
Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea


1
East Sea Fisheries Research Institute, National Institute of Fisheries Science, Gangneung 25435, Korea
2
Department of Marine Science, Incheon National University, Incheon 22012, Korea
*
Author to whom correspondence should be addressed.
Data 2021, 6(5), 44; https://0-doi-org.brum.beds.ac.uk/10.3390/data6050044
Submission received: 18 March 2021
/
Revised: 25 April 2021
/
Accepted: 25 April 2021
/
Published: 26 April 2021
{kind=link}
Abstract
:Marine bacteria are known to play significant roles in marine biogeochemical cycles regarding the decomposition of organic matter. Despite the increasing attention paid to the study of marine bacteria, research has been too limited to fully elucidate the complex interaction between marine bacterial communities and environmental variables. Jinhae Bay, the study area in this work, is the most anthropogenically eutrophied coastal bay in South Korea, and while its physical and biogeochemical characteristics are well described, less is known about the associated changes in microbial communities. In the present study, we reconstructed a metagenomics data based on the 16S rRNA gene to investigate temporal and vertical changes in microbial communities at three depths (surface, middle, and bottom) during a seven-month period from June to December 2016 at one sampling site (J1) in Jinhae Bay. Of all the bacterial data, Proteobacteria, Bacteroidetes, and Cyanobacteria were predominant from June to November, whereas Firmicutes were predominant in December, especially at the middle and bottom depths. These results show that the composition of the microbial community is strongly associated with temporal changes. Furthermore, the community compositions were markedly different between the surface, middle, and bottom depths in summer, when water column stratification and bottom water hypoxia (low dissolved oxygen level) were strongly developed. Metagenomics data contribute to improving our understanding of important relationships between environmental characteristics and microbial community change in eutrophication-induced and deoxygenated coastal areas.
Dataset License: CC-BY.
1. Summary
Marine environmental variables and bacterial data were collected from three different depths at Jinhae Bay (JB), South Korea, in 2016. The obtained dataset comprised temperature, salinity, and dissolved oxygen, as well as data of 324 different bacteria at the species level at three depth layers (surface, middle, and bottom) during a seven-month period from June to December at the J1 sampling site. The obtained bacterial data were quality filtered by removing ambiguous DNA sequences and chimera sequences, and by denoising. In total, 5,418,926 (83.6%) 16S rRNA quality reads were obtained, and approximately 3 million reads were associated with 55,400 operational taxonomic units (OTUs) (97% identity cutoff). At the phylum level, 11 bacterial phyla were detected, of which Proteobacteria (71%), Bacteroidetes (13%), Cyanobacteria (12%), and Actinobacteria (2%) accounted for 98% of all the OTUs in all the samples, except for those taken from the middle and bottom depths in December. With the exception of the month of December, at the class level, Alphaproteobacteria was predominant in all of the samples (accounting for 64%), followed by Flavobacteriia and Gammaproteobacteria. All bacteria sequence data were deposited in the NCBI. The collection of bacterial composition data will aid in understanding the correlation between environmental characteristics and bacterial community composition in eutrophication-induced and deoxygenated coastal areas.
2. Data Description
2.1. Study Area
Jinhae Bay (JB) is located in the southeastern coastal area of South Korea (Figure 1), which, since the 1960s, has suffered from anthropogenic eutrophication due to massive nutrient loading from the adjacent large cities, Changwon and Geoje [1]. As a result, JB has suffered from anthropogenically-derived environmental problems, such as persistent hypoxia, water quality deterioration, and harmful algal blooms [2,3,4,5]. Seasonal hypoxia of JB has developed in bottom waters since the 1970s because of a mixture of eutrophication-derived algal blooming (spring) and water stratification (summer) [6,7].
2.2. Bacterial Community Compositions
To determine bacterial data quality, the QC20 scores of all 21 samples were higher than 97, with a minimum of 97.56 and a maximum of 98.3 (average: 97.92). The relative abundance (%) was calculated using each sample’s OTU count number. For all 21 samples, the total number of sequence reads was 5,418,926 (83.6%) with an average of 258,044 and a mean of 258,552 quality reads per sample, and 55,400 OTUs were associated with roughly 3 million reads. The bacterial data (Supplementary Table S2) showed that Proteobacteria (mean: 71%) was the most abundant at the phylum level, followed by Bacteroidetes (mean: 13%), Cyanobacteria (mean: 12%), and Actinobacteria (mean: 2%). Additionally, at the class level, Alphaproteobacteria was the most dominant in all the samples, accounting for 64% (except in December), followed by Flavobacteriia and Gammaproteobacteria (Supplementary Table S2).
3. Methods
3.1. Sample Collection
Microbial samples were collected at three different depths (surface, middle, and bottom, where the surface is defined as 0.5 m beneath the surface, the middle is 10 m beneath the surface, and the bottom is 1 m above the seabed) at the J1 sampling site in the central area of JB from June to December 2016 using a Niskin water sampler. The water samples were filtered through a sterile 0.22 μm cellulose ester membrane (Millipore, Ireland) to capture microbial cells. The filtered membrane was then immediately placed into a sterile petri dish and stored at −20 °C until analysis.
3.2. Measurement of Marine Environmental Variables
We conducted a hydrographic survey to collect environmental variables and bacterial community composition data at the J1 station in the central area of JB from June to December 2016. The vertical profiles of temperature, salinity, and dissolved oxygen (DO) were measured with a multi-sensor sonde (YSI; 6600 V). Water samples (21 = 3 depths × 7 months) for microbial DNA analysis were collected at the surface (approximately 0.5 m below the surface), middle (10 m below the surface), and bottom depths (approximately 1 m above the seabed) using a Niskin water sampler (Supplementary Table S1).
3.3. DNA Extraction and Sequencing
DNA from the filtered membrane was extracted using the DNeasy PowerWater® DNA Isolation Kit (MO BIO Laboratories Inc., Carlsbad, CA, USA) by a combined chemical and mechanical procedure. Roughly 7–9 filtered membrane pieces were added to the PowerWater® Bead Tubes, and the kit’s protocol was followed, with the exception of the DNA precipitation time, which was prolonged to 30 min. DNA integrity was confirmed by electrophoresis in an agarose gel (1.2%), and the quantity was estimated by the PicoGreen method (Invitrogen) by Victor 3 fluorometry. For microbial biodiversity analysis, the V3–V4 variable region of the 16S rRNA gene was studied. Microbial primer pairs were used to identify the V3‒V4 region [8]. Library quantification was carried out by real-time PCR on a CFX96 real-time system (BioRad, Hercules, CA, USA). After 16S rDNA amplification, the multiplexing step was performed with the Nextera XT Index Kit (Illumina). Verification of the size was carried out using a Bioanalyzer DNA 1000 chip; the expected size was ~300 bp, and the size range of the DNA 1000 kit was 25–1000 bp. After size verification, the libraries were sequenced in a 2 × 300 bp paired-end run (MiSeq Reagent Kit v3, Illumina, CA, USA) on the Illumina MiSeq platform (San Diego, CA, USA).
3.4. Bioinformatics Analysis
Sequencing data were processed using QIIME1.9.1 to assemble paired end reads into tags according to their overlapping relationship [9,10,11]. In the pre-processing step, the primer was removed, and then demultiplexing and quality filtering (Phred ≥ 20) were applied [12]. USEARCH7 was used to perform denoising and chimera detection/filtering in operational taxonomic units (OTUs) grouping [13]. Then, the Silva132 and the NCBI database was used to determine the OTUs with 97% similarity using UCLUST and the open-reference analysis method determined the OTU identifier [14,15,16]. The OTU table was normalized dividing each OTU by the 16S copy number abundance. After filtering the generated OTU table using the Biological Observation Matrix (BIOM) format [17], the resulting sequences were clustered into OTUs based on a similarity threshold of ≥97% using Python Nearest Alignment Space Termination (PyNAST) [18]. We carried out comparative OTU assignment with the database in terms of Phylum, Class, Order, Family, Genus, and Species separately using RDP classifiers [19,20].
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/data6050044/s1, Table S1: Jinhae Bay environmental data, Table S2: Jinhae Bay bacterial data.
Author Contributions
J.-H.L. and I.-N.K. developed the concept and design, and wrote the manuscript. Both authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by a grant from the National Institute of Fisheries Science (R2021032).
Data Availability Statement
The data presented in this study are available within the supplementary material at http://0-doi-org.brum.beds.ac.uk/10.5281/zenodo.4718873 (accessed on 25 April 2021).
Acknowledgments
We thank the crew of the research vessel Tamgu 17 for their valuable support during sampling.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kim, D.; Choi, H.-W.; Choi, S.-H.; Baek, S.H.; Kim, K.-H.; Jeong, J.-H.; Kim, Y. Spatial and seasonal variations in the water quality of Jinhae Bay, Korea. N. Z. J. Mar. Freshwater Res. 2013, 47, 192–207. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-S.; Lee, Y.-H.; Kwon, J.-N.; Choi, H.-G. The effect of low oxygen conditions on biogeo- chemical cycling of nutrients in a shallow seasonally stratified bay in southeast Korea (Jinhae Bay). Mar. Pollut. Bull. 2015, 95, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-K.; Park, T.-G.; Park, Y.-T.; Lim, W.-A. Monitoring and trends in harmful algal blooms and red tides in Korean coastal waters, with emphasis on Cochlodinium polykrikoides. Harmful Algae 2013, 30, S3–S14. [Google Scholar] [CrossRef]
- Lim, H.-S.; Diaz, R.J.; Hong, J.-S.; Schaffner, L.C. Hypoxia and benthic community recovery in Korean coastal waters. Mar. Pollut. Bull. 2006, 52, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Lee, S.H.; Park, J.; Lee, J.; Yoon, J.-E.; Kim, I.-N. Coastal hypoxia in the Jinhae bay, South Korea: Mechanism, spatiotemporal variation, and implications (based on 2011 survey). J. Coast. Res. 2018, 85, 1481–1485. [Google Scholar] [CrossRef]
- Cho, C.H. Mass mortalities of oyster due to red tide in Jinhae Bay in 1978. Korean J. Fish. Aquat. Sci. 1979, 12, 27–33. [Google Scholar]
- Lee, J.; Park, K.-T.; Lim, J.-H.; Yoon, J.-E.; Kim, I.-N. Hypoxia in Korean coastal waters: A case study of the natural Jinhae Bay and artificial Shihwa Bay. Front. Mar. Sci. 2018, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids. Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Navas-Molina, J.A.; Peralta-Sánchez, J.M.; González, A.; McMurdie, P.J.; Vázquez-Baeza, Y.; Xu, Z.; Ursell, L.K.; Lauber, C.; Zhou, H.; Song, S.J.; et al. Chapter Nineteen—Advancing Our Understanding of the Human Microbiome Using QIIME. In Methods in Enzymology; DeLong, E.F., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 531, pp. 371–444. [Google Scholar]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; González, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 10, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Lauber, C.; Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, E0227434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yu, W.-H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford) 2010, 2010, baq013. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Clemente, J.C.; Kuczynski, J.; Rideout, J.R.; Stombaugh, J.; Wendel, D.; Wilke, A.; Huse, S.; Hufnagle, J.; Meyer, F.; et al. The Biological Observation Matrix (BIOM) format or: How I learned to stop worrying and love the ome-ome. GigaScience 2012, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Bittinger, K.; Bushman, F.D.; DeSantis, T.Z.; Andersen, G.L.; Knight, R. PyNAST: A flexible tool for aligning sequences to a template alignment. Bioinformatics 2009, 26, 266–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soergel, D.A.; Dey, N.; Knight, R.; Brenner, S.E. Selection of primers for optimal taxonomic classification of environmental 16S rRNA gene sequences. ISME J. 2012, 6, 1440–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
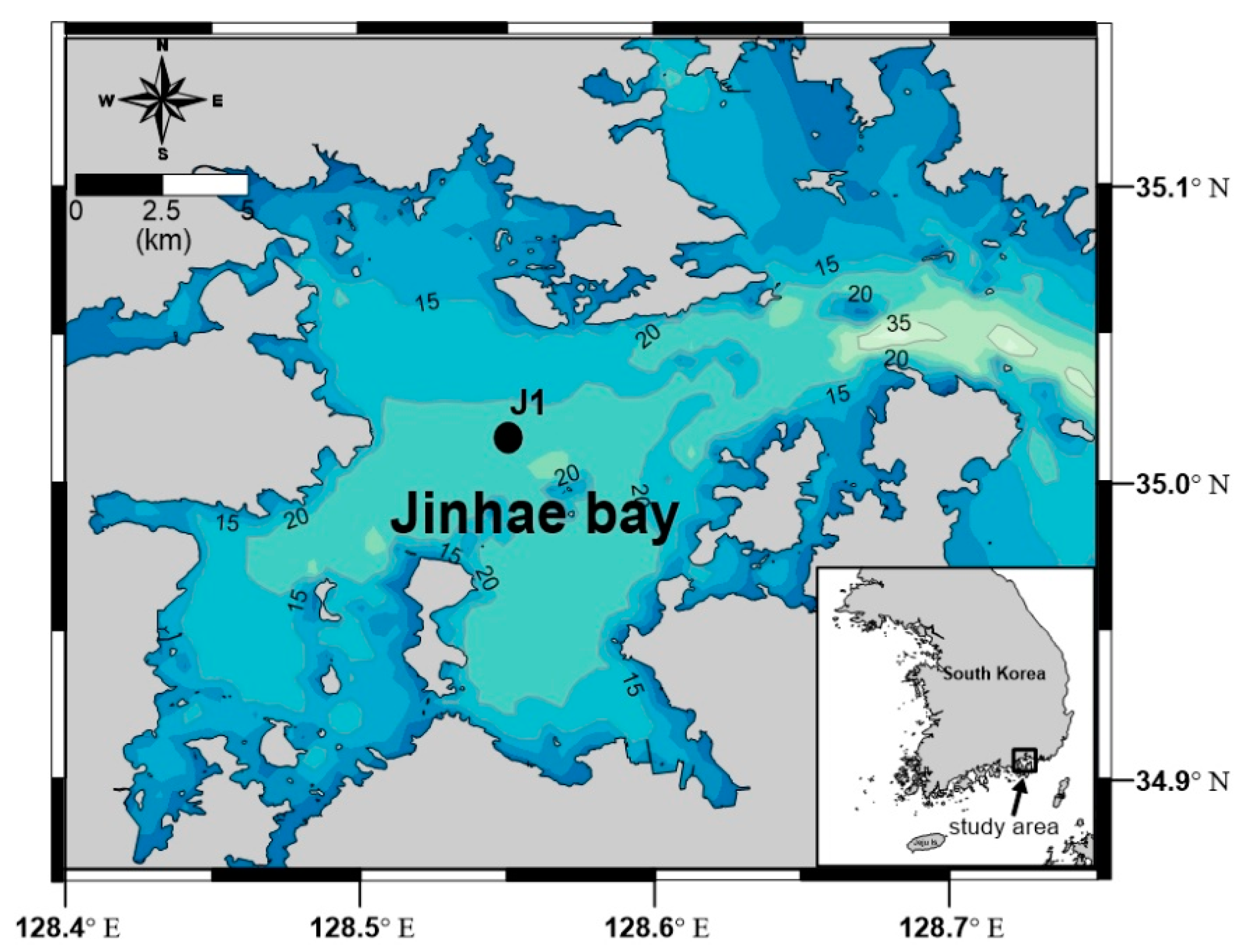
Figure 1.
Study area map of Jinhae Bay, located on the southeastern coast of South Korea. J1 represents a sampling site (latitude: 35°00′ N; longitude: 128°33′ E) in the central part of Jinhae Bay.
Figure 1.
Study area map of Jinhae Bay, located on the southeastern coast of South Korea. J1 represents a sampling site (latitude: 35°00′ N; longitude: 128°33′ E) in the central part of Jinhae Bay.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lim, J.-H.; Kim, I.-N. Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea. Data 2021, 6, 44. https://0-doi-org.brum.beds.ac.uk/10.3390/data6050044
AMA Style
Lim J-H, Kim I-N. Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea. Data. 2021; 6(5):44. https://0-doi-org.brum.beds.ac.uk/10.3390/data6050044
Chicago/Turabian StyleLim, Jae-Hyun, and Il-Nam Kim. 2021. "Collection of a Bacterial Community Reconstructed from Marine Metagenomes Derived from Jinhae Bay, South Korea" Data 6, no. 5: 44. https://0-doi-org.brum.beds.ac.uk/10.3390/data6050044