
Therapeutic Effects of Risperidone against Spinal Cord Injury in a Rat Model of Asphyxial Cardiac Arrest: A Focus on Body Temperature, Paraplegia, Motor Neuron Damage, and Neuroinflammation


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
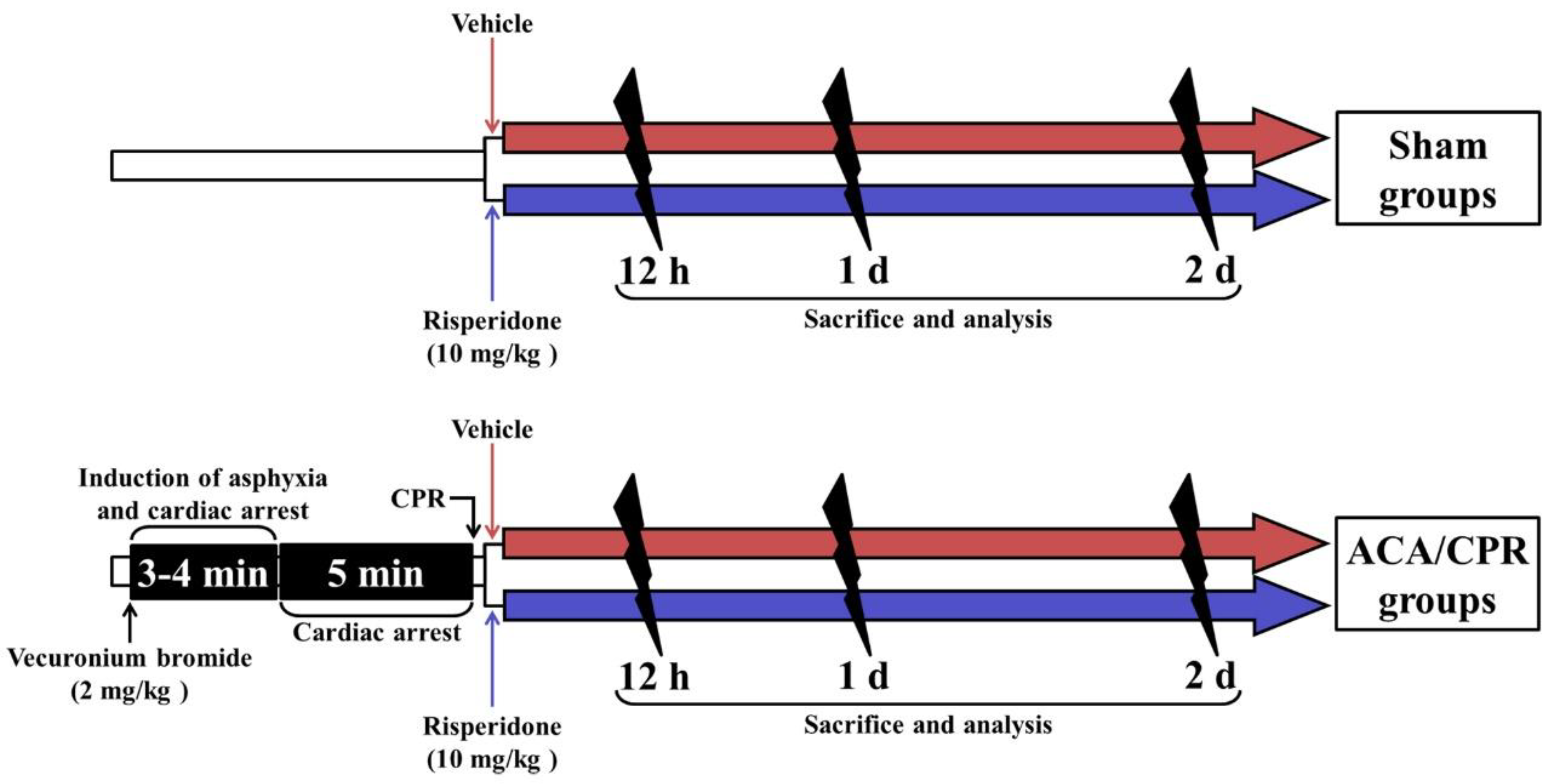
2.1. Rats, Protocol, and Groups for Experiment
2.2. ACA/CPR Operation and RIS Treatment
2.3. Assessment of Physiological Variables and Motor Function
2.4. Preparation of Histological Sections
2.5. Fluoro-Jade B (F-J B) Histofluorescence
2.6. Immunohistochemistry
2.7. Statistical Analysis
3. Results
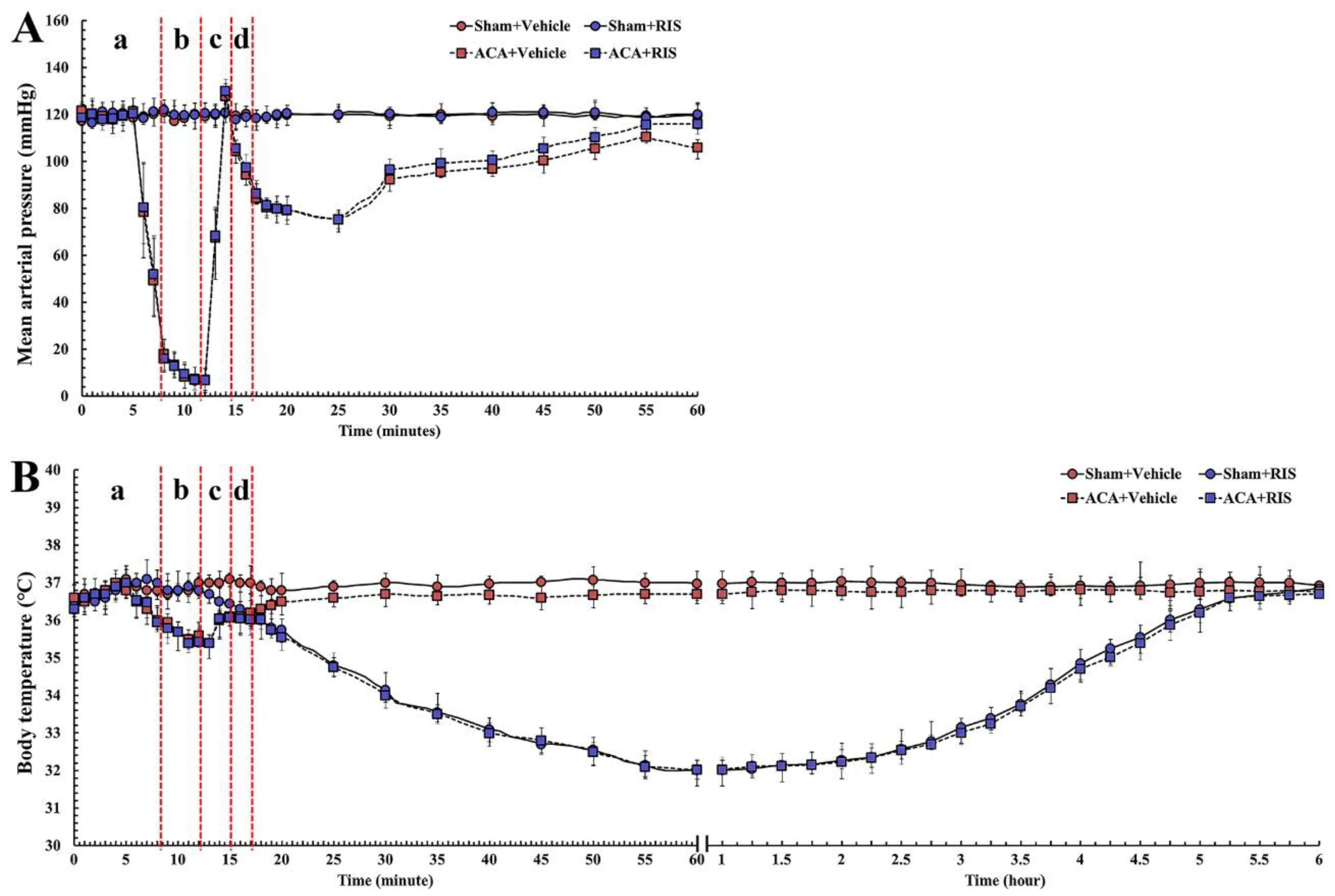
3.1. Changes in Physiological Function and Body Temperature
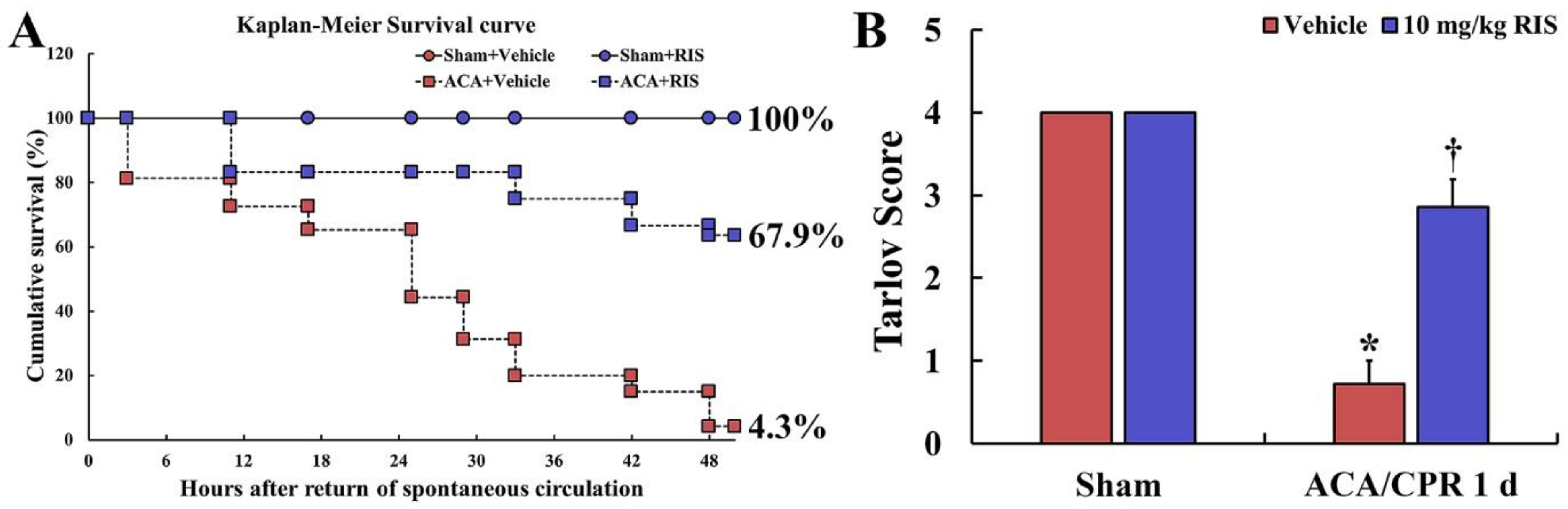
3.2. Survival Rate and Motor Deficit Score
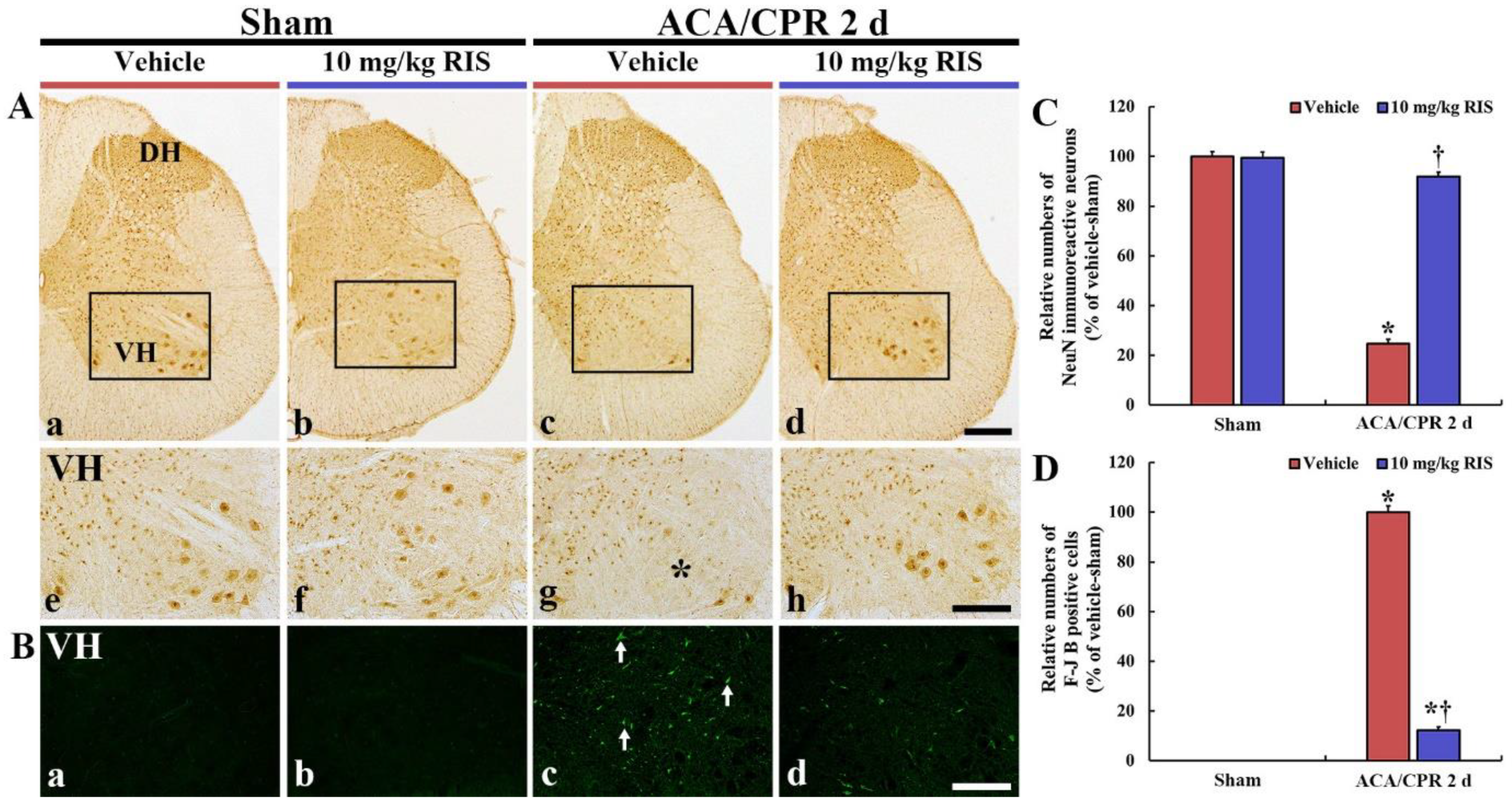
3.3. Neuroprotection by RIS
3.3.1. NeuN Immunoreactive Neurons
3.3.2. F-J B-Positive Cells
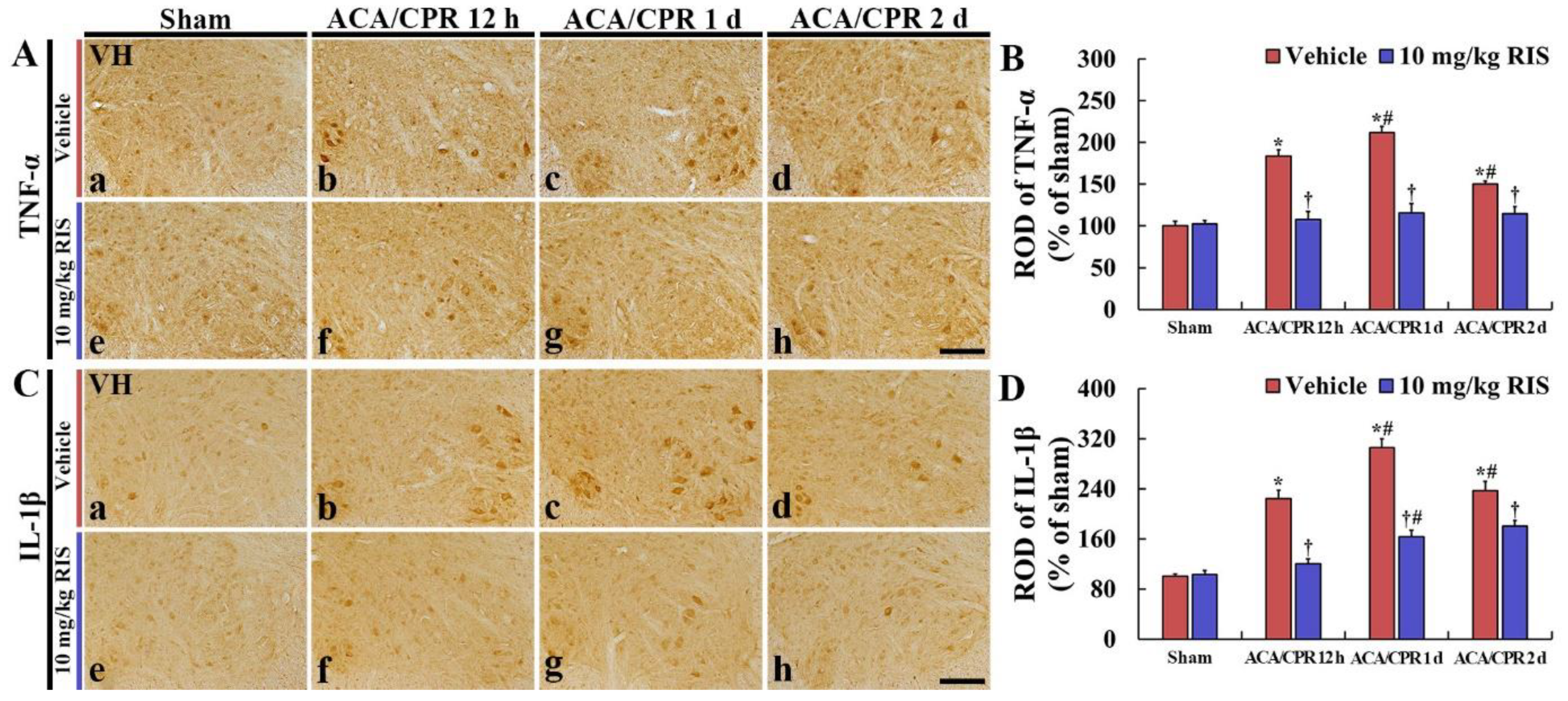
3.4. Decreased Pro-Inflammatory Cytokines by RIS
3.4.1. TNF-α Immunoreactivity
3.4.2. IL-1 β Immunoreactivity
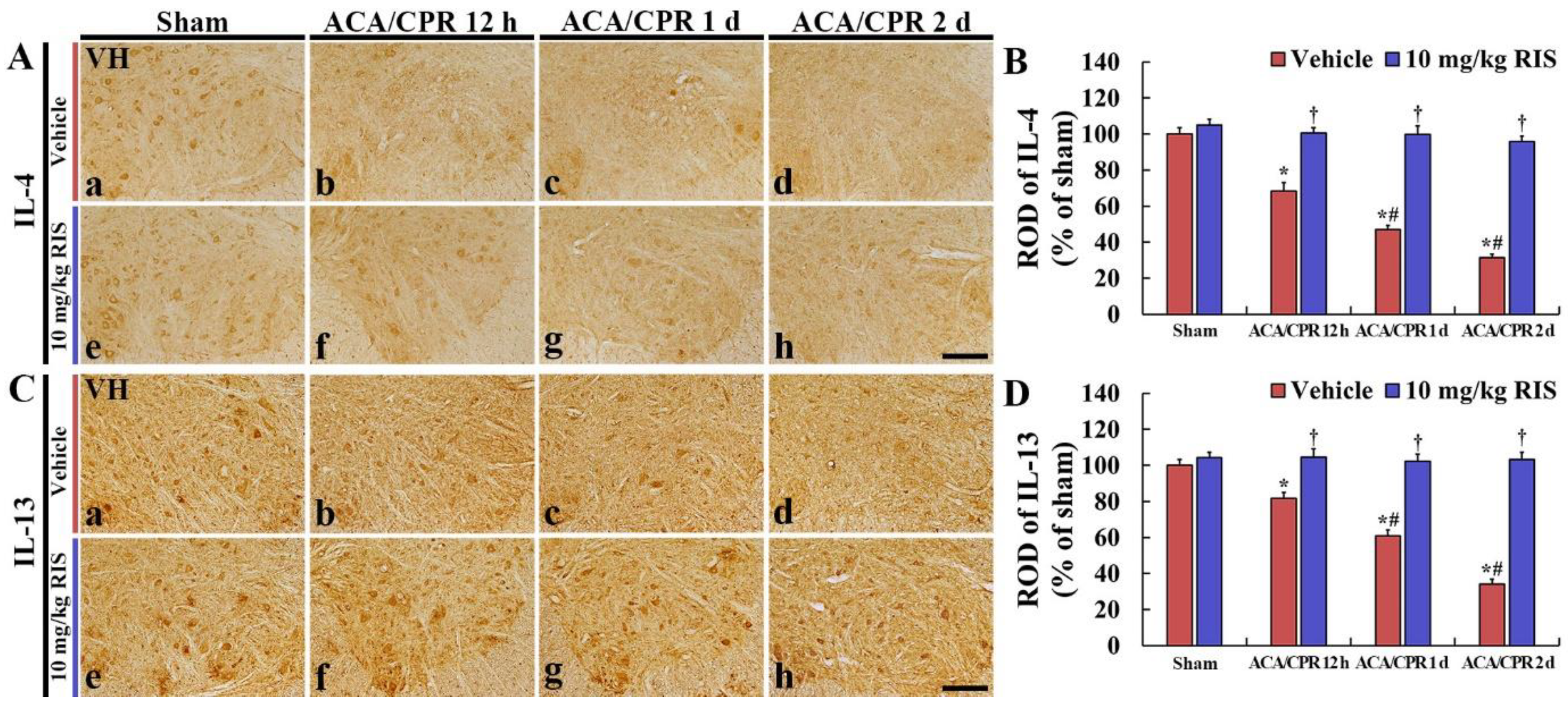
3.5. Increased Anti-Inflammatory Cytokines by RIS
3.5.1. IL-4 Immunoreactivity
3.5.2. IL-13 Immunoreactivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACA | asphyxial cardiac arrest |
| CNS | central nervous system |
| CPR | cardiopulmonary resuscitation |
| F-J B | fluoro-Jade B |
| IL | interleukin |
| NeuN | risperidone |
| RIS | major histocompatibility complex |
| ROSC | return of spontaneous circulation |
| ROD | relative optical density |
| TNF-α | tumor necrosis factor α |
| VH | ventral horn |
References
- Chalkias, A.; Xanthos, T. Post-cardiac arrest brain injury: Pathophysiology and treatment. J. Neurol. Sci. 2012, 315, 1–8. [Google Scholar] [CrossRef]
- Schneider, A.; Bottiger, B.W.; Popp, E. Cerebral resuscitation after cardiocirculatory arrest. Anesth Analg. 2009, 108, 971–979. [Google Scholar] [CrossRef]
- Lopez-Herce, J.; del Castillo, J.; Matamoros, M.; Canadas, S.; Rodriguez-Calvo, A.; Cecchetti, C.; Rodriguez-Nunez, A.; Carrillo, A. Post return of spontaneous circulation factors associated with mortality in pediatric in-hospital cardiac arrest: A prospective multicenter multinational observational study. Crit. Care 2014, 18, 607. [Google Scholar] [CrossRef] [Green Version]
- Mongardon, N.; Dumas, F.; Ricome, S.; Grimaldi, D.; Hissem, T.; Pene, F.; Cariou, A. Postcardiac arrest syndrome: From immediate resuscitation to long-term outcome. Ann. Intensive Care 2011, 1, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Watts, J. Paraplegia and spinal cord ischaemia. Anaesthesia 1995, 50, 745–746. [Google Scholar] [CrossRef] [PubMed]
- Grassner, L.; Klausner, F.; Wagner, M.; McCoy, M.; Golaszewski, S.; Leis, S.; Aigner, L.; Couillard-Despres, S.; Trinka, E. Acute and chronic evolution of mri findings in a case of posterior spinal cord ischemia. Spinal Cord 2014, 52 (Suppl. S1), S23–S24. [Google Scholar] [CrossRef] [PubMed]
- Nedeltchev, K.; Loher, T.J.; Stepper, F.; Arnold, M.; Schroth, G.; Mattle, H.P.; Sturzenegger, M. Long-term outcome of acute spinal cord ischemia syndrome. Stroke 2004, 35, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.H.; Lee, T.K.; Kim, B.; Lee, J.C.; Tae, H.J.; Cho, J.H.; Park, Y.; Shin, M.C.; Ohk, T.G.; Park, C.W.; et al. Therapeutic hypothermia improves hind limb motor outcome and attenuates oxidative stress and neuronal damage in the lumbar spinal cord following cardiac arrest. Antioxidants 2020, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Tae, H.J.; Cho, J.H.; Kim, I.S.; Lee, T.K.; Park, C.W.; Park, Y.E.; Ahn, J.H.; Park, J.H.; Yan, B.C.; et al. Therapeutic hypothermia attenuates paraplegia and neuronal damage in the lumbar spinal cord in a rat model of asphyxial cardiac arrest. J. Therm. Biol. 2019, 83, 1–7. [Google Scholar] [CrossRef]
- Ahn, J.H.; Lee, T.K.; Tae, H.J.; Kim, B.; Sim, H.; Lee, J.C.; Kim, D.W.; Kim, Y.S.; Shin, M.C.; Park, Y.; et al. Neuronal death in the cns autonomic control center comes very early after cardiac arrest and is not significantly attenuated by prompt hypothermic treatment in rats. Cells 2021, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Arrich, J.; Holzer, M.; Havel, C.; Mullner, M.; Herkner, H. Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst. Rev. 2016, 2, CD004128. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.A.; Gray, T.W.; Buist, M.D.; Jones, B.M.; Silvester, W.; Gutteridge, G.; Smith, K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N. Engl. J. Med. 2002, 346, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, K.; Guyette, F.X.; Doshi, A.A.; Callaway, C.W.; Rittenberger, J.C.; Post Cardiac Arrest, S. Prevalence and effect of fever on outcome following resuscitation from cardiac arrest. Resuscitation 2013, 84, 1062–1067. [Google Scholar] [CrossRef] [PubMed]
- Cronberg, T.; Lilja, G.; Horn, J.; Kjaergaard, J.; Wise, M.P.; Pellis, T.; Hovdenes, J.; Gasche, Y.; Aneman, A.; Stammet, P.; et al. Neurologic function and health-related quality of life in patients following targeted temperature management at 33 degrees c vs 36 degrees c after out-of-hospital cardiac arrest: A randomized clinical trial. JAMA Neurol. 2015, 72, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Sterz, F.; Safar, P.; Tisherman, S.; Radovsky, A.; Kuboyama, K.; Oku, K. Mild hypothermic cardiopulmonary resuscitation improves outcome after prolonged cardiac arrest in dogs. Crit. Care Med. 1991, 19, 379–389. [Google Scholar] [CrossRef]
- Kuboyama, K.; Safar, P.; Radovsky, A.; Tisherman, S.A.; Stezoski, S.W.; Alexander, H. Delay in cooling negates the beneficial effect of mild resuscitative cerebral hypothermia after cardiac arrest in dogs: A prospective, randomized study. Crit. Care Med. 1993, 21, 1348–1358. [Google Scholar] [CrossRef]
- Schotte, A.; Janssen, P.F.; Gommeren, W.; Luyten, W.H.; Van Gompel, P.; Lesage, A.S.; De Loore, K.; Leysen, J.E. Risperidone compared with new and reference antipsychotic drugs: In vitro and in vivo receptor binding. Psychopharmacology 1996, 124, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Corena-McLeod, M. Comparative pharmacology of risperidone and paliperidone. Drugs R D 2015, 15, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razaq, M.; Samma, M. A case of risperidone-induced hypothermia. Am. J. Ther. 2004, 11, 229–230. [Google Scholar] [CrossRef] [PubMed]
- Brevik, A.; Farver, D. Atypical antipsychotic induced mild hypothermia. S D J. Med. 2003, 56, 67–70. [Google Scholar] [PubMed]
- Yang, G.E.; Tae, H.J.; Lee, T.K.; Park, Y.E.; Cho, J.H.; Kim, D.W.; Park, J.H.; Ahn, J.H.; Ryoo, S.; Kim, Y.M.; et al. Risperidone treatment after transient ischemia induces hypothermia and provides neuroprotection in the gerbil hippocampus by decreasing oxidative stress. Int. J. Mol. Sci. 2019, 20, 4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 1–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.K.; Kang, I.J.; Kim, B.; Sim, H.J.; Kim, D.W.; Ahn, J.H.; Lee, J.C.; Ryoo, S.; Shin, M.C.; Cho, J.H.; et al. Experimental pretreatment with chlorogenic acid prevents transient ischemia-induced cognitive decline and neuronal damage in the hippocampus through anti-oxidative and anti-inflammatory effects. Molecules 2020, 25, 3578. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, A.G.; Zgavc, T.; Kooijman, R.; Hachimi-Idrissi, S.; Sarre, S.; Michotte, Y. The dual role of the neuroinflammatory response after ischemic stroke: Modulatory effects of hypothermia. J. Neuroinflamm. 2010, 7, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perini, F.; Morra, M.; Alecci, M.; Galloni, E.; Marchi, M.; Toso, V. Temporal profile of serum anti-inflammatory and pro-inflammatory interleukins in acute ischemic stroke patients. Neurol. Sci. 2001, 22, 289–296. [Google Scholar] [CrossRef]
- Park, J.H.; Park, O.; Cho, J.H.; Chen, B.H.; Kim, I.H.; Ahn, J.H.; Lee, J.C.; Yan, B.C.; Yoo, K.Y.; Lee, C.H.; et al. Anti-inflammatory effect of tanshinone i in neuroprotection against cerebral ischemia-reperfusion injury in the gerbil hippocampus. Neurochem. Res. 2014, 39, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.Y.; Kim, I.H.; Cho, J.H.; Ahn, J.H.; Park, J.H.; Lee, J.C.; Tae, H.J.; Kim, D.W.; Kim, J.D.; Hong, S.; et al. Neuroprotection of chrysanthemum indicum linne against cerebral ischemia/reperfusion injury by anti-inflammatory effect in gerbils. Neural Regen. Res. 2016, 11, 270–277. [Google Scholar]
- Martirosyan, N.L.; Patel, A.A.; Carotenuto, A.; Kalani, M.Y.; Bohl, M.A.; Preul, M.C.; Theodore, N. The role of therapeutic hypothermia in the management of acute spinal cord injury. Clin. Neurol. Neurosurg. 2017, 154, 79–88. [Google Scholar] [CrossRef]
- Dietrich, W.D. Therapeutic hypothermia for spinal cord injury. Crit. Care Med. 2009, 37, S238–S242. [Google Scholar] [CrossRef]
- Albus, U. Guide for the Care and Use of Laboratory Animals, 8th ed.; SAGE Publications Sage UK: London, UK, 2012. [Google Scholar]
- Flecknell, P. Laboratory Animal Anaesthesia; Academic Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Lee, C.H.; Hwang, I.K.; Choi, J.H.; Yoo, K.Y.; Han, T.H.; Park, O.K.; Lee, S.Y.; Ryu, P.D.; Won, M.H. Calcium binding proteins immunoreactivity in the rat basolateral amygdala following myocardial infarction. Cell Mol. Neurobiol. 2010, 30, 333–338. [Google Scholar] [CrossRef]
- Lee, J.C.; Park, J.H.; Kim, I.H.; Cho, G.S.; Ahn, J.H.; Tae, H.J.; Choi, S.Y.; Cho, J.H.; Kim, D.W.; Kwon, Y.G.; et al. Neuroprotection of ischemic preconditioning is mediated by thioredoxin 2 in the hippocampal ca1 region following a subsequent transient cerebral ischemia. Brain Pathol. 2017, 27, 276–291. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Ahn, J.H.; Kim, H.; Kim, D.W.; Lee, T.K.; Lee, J.C.; Kim, Y.M.; Lee, C.H.; Hwang, I.K.; Yan, B.C.; et al. Chronic high-fat diet-induced obesity in gerbils increases pro-inflammatory cytokines and mtor activation, and elicits neuronal death in the striatum following brief transient ischemia. Neurochem. Int. 2018, 121, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Turkoz, A.; Gulcan, O.; Kizilkilic, O.; Kocum, A.; Turkoz, R. Spinal cord ischemia caused by cardiac arrest secondary to pericardial effusion. J. Cardiothorac. Vasc. Anesth. 2007, 21, 91–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, M.Y.; Lyu, R.K.; Chang, Y.J.; Chen, C.M.; Chen, S.T.; Wai, Y.Y.; Ro, L.S. Concomitant spinal cord and vertebral body infarction is highly associated with aortic pathology: A clinical and magnetic resonance imaging study. J. Neurol. 2009, 256, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Dublin, A.B.; Latchaw, R.E.; Herrera, D.A.; Dahlin, B.C. Delayed complication after embolotherapy of a vertebral arteriovenous fistula: Spinal cord ischemia. J. Vasc. Interv. Radiol. 2010, 21, 392–393. [Google Scholar] [CrossRef] [PubMed]
- Duggal, N.; Lach, B. Selective vulnerability of the lumbosacral spinal cord after cardiac arrest and hypotension. Stroke 2002, 33, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Liang, C.L.; Chen, H.J.; Chen, S.D.; Hsu, H.C.; Liliang, P.C.; Lin, T.K.; Cho, C.L. Injury severity and cell death mechanisms: Effects of concomitant hypovolemic hypotension on spinal cord ischemia-reperfusion in rats. Exp. Neurol. 2004, 185, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Marsala, M.; Yaksh, T.L. Transient spinal ischemia in the rat: Characterization of behavioral and histopathological consequences as a function of the duration of aortic occlusion. J. Cereb. Blood Flow Metab. 1994, 14, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Lang-Lazdunski, L.; Matsushita, K.; Hirt, L.; Waeber, C.; Vonsattel, J.P.; Moskowitz, M.A.; Dietrich, W.D. Spinal cord ischemia. Development of a model in the mouse. Stroke 2000, 31, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Mazensky, D.; Flesarova, S.; Sulla, I. Arterial blood supply to the spinal cord in animal models of spinal cord injury. A review. Anat. Rec. 2017, 300, 2091–2106. [Google Scholar] [CrossRef] [Green Version]
- Yoo, D.Y.; Cho, S.B.; Jung, H.Y.; Kim, W.; Choi, G.M.; Won, M.H.; Kim, D.W.; Hwang, I.K.; Choi, S.Y.; Moon, S.M. Tat-protein disulfide-isomerase a3: A possible candidate for preventing ischemic damage in the spinal cord. Cell Death Dis. 2017, 8, e3075. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Hwang, I.K.; Yoo, K.Y.; Jung, J.Y.; Cho, J.H.; Moon, S.M.; Kang, T.C.; Kim, W.K.; Kim, Y.S.; Won, M.H. Calbindin d-28k is expressed in the microvascular basal lamina in the ventral horn at early time after transient spinal cord ischemia in the rabbit. Brain Res. 2005, 1047, 123–128. [Google Scholar] [CrossRef]
- Mechirova, E.; Danielisova, V.; Domorakova, I.; Dankova, M.; Stebnicky, M.; Mickova, H.; Burda, J. Bradykinin preconditioning affects the number of degenerated neurons and the level of antioxidant enzymes in spinal cord ischemia in rabbits. Acta Histochem. 2014, 116, 252–257. [Google Scholar] [CrossRef]
- Sengupta, B.; Faisal, A.A.; Laughlin, S.B.; Niven, J.E. The effect of cell size and channel density on neuronal information encoding and energy efficiency. J. Cereb. Blood Flow Metab. 2013, 33, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- van Marum, R.J.; Wegewijs, M.A.; Loonen, A.J.; Beers, E. Hypothermia following antipsychotic drug use. Eur. J. Clin. Pharmacol. 2007, 63, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, W.D.; Atkins, C.M.; Bramlett, H.M. Protection in animal models of brain and spinal cord injury with mild to moderate hypothermia. J. Neurotrauma 2009, 26, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Tian, D.C.; Li, Z.G.; Ducruet, A.F.; Lawton, M.T.; Shi, F.D. Global brain inflammation in stroke. Lancet Neurol. 2019, 18, 1058–1066. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Dugue, R.; Nath, M.; Dugue, A.; Barone, F.C. Roles of pro-and anti-inflammatory cytokines in traumatic brain injury and acute ischemic stroke. Mech. Neuroinflamm. 2017, 211. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Monji, A.; Hashioka, S.; Kanba, S. Risperidone significantly inhibits interferon-gamma-induced microglial activation in vitro. Schizophr. Res. 2007, 92, 108–115. [Google Scholar] [CrossRef] [PubMed]
- MacDowell, K.S.; Garcia-Bueno, B.; Madrigal, J.L.; Parellada, M.; Arango, C.; Mico, J.A.; Leza, J.C. Risperidone normalizes increased inflammatory parameters and restores anti-inflammatory pathways in a model of neuroinflammation. Int. J. Neuropsychopharmacol. 2013, 16, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, F.C.; Arvin, B.; White, R.F.; Miller, A.; Webb, C.L.; Willette, R.N.; Lysko, P.G.; Feuerstein, G.Z. Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 1997, 28, 1233–1244. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Matsuura, N.; Shozuhara, H.; Onodera, H.; Itoyama, Y.; Kogure, K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke 1995, 26, 676–680, discussion 681. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Saito, K.; Hara, A.; Zhu, Y.; Sudo, K.; Niwa, M.; Fujii, H.; Wada, H.; Ishiguro, H.; Mori, H.; et al. Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J. Neurochem. 2005, 93, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Hasturk, A.; Atalay, B.; Calisaneller, T.; Ozdemir, O.; Oruckaptan, H.; Altinors, N. Analysis of serum pro-inflammatory cytokine levels after rat spinal cord ischemia/reperfusion injury and correlation with tissue damage. Turk. Neurosurg. 2009, 19, 353–359. [Google Scholar] [PubMed]
- Gokce, E.C.; Kahveci, R.; Gokce, A.; Sargon, M.F.; Kisa, U.; Aksoy, N.; Cemil, B.; Erdogan, B. Curcumin attenuates inflammation, oxidative stress, and ultrastructural damage induced by spinal cord ischemia-reperfusion injury in rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Li, J.X.; Fujino, M.; Zhuang, J.; Li, X.K. Development and treatments of inflammatory cells and cytokines in spinal cord ischemia-reperfusion injury. Mediat. Inflamm. 2013, 2013, 701970. [Google Scholar] [CrossRef]
- Nakata, T.; Kawachi, K.; Nagashima, M.; Yasugi, T.; Izutani, H.; Ryugo, M.; Okamura, T.; Shikata, F.; Imagawa, H.; Yano, H.; et al. Transient ischemia-induced paresis and complete paraplegia displayed distinct reactions of microglia and macrophages. Brain Res. 2011, 1420, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Hirose, K.; Okajima, K.; Taoka, Y.; Uchiba, M.; Tagami, H.; Nakano, K.; Utoh, J.; Okabe, H.; Kitamura, N. Activated protein c reduces the ischemia/reperfusion-induced spinal cord injury in rats by inhibiting neutrophil activation. Ann. Surg. 2000, 232, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Reece, T.B.; Okonkwo, D.O.; Ellman, P.I.; Warren, P.S.; Smith, R.L.; Hawkins, A.S.; Linden, J.; Kron, I.L.; Tribble, C.G.; Kern, J.A. The evolution of ischemic spinal cord injury in function, cytoarchitecture, and inflammation and the effects of adenosine a2a receptor activation. J. Thorac. Cardiovasc. Surg. 2004, 128, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.D.; Puskas, F.; Meng, X.; Lee, J.H.; Cleveland, J.C., Jr.; Weyant, M.J.; Fullerton, D.A.; Reece, T.B. The evolution of chemokine release supports a bimodal mechanism of spinal cord ischemia and reperfusion injury. Circulation 2012, 126, S110–S117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.T.; Lee, C.H.; Yoo, K.Y.; Choi, J.H.; Li, H.; Park, O.K.; Yan, B.; Hwang, I.K.; Kwon, Y.G.; Kim, Y.M.; et al. Maintenance of anti-inflammatory cytokines and reduction of glial activation in the ischemic hippocampal ca1 region preconditioned with lipopolysaccharide. J. Neurol. Sci. 2010, 296, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.C.; Kim, S.K.; Park, J.H.; Ahn, J.H.; Lee, C.H.; Yoo, K.Y.; Choi, J.H.; Lee, D.S.; Kim, M.J.; Kim, Y.M.; et al. Comparison of inflammatory cytokines changes in the hippocampal ca1 region between the young and adult gerbil after transient cerebral ischemia. Brain Res. 2012, 1461, 64–75. [Google Scholar] [CrossRef] [PubMed]
- de Waal Malefyt, R.; Figdor, C.G.; Huijbens, R.; Mohan-Peterson, S.; Bennett, B.; Culpepper, J.; Dang, W.; Zurawski, G.; de Vries, J.E. Effects of il-13 on phenotype, cytokine production, and cytotoxic function of human monocytes. Comparison with il-4 and modulation by ifn-gamma or il-10. J. Immunol. 1993, 151, 6370–6381. [Google Scholar] [PubMed]
- te Velde, A.A.; Huijbens, R.J.; Heije, K.; de Vries, J.E.; Figdor, C.G. Interleukin-4 (il-4) inhibits secretion of il-1 beta, tumor necrosis factor alpha, and il-6 by human monocytes. Blood 1990, 76, 1392–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.-K.; Lee, J.-C.; Tae, H.-J.; Kim, H.-I.; Shin, M.C.; Ahn, J.H.; Park, J.H.; Kim, D.W.; Hong, S.; Choi, S.Y.; et al. Therapeutic Effects of Risperidone against Spinal Cord Injury in a Rat Model of Asphyxial Cardiac Arrest: A Focus on Body Temperature, Paraplegia, Motor Neuron Damage, and Neuroinflammation. Vet. Sci. 2021, 8, 230. https://0-doi-org.brum.beds.ac.uk/10.3390/vetsci8100230
Lee T-K, Lee J-C, Tae H-J, Kim H-I, Shin MC, Ahn JH, Park JH, Kim DW, Hong S, Choi SY, et al. Therapeutic Effects of Risperidone against Spinal Cord Injury in a Rat Model of Asphyxial Cardiac Arrest: A Focus on Body Temperature, Paraplegia, Motor Neuron Damage, and Neuroinflammation. Veterinary Sciences. 2021; 8(10):230. https://0-doi-org.brum.beds.ac.uk/10.3390/vetsci8100230
Chicago/Turabian StyleLee, Tae-Kyeong, Jae-Chul Lee, Hyun-Jin Tae, Hyung-Il Kim, Myoung Cheol Shin, Ji Hyeon Ahn, Joon Ha Park, Dae Won Kim, Seongkweon Hong, Soo Young Choi, and et al. 2021. "Therapeutic Effects of Risperidone against Spinal Cord Injury in a Rat Model of Asphyxial Cardiac Arrest: A Focus on Body Temperature, Paraplegia, Motor Neuron Damage, and Neuroinflammation" Veterinary Sciences 8, no. 10: 230. https://0-doi-org.brum.beds.ac.uk/10.3390/vetsci8100230