In Silico Estimation of the Abundance and Phylogenetic Significance of the Composite Oct4-Sox2 Binding Motifs within a Wide Range of Species



Abstract
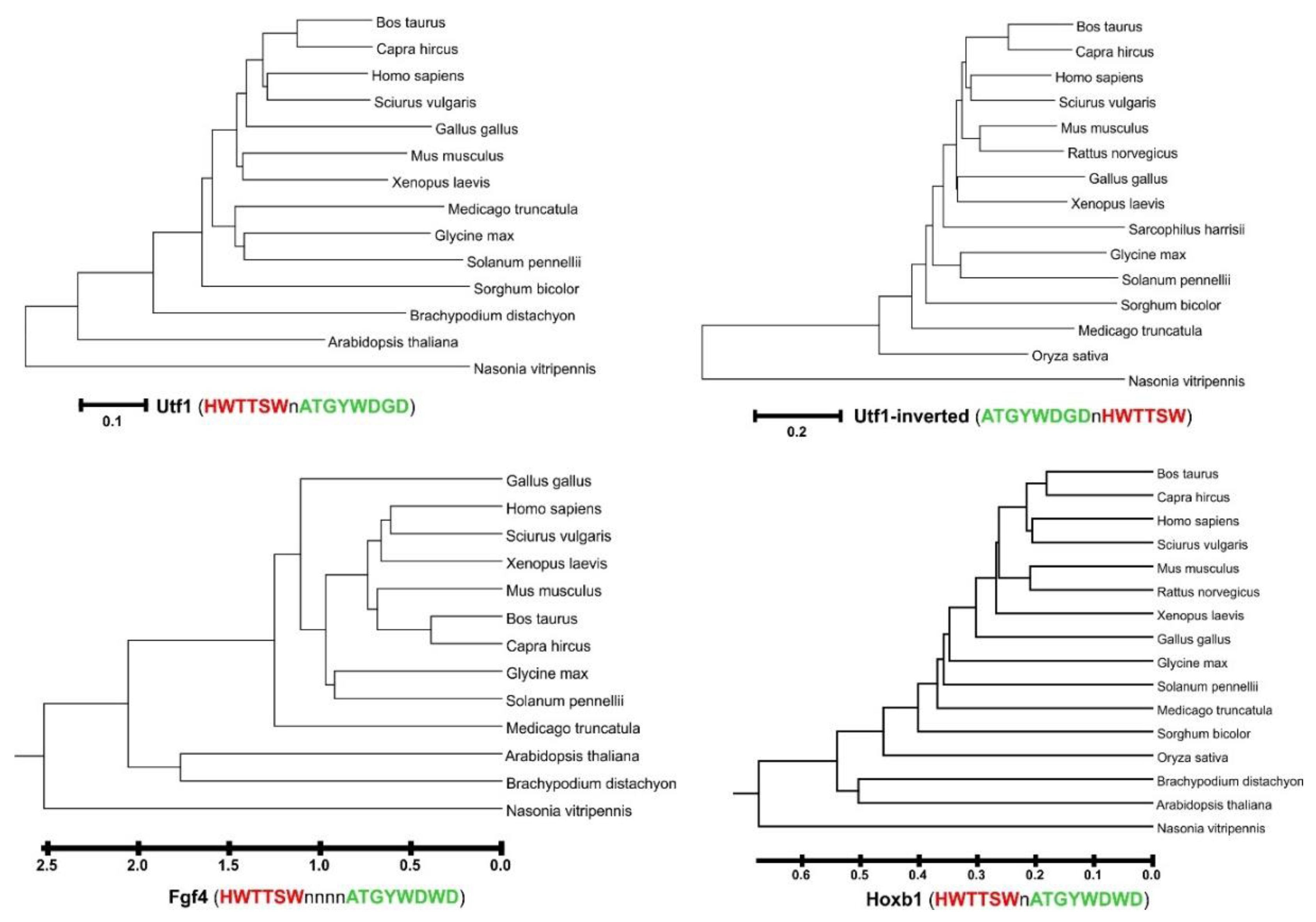
:1. Introduction
2. Materials and Methods
2.1. Bioinformatic Identification of Sox2/Oct4 Motif Sequences
2.2. A Phylogenetic Tree of Sox2/Oct4 Motif Sequences
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BAP | Biotin Acceptor Peptide |
| BirA | Biotinylating ligase and repressor of biotin biosynthesis of Escherichia coli |
| ChIP | Chromatin immunoprecipitation |
| ESCs | Embryonic stem cells |
| Fgf4 | Fibroblast growth factor 4 |
| HMG | High-mobility group |
| Hox-B1 | Homeobox protein |
| iPSCs | Induced pluripotent stem cells |
| NMR | Nuclear magnetic resonance spectroscopy |
| Oct4 | Octamer-binding transcription factor 4 |
| POU’HD’ | POU homeodomain |
| POU’S’ | POU-specific domain |
| PPI | Protein-protein interactions |
| PUB | Proximity Utilizing Biotinylation |
| Sox2 | SRY-box 2 |
| TF | Transcription factors |
| Utf1 | Undifferentiated embryonic cell transcription factor 1 |
References
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Belmonte, J.C. Ground rules of the pluripotency gene regulatory network. Nat. Rev. Genet. 2017, 18, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbay, J.; Collignon, J.; Koopman, P.; Capel, B.; Economou, A.; Munsterberg, A.; Vivian, N.; Goodfellow, P.; Lovellbadge, R. A gene-mapping to the sex-determining region of the mouse y-chromosome is a member of a novel family of embryonically expressed genes. Nature 1990, 346, 245–250. [Google Scholar] [CrossRef]
- Sinclair, A.H.; Berta, P.; Palmer, M.S.; Hawkins, J.R.; Griffiths, B.L.; Smith, M.J.; Foster, J.W.; Frischauf, A.M.; Lovell-Badge, R.; Goodfellow, P.N. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 1990, 346, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Bowles, J.; Schepers, G.; Koopman, P. Phylogeny of the sox family of developmental transcription factors based on sequence and structural indicators. Dev. Biol. 2000, 227, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Stros, M.; Launholt, D.; Grasser, K.D. The hmg-box: A versatile protein domain occurring in a wide variety of DNA-binding proteins. Cell Mol. Life Sci. 2007, 64, 2590–2606. [Google Scholar] [CrossRef]
- Yesudhas, D.; Batool, M.; Anwar, M.A.; Panneerselvam, S.; Choi, S. Proteins recognizing DNA: Structural uniqueness and versatility of DNA-binding domains in stem cell transcription factors. Genes 2017, 8, 192. [Google Scholar] [CrossRef] [Green Version]
- Kamachi, Y.; Kondoh, H. Sox proteins: Regulators of cell fate specification and differentiation. Development 2013, 140, 4129–4144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, K.; Luisi, B. The virtuoso of versatility: Pou proteins that flex to fit. J. Mol. Biol. 2000, 302, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Tantin, D. Oct transcription factors in development and stem cells: Insights and mechanisms. Development 2013, 140, 2857–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerabek, S.; Merino, F.; Scholer, H.R.; Cojocaru, V. Oct4: Dynamic DNA binding pioneers stem cell pluripotency. Biochim. Biophys. Acta 2014, 1839, 138–154. [Google Scholar] [CrossRef]
- Gold, D.A.; Gates, R.D.; Jacobs, D.K. The early expansion and evolutionary dynamics of pou class genes. Mol. Biol. Evol. 2014, 31, 3136–3147. [Google Scholar] [CrossRef] [Green Version]
- Malik, V.; Zimmer, D.; Jauch, R. Diversity among pou transcription factors in chromatin recognition and cell fate reprogramming. Cell Mol. Life Sci. 2018, 75, 1587–1612. [Google Scholar] [CrossRef]
- Esch, D.; Vahokoski, J.; Groves, M.R.; Pogenberg, V.; Cojocaru, V.; Vom Bruch, H.; Han, D.; Drexler, H.C.; Arauzo-Bravo, M.J.; Ng, C.K.; et al. A unique oct4 interface is crucial for reprogramming to pluripotency. Nat. Cell Biol. 2013, 15, 295–301. [Google Scholar] [CrossRef]
- Jauch, R.; Aksoy, I.; Hutchins, A.P.; Ng, C.K.; Tian, X.F.; Chen, J.; Palasingam, P.; Robson, P.; Stanton, L.W.; Kolatkar, P.R. Conversion of sox17 into a pluripotency reprogramming factor by reengineering its association with oct4 on DNA. Stem Cells 2011, 29, 940–951. [Google Scholar] [CrossRef]
- Lloyd, S.M.; Bao, X. Pinpointing the genomic localizations of chromatin-associated proteins: The yesterday, today, and tomorrow of chip-seq. Curr. Protoc. Cell Biol. 2019, 84, e89. [Google Scholar] [CrossRef]
- Lai, X.; Stigliani, A.; Vachon, G.; Carles, C.; Smaczniak, C.; Zubieta, C.; Kaufmann, K.; Parcy, F. Building transcription factor binding site models to understand gene regulation in plants. Mol. Plant 2019, 12, 743–763. [Google Scholar] [CrossRef]
- Robertson, G.; Hirst, M.; Bainbridge, M.; Bilenky, M.; Zhao, Y.; Zeng, T.; Euskirchen, G.; Bernier, B.; Varhol, R.; Delaney, A.; et al. Genome-wide profiles of stat1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 4, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Valouev, A.; Johnson, D.S.; Sundquist, A.; Medina, C.; Anton, E.; Batzoglou, S.; Myers, R.M.; Sidow, A. Genome-wide analysis of transcription factor binding sites based on chip-seq data. Nat. Methods 2008, 5, 829–834. [Google Scholar] [CrossRef] [Green Version]
- Kalendar, R.; Khassenov, B.; Ramankulov, Y.; Samuilova, O.; Ivanov, K.I. Fastpcr: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 2017, 109, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Lee, D.; Schulman, A.H. Java web tools for pcr, in silico pcr, and oligonucleotide assembly and analysis. Genomics 2011, 98, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Stecher, G.; Tamura, K.; Kumar, S. Molecular evolutionary genetics analysis (mega) for macos. Mol. Biol. Evol. 2020, 37, 1237–1239. [Google Scholar] [CrossRef] [PubMed]
- Kulyyassov, A.; Ogryzko, V. In vivo quantitative estimation of DNA-dependent interaction of sox2 and oct4 using bira-catalyzed site-specific biotinylation. Biomolecules 2020, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Tapia, N.; MacCarthy, C.; Esch, D.; Gabriele Marthaler, A.; Tiemann, U.; Arauzo-Bravo, M.J.; Jauch, R.; Cojocaru, V.; Scholer, H.R. Dissecting the role of distinct oct4-sox2 heterodimer configurations in pluripotency. Sci. Rep. 2015, 5, 13533. [Google Scholar] [CrossRef]
- Scholer, H.R.; Ruppert, S.; Suzuki, N.; Chowdhury, K.; Gruss, P. New type of pou domain in germ line-specific protein oct-4. Nature 1990, 344, 435–439. [Google Scholar] [CrossRef]
- Remenyi, A.; Lins, K.; Nissen, L.J.; Reinbold, R.; Scholer, H.R.; Wilmanns, M. Crystal structure of a pou/hmg/DNA ternary complex suggests differential assembly of oct4 and sox2 on two enhancers. Genes Dev. 2003, 17, 2048–2059. [Google Scholar] [CrossRef] [Green Version]
- Merino, F.; Ng, C.K.L.; Veerapandian, V.; Scholer, H.R.; Jauch, R.; Cojocaru, V. Structural basis for the sox-dependent genomic redistribution of oct4 in stem cell differentiation. Structure 2014, 22, 1274–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Srivastava, Y.; Jauch, R. Molecular basis for the genome engagement by sox proteins. Semin Cell Dev. Biol. 2017, 63, 2–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.C., Jr.; Cai, M.; Clore, G.M. Molecular basis for synergistic transcriptional activation by oct1 and sox2 revealed from the solution structure of the 42-kda oct1.Sox2.Hoxb1-DNA ternary transcription factor complex. J. Biol. Chem. 2004, 279, 1449–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parslow, T.G.; Blair, D.L.; Murphy, W.J.; Granner, D.K. Structure of the 5′ ends of immunoglobulin genes—A novel conserved sequence. Proc. Natl. Acad. Sci. USA 1984, 81, 2650–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Shlyueva, D.; Stampfel, G.; Stark, A. Transcriptional enhancers: From properties to genome-wide predictions. Nat. Rev. Genet. 2014, 15, 272–286. [Google Scholar] [CrossRef]
- Kamachi, Y.; Uchikawa, M.; Tanouchi, A.; Sekido, R.; Kondoh, H. Pax6 and sox2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001, 15, 1272–1286. [Google Scholar] [CrossRef] [Green Version]
- Ambrosetti, D.C.; Basilico, C.; Dailey, L. Synergistic activation of the fibroblast growth factor 4 enhancer by sox2 and oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol. Cell. Biol. 1997, 17, 6321–6329. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.B.; Corbi, N.; Basilico, C.; Dailey, L. Developmental-specific activity of the fgf-4 enhancer requires the synergistic action of sox2 and oct-3. Gene Dev. 1995, 9, 2635–2645. [Google Scholar] [CrossRef] [Green Version]
- Mistri, T.K.; Arindrarto, W.; Ng, W.P.; Wang, C.; Lim, L.H.; Sun, L.; Chambers, I.; Wohland, T.; Robson, P. Dynamic changes in sox2 spatio-temporal expression promote the second cell fate decision through fgf4/fgfr2 signaling in preimplantation mouse embryos. Biochem. J. 2018, 475, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Chew, J.L.; Loh, Y.H.; Zhang, W.; Chen, X.; Tam, W.L.; Yeap, L.S.; Li, P.; Ang, Y.S.; Lim, B.; Robson, P.; et al. Reciprocal transcriptional regulation of pou5f1 and sox2 via the oct4/sox2 complex in embryonic stem cells. Mol. Cell Biol. 2005, 25, 6031–6046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosetti, D.C.; Scholer, H.R.; Dailey, L.; Basilico, C. Modulation of the activity of multiple transcriptional activation domains by the DNA binding domains mediates the synergistic action of sox2 and oct-3 on the fibroblast growth factor-4 enhancer. J. Biol. Chem. 2000, 275, 23387–23397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamachi, Y.; Uchikawa, M.; Kondoh, H. Pairing sox off: With partners in the regulation of embryonic development. Trends Genet. 2000, 16, 182–187. [Google Scholar] [CrossRef]
- Jung, M.; Peterson, H.; Chavez, L.; Kahlem, P.; Lehrach, H.; Vilo, J.; Adjaye, J. A data integration approach to mapping oct4 gene regulatory networks operative in embryonic stem cells and embryonal carcinoma cells. PLoS ONE 2010, 5, e10709. [Google Scholar] [CrossRef]
- Nishimoto, M.; Miyagi, S.; Katayanagi, T.; Tomioka, M.; Muramatsu, M.; Okuda, A. The embryonic octamer factor 3/4 displays distinct DNA binding specificity from those of other octamer factors. Biochem. Biophys. Res. Commun. 2003, 302, 581–586. [Google Scholar] [CrossRef]
- Tokuzawa, Y.; Kaiho, E.; Maruyama, M.; Takahashi, K.; Mitsui, K.; Maeda, M.; Niwa, H.; Yamanaka, S. Fbx15 is a novel target of oct3/4 but is dispensable for embryonic stem cell self-renewal and mouse development. Mol. Cell Biol. 2003, 23, 2699–2708. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Motif | Sequence | Reference | Number of Hits Found on Ensembl |
|---|---|---|---|
| SoxOct | |||
| Fgf4 | CTTTGTTTGAATGCTAAT | [11] | 32 |
| Fgf4 | CTTTGTTTGGATGCTAAT | [30,37,40,41,42] | 37 |
| Pou5f1 | CTTTGTTATGCATCT | [11,40,41] | 12 |
| Sox2 | CATTGTGATGCATAT | [11,40,41,43] | 8 |
| Nanog | CATTGTAATGCAAAA | [11,40,41,44] | 13 |
| Utf1 | CATTGTTATGCTAGT | [11,29,30,40,41,43,45] | 4 |
| Utf1 | CATTGTTATGCTAGA | [18] | 1 |
| HoxB1 | CTTTGTCATGCTAAT | [18] | 14 |
| Fbxo15 | CATTGTTATGATAAA | [11,41,46] | 32 |
| Dppa4 | ATTTGTAaATGCTAAA | [11] | 47 |
| Gsh2 | CTTTGTCATGCAGAG | [18] | 16 |
| Nes | ATGCTAATtattgccTTTTGTC | [11] | 62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulyyassov, A.; Kalendar, R. In Silico Estimation of the Abundance and Phylogenetic Significance of the Composite Oct4-Sox2 Binding Motifs within a Wide Range of Species. Data 2020, 5, 111. https://0-doi-org.brum.beds.ac.uk/10.3390/data5040111
Kulyyassov A, Kalendar R. In Silico Estimation of the Abundance and Phylogenetic Significance of the Composite Oct4-Sox2 Binding Motifs within a Wide Range of Species. Data. 2020; 5(4):111. https://0-doi-org.brum.beds.ac.uk/10.3390/data5040111
Chicago/Turabian StyleKulyyassov, Arman, and Ruslan Kalendar. 2020. "In Silico Estimation of the Abundance and Phylogenetic Significance of the Composite Oct4-Sox2 Binding Motifs within a Wide Range of Species" Data 5, no. 4: 111. https://0-doi-org.brum.beds.ac.uk/10.3390/data5040111