support—Phase I study with primary porcine liver cells. Xenotransplantation 2003, 10, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Agoram, B.; Woltosz, W.S.; Bolger, M. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 2001, 50, S41–S67. [Google Scholar] [CrossRef]
- Nibourg, G.A.; Boer, J.D.; van der Hoeven, T.V.; Ackermans, M.T.; van Gulik, T.M.; Chamuleau, R.A.; Hoekstra, R. Perfusion flow rate substantially contributes to the performance of the HepaRG-AMC-bioartificial liver. Biotechnol. Bioeng. 2012, 109, 3182–3188. [Google Scholar] [CrossRef]
- Soman, S.; Yoo, M.J.; Jang, Y.J.; Hage, D.S. Analysis of Lidocaine Interactions with Serum Proteins Using High-Performance Affinity Chromatography. J. Chromatogr. B 2010, 878, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Unger, J.K.; Kuehlein, G.; Schroers, A.; Gerlach, J.C.; Rossaint, R. Adsorption of xenobiotics to plastic tubing incorporated into dynamic in vitro systems used in pharmacological research—Limits and progress. Biomaterials 2001, 22, 2031–2037. [Google Scholar] [CrossRef]
- Gerlach, J.C.; Encke, J.; Hole, O.; Mueller, C.; Ryan, C.J.; Neuhaus, P. Bioreactor for a larger scale hepatocyte in vitro perfusion. Transplantation 1994, 58, 984–988. [Google Scholar] [CrossRef]
- Gerlach, J.; Brombacher, J.; Smith, M.; Neuhaus, P. High yield hepatocyte isolation from pig livers for investigation of hybrid liver support systems: Influence of collagenase concentration and body weight. J. Surg. Res. 1996, 62, 85–89. [Google Scholar] [CrossRef]
- Gerlach, J.C.; Botsch, M.; Kardassis, D.; Lemmens, P.; Schön, M.; Janke, J.; Puhl, G.; Unger, J.; Kraemer, M.; Busse, B.; et al. Experimental Evaluation of a cell module for hybrid liver support. Int. J. Artif. Organs 2001, 24, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. The carbon monoxide-binding pigment of liver microsomes II. Solubilization, purification, and properties. J. Biol. Chem. 1964, 239, 2379–2385. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [CrossRef]
- Unger, J.K.; Catapano, G.; Horn, N.A.; Schroers, A.; Gerlach, J.C.; Rossaint, R. Comparative analysis of metabolism of medium- and plasma perfused primary pig hepatocytes cultured around a 3-D membrane network. Int. J. Artif. Organs 2000, 23, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Routledge, P.A.; Barchowsky, A.; Bjornsson, T.D.; Kitchell, B.B.; Shand, D.G. Lidocaine plasma protein binding. Clin. Pharmacol. Ther. 1980, 27, 347–351. [Google Scholar] [CrossRef]
- Fogler, H. Elements of Chemical Reaction Engineering, 4th ed; Pearson Education, Inc.: Upper Saddle River, NJ, USA, 2006. [Google Scholar]
- Banks, H.; Joyner, M. AIC under the Framework of Least Squares Estimation. Appl. Math. Lett. 2017, 74, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.T.; Castell, J.V.; Gomez-Lechon, M. Characterization of drug metabolizing activities in pig hepatocytes for use in bioartificial liver devices: Comparison with other hepatic cellular models. J. Hepatol. 1999, 31, 542–549. [Google Scholar] [CrossRef]
- Myers, M.J.; Farrell, D.E.; Howard, K.D.; Kawalek, J. Identification of multiple constitutive and inducible hepatic cytochrome P450 enzymes in market weight swine. Drug Metab. Dispos. 2001, 29, 908–915. [Google Scholar]
- Gonzalez, G. Human cytochromes P450: Problems ans prospects. Trends Pharmacol. Sci. 1992, 13, 346–352. [Google Scholar] [CrossRef]
- Riordan, S.M.; Skouteris, G.G.; Williams, R. Metabolic activity and clinical efficacy of animal and human hepatocytes in bioartificial support for acute liver failure. Int. J. Artif. Organs 1998, 21, 312–318. [Google Scholar] [CrossRef]
- Desille, M.; Cocos, L.; L’Helgoualch, A.; Fremond, B.; Campion, J.P.; Guillouzo, A. Detoxifying activity in pig livers and hepatocytes intended for xenotherapy. Transplantation 1999, 68, 1437–1443. [Google Scholar] [CrossRef]
- Jurima-Romet, M.; Casley, W.L.; Leblanc, C.A.; Nowakowska, M. Evidence for the catalysis of dextromethorphan O-demethylation by a CYP2D6-like enzyme in pig liver. Toxicol. In Vitro 2000, 14, 253–263. [Google Scholar] [CrossRef]
- Gerlach, J.; Unger, J.K.; Hole, O.; Encke, J.; Müller, C.; Neuhaus, P. Bioreactor for long-term maintenance of differentiated hepatic cell functions. Altex 1994, 11, 207–215. [Google Scholar] [PubMed]
- Lazar, A.; Mann, H.J.; Remmel, R.P.; Shatford, R.A.; Cerra, F.B.; Hu, W. Extended liver-specific functions of porcine hepatocyte spheroids entrapped in collagen gel. In Vitro Cell. Dev. Biol. Anim. 1994, 31, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Sielaff, T.D.; Hu, M.Y.; Rao, S.; Groehler, K.; Olson, D.; Mann, H.J.; Remmel, R.P.; Shatford, R.A.; Amiot, B.; Hu, W.S.; et al. A technique for porcine hepatocyte harvest and description of differentiated metabolic functions in static culture. Transplantation 1995, 59, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Behnia, K.; Bhatia, S.; Jastromb, N.; Balis, U.; Sullivan, S.; Yarmush, M.; Toner, M. Xenobiotic metabolism by cultured primary porcine hepatocytes. Tissue Eng. 2000, 6, 467–479. [Google Scholar] [CrossRef]
- Koebe, H.G.; Schildberg, F. Isolation of porcine hepatocytes from slaughterhouse organs. Int. J. Artif. Organs 1996, 19, 53–60. [Google Scholar] [CrossRef]
- Witkamp, R.F.; Monshouwer, M. Pharmacokinetics in vivo and in vitro in swine. Scand. J. Lab. Anim. Sci. 1998, 25, 45–56. [Google Scholar]
- Hosagrahara, V.; Hansen, L.; Remmel, R. Induction of the metabolism of midazolam by rifampin in cultured porcine hepatocytes: Preliminary evidence for CYP3A isoforms in pigs. Drug Metab. Dispos. 1999, 27, 1512–1518. [Google Scholar]
- Gerlach, J.; Zeilinger, K.; Sauer, I.M.; Mieder, T.; Nauman, G.; Grünwald, A.; Pless, G.; Holland, G.; Mas, A.; Vienken, J.; et al. Extracorporeal liver support: Porcine or human cell based systems? Int. J. Artif. Organs 2002, 25, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Rozga, J.; Williams, F.; Ro, M.S.; Neuzil, D.F.; Giorgio, T.D.; Backfisch, G.; Moscioni, A.D.; Hakim, R.; Demetriou, A.A. Development of a bioartificial liver: Properties and function of a hollow-fiber module inoculated with liver cells. Hepatology 1993, 17, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.; Knop, E.; Boeker, K.; Fruehauf, N.; Schuettler, W.; Oldhafer, K.; Burkhard, R.; Pichlmayr, R.; Sewing, K.F. A novel bioreactor design for in vitro reconstruction of in vivo liver characteristics. Artif. Organs 1995, 19, 368–374. [Google Scholar] [CrossRef]
- Flendrig, L.M.; la Soe, J.W.; Jorning, G.G.; Steenbeek, A.; Karlsen, O.T.; Bovee, W.M.; Ladiges, N.C.; te Velde, A.A.; Chamuleau, R.A. In vitro evaluation of a novel bioreactor based on an integral oxygenator and a spirally wound nonwoven polyester matrix for hepatocyte culture as small aggregates. J. Hepatol. 1997, 26, 1379–1392. [Google Scholar] [CrossRef] [Green Version]
- Zeilinger, K.; Auth, S.; Unger, J.; Grebe, A.; Mao, L.; Petrik, M.; Holland, G.; Appel, K.; Nüssler, A.; Neuhaus, P.; et al. Liver cell culture in bioreactors for drug studies as an alternative to animal testing. Altex 2000, 1, 3–10. [Google Scholar]
- Monga, S.; Gerlach, J. Human fetal hepatocyte behavior in dynamic 3D perfusion culture bioreactors. J. Organ. Dysfunct. 2007, 3, 183–192. [Google Scholar] [CrossRef]
- Ring, A.; Gerlach, J.C.; Peters, G.; Pazin, B.J.; Minervini, C.F.; Turner, M.E.; Thompson, R.L.; Triolo, F.; Gridelli, B.; Miki, T. Hepatic maturation of human fetal hepatocytes in four-compartment three-dimensional perfusion culture. Tissue Eng. Part C 2010, 16, 835–845. [Google Scholar] [CrossRef]
- Zeilinger, K.; Schreiter, T.; Darnell, M.; SÖderdahl, T.; Lübberstedt, M.; Dillner, B.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; Andersson, T.B. Scaling down of a clinical three-dimensional perfusion multicompartment hollow fiber liver bioreactor developed for extracorporeal liver support to an analytical scale device useful for hepatic pharmacological in vitro studies. Tissue Eng. Part C 2011, 17, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Pekor, C.; Gerlach, J.C.; Nettleship, I.; Schmelzer, E. Induction of hepatic and endothelial differentiation by perfusion in a three-dimensional cell culture model of human fetal live. Tissue Eng. Part C 2015, 21, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.A.; Biemel, K.; Knobeloch, D.; Heydel, S.; Lübberstedt, M.; Nüssler, A.K.; Andersson, T.B.; Gerlach, J.C.; Zeilinger, K. Analysis of drug metabolism activities in a miniaturized liver cell bioreactor for use in pharmacological studies. Biotechnol. Bioeng. 2012, 109, 3172–3181. [Google Scholar] [CrossRef]
- Lübberstedt, M.; Müller-Vieira, U.; Biemel, K.; Darnell, M.; Hoffmann, S.; Knospel, F.; Wönne, E.C.; Knobeloch, D.; Nüssler, A.K.; Gerlach, J.C.; et al. Serum-free culture of primary human hepatocytes in a miniaturized hollow-fibre membrane bioreactor for pharmacological in vitro studies. J. Tissue Eng. Regen. Med. 2015, 9, 1017–1026. [Google Scholar] [CrossRef]
- Zeilinger, K.; Holland, G.; Sauer, I.M.; Efimova, E.; Kardassis, D.; Obermayer, N.; Obermayer, N.; Liu, M.; Neuhaus, P.; Gerlach, J.C. Time course of primary liver cell reorganization in three-dimensional high-density bioreactors for extracorporeal liver support: An immunohistochemical and ultrastructural study. Tissue Eng. 2004, 10, 1113–1124. [Google Scholar] [CrossRef]
- Benowitz, N.L.; Meister, W. Clinical pharmacokinetics of lignocaine. Clin. Pharamacokinet. 1978, 3, 177–201. [Google Scholar] [CrossRef]
- Hermansonn, J.Y.; Glaumann, H.; Karlen, B.; Van Bahr, C. Metabolism of lidocaine in human liver in vitro. Acta Pharmacol. Toxicol. 1980, 47, 49–52. [Google Scholar] [CrossRef]
- Bargetzi, M.J.; Aoyama, T.; Gonzalez, F.J.; Meyer, U. Lidocaine metabolism in human liver microsomes by cytochrome P450 III A4. Clin. Pharmacol. Ther. 1989, 46, 521–557. [Google Scholar] [CrossRef]
- Furlan, V.; Demirdjian, S.; Bourdon, O.; Magdalou, J.; Taburet, A.-M. Glucuronidation of drugs by hepatic microsomes derived from healthy and cirrothic human livers. J. Pharmacol. Exp. Ther. 1999, 289, 1169–1175. [Google Scholar] [PubMed]
- Oellerich, M.; Burdelski, M.; Lautz, H.U.; Schueltz, M.; Schmidt, F.W.; Hermann, H. Lidocaine metabolite formation as a measure of liver function in patients with cirrhosis. Ther. Drug Monit. 1990, 12, 219–226. [Google Scholar] [CrossRef]
- Huang, Y.S.; Lee, S.D.; Deng, J.F.; Wu, J.C.; Lu, R.H.; Lin, Y.F.; Wang, Y.J.; Lo, K.J. Measuring lidocaine metabolite monoethylglycinexylidide as a quantitative index of hepatic function in adults with chronic hepatitis and cirrhosis. J. Hepatol. 1993, 19, 140–147. [Google Scholar] [CrossRef]
- Pea, F.; Licari, M.; Baldassarre, M.; Furlanut, M. MEGX disposition in critically-ill trauma patients: Subsequent assessments during the first week following trauma. Fundam. Clin. Pharmacol. 2002, 16, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.J.; Farrell, D.E.; Howard, K.D. Pig and minipig cytochromes P450—Letter to the Editor. Drug Metab. Dispos. 2002, 30, 101–102. [Google Scholar]
- Balis, U.J.; Behnia, K.; Dwarakanath, B.; Bhatia, S.N.; Sullivan, S.J.; Yarmush, M.L.; Toner, M. Oxygen consumption characteristics of porcine hepatocytes. Metab. Eng. 1999, 1, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.M.; Berko, R.S.; Gross, J.B.; Kagle, D.M.; Shaw, L. The effect of changes in arterial CO. Can. J. Anaesth. 1987, 34, 343–345. [Google Scholar] [CrossRef] [Green Version]
- Orlando, R.; Piccoli, P.; De Martin, S.; Padrini, R.; Palatini, P. Effect of the CYP3A4 inhibitor erythromycin on the pharmacokinetics of lignocaine and its pharmacologically active metabolites in subjects with normal and impaired liver function. Br. J. Clin. Pharmacol. 2003, 55, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanns, H.; Hollmann, M.W.; Stevens, M.F.; Lirk, P.; Brandenburger, T.; Piegeler, T.; Werdehausen, R. Molecular mechanisms of action of systemic lidocaine in acute and chronic pain: A narrative review. Br. J. Anaesth. 2019, 123, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Inoue, K.; Shaw, P.M.; Checovich, W.J.; Guegerich, F.P.; Shimada, T. Different contributions of cytochrome P450 2C19 and 3A4 in the oxidation of omeoprazole by human liver microsomes: Effects of contents of these two forms in individual human samples. J. Pharmacol. Exp. Ther. 1997, 283, 434–442. [Google Scholar]
- Venkatakrishnan, K.; von Moltke, L.L.; Greenblatt, D. Nortriptyline E-10-hydroxylation in vitro is mediated by human CYP2D6 (high affinity) and CYP3A4 (low affinity): Implications for interactions with enzyme-inducing drugs. J. Clin. Pharmacol. 1999, 39, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Catapano, G.; Euler, M.; Gaylor, J.; Gerlach, J. Characterization of the distribution of matter in hybrid liver support devices where cells are cultured in a 3-D membrane network or on flat substrata. Int. J. Artif. Organs 2001, 24, 102–109. [Google Scholar] [CrossRef]
- Obach, R. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27, 1350–1359. [Google Scholar] [PubMed]
- Shibata, Y.; Takahashi, H.; Ishii, Y.A. convenient in vitro screening method for predicting in vivo drug metabolic clearance using isolated hepatocytes suspended in serum. Drug Metab. Dispos. 2000, 28, 1518–1523. [Google Scholar] [PubMed]
- Heydari, Z.; Najimi, M.; Mirzaei, H.; Shpichka, A.; Ruoss, M.; Farzaneh, Z.; Montazeri, L.; Piryaei, A.; Timashev, P.; Gramignoli, R.; et al. Tissue Engineering in Liver Regenerative Medicine: Insights into Novel Translational Technologies. Cells 2020, 9, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mets, B.; Hickman, R.; Allin, R.; van Dyk, J.; Lotz, Z. Effect of hypoxia on the hepatic metabolism of lidocaine in the isolated perfused pig liver. Hepatology 1993, 17, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Sohlenius-Sternbeck, A. Determination of the hepatocellularity number for human, dog, rabbit, rat and mouse livers from protein concentration measurements. Toxicol. In Vitro 2006, 20, 1582. [Google Scholar] [CrossRef]
- Saville, B.A.; Gray, M.R.; Tam, Y. Experimental studies of transient mass transfer and reaction in the liver: Interpretation with a heterogeneous compartment model. J. Pharm. Sci. 1992, 81, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Catapano, G.; De Bartolo, L.; Lombardi, C.P.; Drioli, E. The effect of oxygen transport resistances on the viability and functions of isolated rat hepatocytes. Int. J. Artif. Organs 1996, 19, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Richert, L.; Binda, D.; Hamilton, G.; Viollon-Abadie, C.; Alexandre, E.; Bigot-Lasserre, D.; Coassolo, P.; Le Cluyse, E. Evaluation of the effect of culture configuration on morphology, survival time, antioxidant status and metabolic capacities of cultured rat hepatocytes. Toxicol. In Vitro 2002, 16, 89–99. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catapano, G.; Unger, J.K.; Zanetti, E.M.; Fragomeni, G.; Gerlach, J.C. Kinetic Analysis of Lidocaine Elimination by Pig Liver Cells Cultured in 3D Multi-Compartment Hollow Fiber Membrane Network Perfusion Bioreactors. Bioengineering 2021, 8, 104. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8080104
Catapano G, Unger JK, Zanetti EM, Fragomeni G, Gerlach JC. Kinetic Analysis of Lidocaine Elimination by Pig Liver Cells Cultured in 3D Multi-Compartment Hollow Fiber Membrane Network Perfusion Bioreactors. Bioengineering. 2021; 8(8):104. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8080104
Chicago/Turabian StyleCatapano, Gerardo, Juliane K. Unger, Elisabetta M. Zanetti, Gionata Fragomeni, and Jörg C. Gerlach. 2021. "Kinetic Analysis of Lidocaine Elimination by Pig Liver Cells Cultured in 3D Multi-Compartment Hollow Fiber Membrane Network Perfusion Bioreactors" Bioengineering 8, no. 8: 104. https://0-doi-org.brum.beds.ac.uk/10.3390/bioengineering8080104