
Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine
by
 and
and
Spencer R. Marsh
1,2, 
Zachary J. Williams
1,2,3,
Kevin J. Pridham
1,2 and
Robert G. Gourdie
1,2,3,4,5,* 1
Fralin Biomedical Research Institute at VTC, Virginia Tech, Roanoke, VA 24016, USA
2
Center for Heart and Reparative Medicine Research, Virginia Tech, Roanoke, VA 24016, USA
3
Translational Biology Medicine and Health Graduate Program, Virginia Tech, Roanoke, VA 24016, USA
4
Department of Biomedical Engineering and Mechanics, Virginia Tech, Blacksburg, VA 24061, USA
5
Department of Emergency Medicine, Virginia Tech Carilion School of Medicine, Virginia Tech, Roanoke, VA 24016, USA
*
Author to whom correspondence should be addressed.
J. Cardiovasc. Dev. Dis. 2021, 8(5), 52; https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050052
Submission received: 2 March 2021
/
Revised: 19 April 2021
/
Accepted: 1 May 2021
/
Published: 5 May 2021
/
Corrected: 16 April 2022
(This article belongs to the Special Issue Cardiomyopathy at the Sub-Cellular Level)
Abstract
:Connexin (Cx43)-formed channels have been linked to cardiac arrhythmias and diseases of the heart associated with myocardial tissue loss and fibrosis. These pathologies include ischemic heart disease, ischemia-reperfusion injury, heart failure, hypertrophic cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and Duchenne muscular dystrophy. A number of Cx43 mimetic peptides have been reported as therapeutic candidates for targeting disease processes linked to Cx43, including some that have advanced to clinical testing in humans. These peptides include Cx43 sequences based on the extracellular loop domains (e.g., Gap26, Gap 27, and Peptide5), cytoplasmic-loop domain (Gap19 and L2), and cytoplasmic carboxyl-terminal domain (e.g., JM2, Cx43tat, CycliCX, and the alphaCT family of peptides) of this transmembrane protein. Additionally, RYYN peptides binding to the Cx43 carboxyl-terminus have been described. In this review, we survey preclinical and clinical data available on short mimetic peptides based on, or directly targeting, Cx43, with focus on their potential for treating heart disease. We also discuss problems that have caused reluctance within the pharmaceutical industry to translate peptidic therapeutics to the clinic, even when supporting preclinical data is strong. These issues include those associated with the administration, stability in vivo, and tissue penetration of peptide-based therapeutics. Finally, we discuss novel drug delivery technologies including nanoparticles, exosomes, and other nanovesicular carriers that could transform the clinical and commercial viability of Cx43-targeting peptides in treatment of heart disease, stroke, cancer, and other indications requiring oral or parenteral administration. Some of these newly emerging approaches to drug delivery may provide a path to overcoming pitfalls associated with the drugging of peptide therapeutics.
1. Connexin43-Formed Channels
The subunit of gap junction channels, connexin proteins, are widely expressed in the heart, as well as throughout the body [1,2,3,4]. Connexins are crucially important to cardiac electrophysiology, having direct and indirect assignments in facilitating the propagation of action potentials between cardiomyocytes [5,6]. Connexin43 (Cx43 gene name GJA1) is the main cardiac connexin, being largely expressed by cardiomyocytes, but also expressed by fibroblasts, myofibroblasts, and vascular cells in the heart [7,8,9,10]. The half-life of Cx43 is ~1.5 h, which is approximately 1000 times shorter than that of cardiac collagen—a prodigious rate of turnover hinting at the substantial and still not as yet fully understood functions of Cx43 [6,11,12]. As is the case with all 21 expressed connexins that are encoded by the human genome [13], Cx43 is a transmembrane protein, with four membrane-spanning domains and cytoplasmically located loop, amino- and carboxyl-terminal domains [14].
Six connexin subunits oligomerize during trafficking to the cell membrane to form a connexon channel, which when open is capable of transferring ions and other small molecules (typically < 1000 daltons) in a gated and relatively nonselective manner (Figure 1) [5,6]. Connexons formed by most connexins can occur in two states in the cell membrane, either as undocked hemichannels (HCs) or docked with another connexon from an adjacent cell to form a gap junction (GJ) channel. Consequently, connexons can perform two distinct information exchange functions: first, as undocked connexon HCs, which enable two-way exchange between the cell interior and the extracellular milieu; and second, as docked connexons in GJ channels providing a regulated pathway for cytoplasmic exchange between cells without recourse to the extracellular space. Undocked HCs in the cell membrane comprised of Cx43 are typically closed but can open in response to various prompts including ischemic injury [15,16,17,18,19,20,21,22].
The biology of connexin structure and function and particularly that of Cx43 and its functions in intercellular communication have been covered extensively by us and others [6,7,9,10,23,24]. A focus of this review is the biology and pathophysiology of HCs formed by Cx43 in the myocardium, as well as the growing literature on the opportunities and barriers to pharmacological targeting of these channels in the treatment of heart disease. There is a wealth of preclinical data indicating the potential for therapeutic benefit from targeting HC activity by drugs mimicking the sequence of Cx43 in experimental models of human pathology, including those involving injury to the skin, heart, and brain [4,24,25,26,27,28]. If translation of these Cx43-targeting drugs to the clinic is to occur, careful attention to addressing questions of how to safely and effectively deliver drugs based on short peptides is required.
2. Connexin43 and Myocardial Pathology
Numerous cardiac disease processes and pathologic mechanisms have been linked to Cx43 (recent reviews of this topic include [9,24,28,29,30,31]). Of these, the most studied and clinically significant are myocardial infarction (MI) and ischemia reperfusion (I/R) injuries of the heart linked to this syndrome [6]. In the post-MI environment, Cx43 expression is reduced and localization of Cx43 is disturbed (lateralized) in myocytes at the infarct border from as soon as 1 h following ischemic insult [32]. Long-term Cx43 downregulation and lateralization are thought to contribute to the increased potential for electrical conduction disturbance in hearts subject to ischemic injury, particularly re-entrant arrhythmias triggered in tissues surrounding a MI—the infarct border zone (IBZ) [33,34].
Among the post-translational changes that appear to be significant to the injury status of the heart are alterations in the phospho-status of Cx43. Ek-Vitorin and colleagues found that Cx43 phosphorylated at a consensus PKC (protein kinase C) site, serine 368, was retained at intercalated disks during early ischemia and that this retention was associated with cardioprotection [35]. Pertinently, phosphorylation of Cx43 at S368 (Cx43 pS368) is correlated with reduced activity of Cx43-formed HCs [35,36,37]. Phosphorylation of Cx43 associated with mitochondrial membranes has also been linked to I/R injury [38]. Phosphorylation at S368 and certain other serines in CT (e.g., Serines 262 and 373) are dependent upon dephosphorylation of a serine 365, “gatekeeper” amino acid [39]. The downstream changes to Cx43 that ensue following S365 dephosphorylation result in changes to the gating and perm-selectivity of Cx43-formed channels, as well as the dysregulation of Cx43-ZO-1 interactions [40]. Another post-translational modification significantly affecting Cx43 is ubiquitination [41]. One of first pieces of evidence that Cx43 is ubiquitinated was provided by Laing and Beyer [42]. Cx43 ubiquination acts as a signal for gap junction endocytosis by recruiting the ubiquitin binding protein Eps15 (epidermal growth factor receptor substrate-15) [43]. Significantly, Cx43 ubiquitination has been proposed to control its post-endocytic sorting from early endosomes to lysosomes, controlling degradation of Cx43 [44]. Ubiquitinated forms of Cx43 are recognized by the endosomal sorting complex required for transport (ESCRT) sorting system, which is responsible for sorting into endosomes and exosomes.
Post-translational modifications to Cx43 appear to be of particular significance at the edge of GJs—in the perinexus—a specialized nanodomain where Cx43 HCs are concentrated (Figure 1) [6,45]. During the acute phase of an MI, large-scale opening of Cx43 HCs occurs [18,46]. HC opening may be also exacerbated during the reperfusion phase of an I/R injury. Pro-inflammatory and injury-spread signals resulting from unregulated opening of Cx43 HCs are likely crucial to the generation and severity of I/R damage [6,10,47,48]. The flux of cytoplasmic contents released by HCs is thought to contribute to a “bystander effect”, wherein cell loss and necrotic damage are caused in otherwise healthy tissue adjacent to the primary site of injury. Intercellular coupling by GJs may also contribute to injury spread via the bystander effect [49,50,51]. Examples of tissues susceptible to the “bystander effect” include myocardium-at-risk [52], cardiac muscle tissue surrounding the ischemic core of an MI. Adenosine triphosphate (ATP) is thought to be one of the more consequential pro-inflammatory molecules released by HCs [17,53,54,55]. Extracellular gradients of ATP facilitate directed migration by neutrophils to the sites of injury, disease, or infection [56]. This purinergic signal also induces production of extracellular nets—a lethal behavior elicited by inflammatory cells that likely contributes to myocyte death and the extent of myocardium-at-risk following MI [57].
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a pathology that is likely directly impacted by Cx43 expression and phosphorylation; ARVC patients experience a loss of desmosomes, a specialized cell–cell junction [58,59,60]. Among key proteins in desmosomes is desmoplakin—the gene-encoding desmoplakin was the first desmosomal gene to be linked to ARVC [61]. When this protein is deleted from cardiomyocytes, the cells exhibit large reductions in GJs and Cx43, as well as changes to Cx43 phospho-status, associated with loss of intercellular communication [62]. A mutation of another desmosomal protein, plakophilin-2 (PKP2) has shed further light on ARVC disease mechanisms [60]. Cardiomyocyte-specific knockout of PKP2 in mice results in generation of an arrhythmogenic substrate (Ca2+ dysregulation) that was apparent at time points before overt structural changes in myocardium associated with ARVC-like disease (e.g., fibrosis). Interestingly, Cx43 ablation relieved these functional deficits in the ventricle. Moreover, treatment with a selective peptide-based blocker of Cx43 HCs normalized arrhythmogenic Ca2+ dynamics—suggestive of a role for HCs in the formative stages of ARVC pathogenesis.
Duchenne muscular dystrophy (DMD) is an X-linked recessive disorder resulting from loss-of-function of dystrophin [63]. In skeletal and cardiac myocytes cells, dystrophin is a membrane-associated protein that links the cytoskeleton and the surrounding extracellular matrix, thereby providing mechanical integrity during muscle cell work [64]. The absence of dystrophin triggers a cascade of biochemical changes that ultimately leads to the death of muscle cells and fibrosis associated with myofibroblast proliferation. The primary culprit in muscle loss appears to be intracellular calcium overload and the triggering of oxidative stress pathways leading to cell death. This chain of events results in over 50% of DMD patients experiencing cardiomyopathy by the age of 10, with over 90% of patients experiencing cardiac dysfunction by the age of 18 [65]. The particular danger in DMD in relation to Cx43 lies in progression to dystrophic cardiomyopathy, a pathology strongly affecting Cx43 expression and function [66]. Dystrophic cardiomyopathy patients experience Cx43 remodeling away from intercalated discs to lateralized positions, increased Cx43 expression, and a propensity to develop disturbances to cardiac conduction. Recent evidence suggests that unregulated opening of HCs is central to the role of Cx43 in the pathogenesis of DMD [67,68,69]. S-nitrosylation and reduced phosphorylation of Cx43 at serines S325/S328/S330 have been reported to be associated with HC activation, arrhythmogenesis, and ventricular remodeling in the Mdx mouse model of DMD [67]. A recent study has suggested that inhibition of aberrant HC activity in skeletal muscle macrophages neighboring Cx43 nonexpressing fibers in symptomatic DMD mice leads to prevention of Cx43 remodeling in the heart and protection from the loss of skeletal and cardiac muscle cells that characterize this disease [70].
Yet another example of probable involvement of Cx43 HCs in cardiac disease processes comes from a study of myocytes expressing altered nuclear lamin A/C proteins, mutations associated with laminopathy—a disease in humans leading to heart failure and arrhythmias [71]. Cultured neonatal rat cardiomyocytes expressing pathologic human lamin A/C mutations exhibit altered microtubule structure, hemichannel localization and beating force, frequency, and contractile amplitude. The common endpoints of diseases such as ARVC, DMD, and laminopathy are muscle cell death and irreversible replacement of lost myocardium with scar tissue. The mounting evidence suggests that HC activation is a precursor to this fibrotic replacement, often in association with oxidative stress, dysregulated intracellular Ca2+ dynamics, disturbances to cardiac conduction, and arrhythmias.
3. Cx43-Targeting Therapeutic Peptides
Prior to the new millennium, connexins were not considered well suited for pharmacological targeting for reasons that included their minimal extracellular profile and, thus, absence of an obvious external receptor for ligand binding. However, over the last decade, there has been growing interest in the potential for connexin pharmacology. This has occurred as a result of growing evidence of roles for connexin-formed channels in clinically relevant phenomena including arrhythmias, cancer, wound healing, inflammation, and tissue injury responses, as well as the growing understanding of the potential contribution of HCs and GJs to the “bystander effect”, such as what occurs in myocardium-at-risk following MI. Additionally, a path to drugging Cx43 has been illuminated by the development of peptides that mimic or bind to Cx43 [4,6,26,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94]. Figure 2 provides a summary of these peptidic therapeutic candidates, and Table 1 shows results to date pertinent to heart disease as reported in preclinical studies, each of which is discussed in the following sections.
3.1. Mimetic Peptides Based on Cx43 Extracellular Domains
3.1.1. Gap26 and Gap27
The first connexin mimetic peptides discovered to have biological activity were Gap26 and Gap27, which contain segments of extracellular loops (EL) 1 and 2, of Cx43, respectively [86,87,88,89,90,91,92]. In initial reports, these peptides were found to inhibit the synchrony of contractions between pairs of cell aggregates prepared from embryonic chick hearts [89]. These results led the authors to propose that Gap26 and Gap27 may be inhibiting gap junctional coupling. The mechanisms by which Gap26/27 interfere with connexin-formed channels remain to be fully understood. A review characterizing the possible mechanism of action for extracellular loop peptides is found in the literature by Berthoud et al. [90]. It is suggested that these peptides may reversibly interact with the EL domain of HCs, thereby preventing the interaction and docking of HCs to form GJ channels [90,91]. Evidence supporting this hypothesis comes from atomic force microscopy experiments in which Gap26 was covalently bonded to the scanning tip and exhibited binding interactions with Cx43 EL domains [92]. The data indicate that peptides derived from the second Cx43 EL (EL2) may interact with EL2 and not EL1, while EL1 peptides synergistically increase inhibition of Cx43 channel formation when co-administered with an EL2-based peptide [93]. Peptide mimetics of connexin extracellular loop domains have also been reported to inhibit the activity of channels formed by other connexins, including Cx40 and Cx37 [94,95,96,97,98].
Another possibility for the effects of Gap26/27 mode of action may be direct interactions of peptides with existing GJs, resulting in separation of docked channels or in blocking of GJ gating. While binding with existing GJs is possible in principle and has been proposed, it has yet to be proven. A theory that has gained more traction is direct interaction of Gap26/27 with HCs, which is presumed to block the formation of GJs. The most robust evidence for this proposal comes from the short time course in which EL peptides block HC activity, relative to GJs [88]. HCs are inhibited within a few minutes, whereas GJ inhibition occurs over time scales of an hour or more. A recent advance in both the administration and mechanism of EL peptides has been the development of a lipidated version of a Gap27-like peptide (SRPTEKT-Hdc) by Burt and colleagues [99]. This chemical modification increased the potency of EL-peptide with respect to effects on Ca2+-wave propagation, dye coupling, and HC-mediated dye uptake in cultured cells, wherein it showed activity at nanomolar concentrations, rather than in the micromolar range, as occurred for nonlipidated peptide. Interestingly, SRPTEKT-Hdc did not have similar effects on cells expressing Cx43 with mutations at S368, suggesting that phosphorylation at this locus may be involved in the mode of action of Gap26/27-like peptides [99].
Some of the earliest studies on Cx43 EL mimetic peptide effects in heart cells were carried out in vitro on monolayers of cultured neonatal rat ventricular myocytes subject to simulated ischemia (oxygen-glucose deprivation)—a challenge that results in upregulated HC opening within an hour [46]. Treatment of myocyte monolayers that were subject to simulated ischemic injury with Gap26 reduced HC opening and improved myocyte viability up to 80% of control levels. Gap26 and 27 have also demonstrated cardioprotective effects in intact hearts in ischemia reperfusion (IR) injury models in rodents [75,76,77,78]. Hawat and coworkers found that single bolus injections of Gap26 or Gap27 at 1 μg/kg into the jugular vein of rats subjected to MI in vivo, either prior to or following the ischemic episode, caused infarct size reductions in injured ventricles of up to 61% relative to control rats [77]. In addition to acute treatments, there is also evidence that repeated dosing of EL-based peptides can have beneficial effects in chronic models of heart disease. Lucero et al. [79] administered Gap27 continuously via osmotic minipumps in a high-output heart failure model in adult male rats for 4 weeks. This recurring and sustained treatment regime resulted in significant improvements in heart mechanical function, lowered arrhythmia burden and reduced cardiac hypertrophy, as compared to control hearts—supporting further investigations of the therapeutic use of EL-based peptides in mitigating progression to heart failure.
3.1.2. Peptide5
Peptide5, developed by Green and coworkers, is another well-studied Cx43 EL mimetic, incorporating the SRPTEK domain found within Gap27, with the full sequence VDCFLSRPTEKT [100]. This peptide was developed by screening various sequences from Cx43 EL domains for optimized potential to inhibit neuronal death in an in vitro model of spinal cord injury. Peptide5 inhibits HCs at a concentration of 5 uM, while concentrations of two orders of magnitude greater are required for GJ inhibition. Follow-up work has demonstrated inflammatory dampening effects, as well as neuroprotective capabilities [101,102,103]. This was supported by research which utilized Peptide5 in L3-L5 lumbar spinal injury abatement; administration of Peptide5 reduced NOD-like receptor protein 3 (NLRP3) inflammasome complex, a key mediator of neuroinflammation [104]. Ongoing studies have indicated that Cx43 HC activity has a direct role in inflammation and inflammasome activation, while Peptide5 blocks the development of these inflammatory signals [105]. Recent work by Kim and coworkers determined that the SRPTEKT region is likely the operational stretch of Peptide5 with respect to HC inhibition [93]. Other investigations of Peptide5 in preclinical models have included elucidation of its salutary effects on retinal pigment epithelial cell barrier function [106] and chronic kidney disease [107]. To date, there are no publications on Peptide5 efficacy in preclinical models of heart disease.
3.2. Mimetic Peptides Based on Cx43 Cytoplasmic Loop Domains
Gap19 and L2
Gap19 is a peptide mimicking a nine amino acid sequence (KQIEIKKFK) within the cytoplasmic CL domain of Cx43 [20,80]. The Gap19 sequence is located within a sub-region of the CL referred to as L2, which has been shown to be involved in Cx43 self-interactions with domains on the CT of Cx43, with the balance of data suggesting the molecular mechanism of Gap19 involves inhibition of Cx43 CL-CT interactions. This mode-of-action is further supported by studies of the effects of Gap19 on Ca2+ sensitive gating of Cx43 CT truncation mutants [108]. Gap19 also seems likely to modify interaction with interacting partners such as ZO-1, whose PDZ2 also has strong affinity for the portion of the Cx43 CT that interacts with Gap19 [85]. Importantly, Gap19 appears to show selectivity for Cx43s HCs, only affecting GJ activity at high concentrations [20,109]. Recent studies have also indicated that Gap19 may have a bimodal effect on gating, decreasing gating of a 210 pS conductance state but also significantly increasing the open probability of an 80 pS substate of HCs formed by Cx43 [110].
The first demonstration that Gap19 may have effects in a preclinical model of cardiac disease was an investigation of its inhibitory actions on metabolic inhibition-enhanced HC opening, protecting myocytes against volume overload and cell death following IR injury in vitro, as well as modestly decreasing MI size after myocardial IR injury to mouse hearts [80]. There is evidence the cardioprotective effects of Gap19 may relate to inhibition of mitochondrial Cx43 [81]. Treatment of mitochondria isolated from subsarcolemmal zones of ventricular myocytes with Gap19 caused reductions in the velocity of potassium influx– a significant aspect of oxygen consumption in these organelles. The facility of Gap19 to act as a modifier of HC gating also provided a tool in studies of the role of Cx43 HCs in the pathogenesis of ARVC and DMD, wherein Gap19 was found to have anti-arrhythmic effects in mouse models of these diseases [60,82].
As Gap19 targets cytoplasmic domains of Cx43 the peptide must gain entry to cells for activity—an ability that was achieved in many past studies via conjugation to TAT cell penetration peptide [80,111]. In a recent innovation, Rupenthal et al. linked Gap19 to the cell-penetrating peptide Xentry (XG19), determining that the novel peptide exhibited increased levels of cellular uptake [112]. The authors also reported preferential uptake of XG19 at sites of hypoxic injury, leading to increased delivery to injured cells. Given the cardioprotective effects of Gap19 [80], as well as reports that this peptide decreases oxidative stress, inflammatory cytokine levels, and senescence in endothelial cells [113], the facility of Gap19 to selectively target hypoxic cells and abrogate ischemia-related pathology supports the case for ongoing research on this peptide as a candidate therapeutic.
3.3. Mimetic Peptides Based on Cx43 Cytoplasmic Terminal Domain
3.3.1. alphaCT/αCT Peptides
The first peptide incorporating a sequence from the cytoplasmic CT of Cx43 demonstrating biological activity was reported by the Gourdie lab in 2005 [85]. The peptide, known as alphaCT1 (αCT1), includes an antennapedia cell penetration sequence and the CT-most nine amino acids of Cx43, mimicking its ZO-1 PDZ2 binding ligand. In cell culture models, αCT1 was shown to increase GJ size and coupling, as well as reduce levels of HC activity [85,114]. The peptide was later found to also bind domains within Cx43 itself [83,115], including the CT H2 domain [116]. At present, it is unclear whether these interactions occur within the same Cx43 molecule or involve dimeric CT-CT interactions between different Cx43 molecules in the same connexon. The interaction of αCT1 with the H2 domain is associated with the increase in a PKCe-mediated phosphorylation of Cx43 at S368 [83,117] a post-translational modification that has been linked to reduced activity of Cx43-formed channels [36,37,39].
Whilst initially developed as a research tool, αCT1 was subsequently found to increase closure rate and reduce inflammation and scarring in skin and corneal wound healing studies in rodents [4,26,118,119]. Based on such results, Ghatnekar and colleagues at FirstString Research Inc. have advanced the peptide to testing in humans, reporting outcomes from Phase II clinical trials on the safe and efficacious use of αCT1 in promoting faster closure of venous leg ulcers and diabetic foot ulcers, as well as in surgical scar mitigation [120,121,122]. Preclinical studies of αCT1 also indicate potential for its use in treatment of heart disease. αCT1 has been shown to have anti-arrhythmic effects [117], as well as demonstrating cardioprotective properties in mouse hearts subject to ischemia reperfusion injuries [83]. A short version of αCT1 25 mer called αCT11 (nine amino acids) was found to be significantly more potent in reducing ischemia reperfusion injury in Langendorff-perfused mouse hearts than αCT1 [83]. Moreover, when αCT11 was administered postmyocardial infarction in vivo, it reduced infarct size by nearly 50%, raising the prospect that this short peptide could have potential for clinical translation [84].
Of further interest with respect to the activity and mechanism of Cx43 CT peptide mimetics is that Shaw and coworkers recently demonstrated that exogenous overexpression of a naturally occurring 20 kDa isoform of Cx43 corresponding to its CT domain (GJA1-20k) reduces myocardial infarct size in mouse hearts subject to ischemia reperfusion injury [123]. The effects reported appear to be similar to the cardioprotective activity observed for αCT1 and αCT11 [83,84]. GJA1-20k comprises considerably more of the Cx43 CT sequence than αCT11 and αCT11 and thus care must be taken to avoid over extending interpretation beyond available data. Nonetheless, it may be material to the cardioprotective mechanism of CT mimetics that the GJA1-20k Cx43 isoform associates with the outer mitochondrial membrane and affects mitochondrial motility and biogenesis [123].
3.3.2. CT9
In 2010, Leybaert and colleagues published studies of peptides containing sequences identical to αCT1, including a molecule called CT9 based on the CT-most nine amino acids of Cx43 (RPRPDDLEI) [124]. This group further reported that Cx43 CT-based peptides promoted thrombin-induced Cx43 HC opening. A more recent study showed the potential for CT9 to acutely activate HCs in the presence of TNF alpha or low extracellular Ca2+ concentrations—though CT9 had no effect on HCs in the absence of lowered [Ca2+] or TNFα [125]. These data raise a paradox, as the results from αCT1 and αCT11 suggest that under some circumstances the effects of these peptides are consistent with reduced HC activity—including reductions in HC density within the perinexus and HC permeation [114], together with increased S368 phosphorylation of Cx43 [83,117,126]. The context of measurement and other experimental details may be considerations in addressing these differing results. First, it is worth noting that generally CT9 has not been shown to promote HC opening in its own right—experimental accounts indicate that CT9-induced HC activation typically occurs in the presence of cofactors, e.g., a HC-opening treatment such as lowered extracellular [Ca2+] [124,125]. In an interesting parallel, Palatinus et al. found that αCT1-enhancement of PKC-mediated phosphorylation of the Cx43 was dependent on peptide treatment being a cofactor in accompanying a cellular injury [126]. Second, descriptions of the direct HC-activating effects of CT9 have thus far come from studies of isolated cells. Given the perinexal nanodomain is not present, or is disrupted in unitary cells, it may be that regulatory elements normally at-hand in this key HC niche are not available to exert modulatory effects on HC activity. Third, and probably most important of all, reports of the time course of activation of HCs by CT9 have thus far been confined to recording of less than a few seconds [125]. The HC-opening effects of CT9 over time intervals extending for minutes or hours are presently unknown. The induction of channel inhibitory phosphorylations of Cx43 or changes to GJ/HC organization reported following αCT1/αCT11 treatment were measured over time intervals considerably longer than a few seconds, i.e., many minutes to multiple hours and days [83,84,114,117]. Thus, one path to reconciling differing interpretations on CT9 vs. αCT1/αCT11 could be that the phenomena observed in response to isolated Cx43 CT fragments depend on the time frame of measurement—namely, that there may be a short-term HC-activating effect and longer-term HC downregulation. A pertinent consideration here may be the salutary effect of ischemic preconditioning by short bursts of hypoxia. Acute ischemia is a known HC-opening prompt [46], but longer-term preconditioning results in reduced activity of Cx43-formed channels and enhanced cardioprotection from ischemia reperfusion injury [48]. Thus, the apparently differing results on CT9 and αCT1/αCT11 provide a fruitful dilemma. Ongoing research of the paradoxes inherent in the findings from different groups may lead to new approach to treatment of diseases of the heart.
3.3.3. Other Cx43 CT Mimetic or Targeting Peptides
A number of other peptides based on or directly binding to the Cx43 CT have been reported. The JM2 peptide mimicking 15 amino acids located in the juxtamembrane region of Cx43 CT (VFFKGVKDRVKGRSD) has been shown to inhibit ATP release triggered by low extracellular Ca2+ concentrations in cultures of human microvascular endothelial cells [127]—as such, JM2 appears to show Cx43 HC inhibitory activity. The JM2 peptide incorporates a sequence of Cx43 thought to function as a microtubule binding domain [128]. JM2 has demonstrated utility in preclinical studies as an anti-inflammatory agent in the foreign body response [127] and in the targeted killing of glioblastoma stem cells [129]. Interestingly, another peptide based on sequence adjacent to juxtamembrane-located JM2 on the Cx43 CT, Tat-Cx43 266-283 has also shown efficacy in in vitro and in vivo models in decreasing glioblastoma stem cell viability [130].
Delmar and coworkers used biophysical techniques to identify novel peptide sequences that bind to the CT of Cx43 [131]. One molecule isolated via this method named RXP-E was found to prevent acidification-induced GJ uncoupling and preserve action potential propagation in cultured myocytes, suggesting potential as an anti-arrhythmic [132]. Whilst neither mimicking nor known to interact with the Cx43 CT, AAP10 is a short peptide characterized by affecting Cx43 structure and function, including modulating Cx43 phosphorylation. AAP10 and a group of related molecules have shown preclinical potential in treating cardiac disease [133]. This group includes Rotigaptide (ZP123) and Danegaptide (ZP1609), which were developed in an effort to chemically engineer more stable and potent variants of the parent AAP10 molecule [133]. The mode-of-action of these peptide-based molecules remains to be characterized, though AAP10 has been shown to increase intercellular communication between myocytes [134]. Studies in porcine and canine models have demonstrated that Rotigaptide has anti-arrhythmic properties [135,136,137]. Work in pigs have also determined that when Danegaptide was given prior to an IR injury it reduced MI severity to levels comparable to ischemic postconditioning [138]. Regrettably, accounts of the Phase II clinical trials on Danegaptide suggests that this drug candidate provides no significant cardioprotective effect in humans when administered acutely following ischemic injury [139]. Clinical studies of Rotigaptide have also been undertaken for indications including vascular disease, coronary artery disease, and heart rhythm, though these studies were terminated without follow-up reports in the primary literature [140].
4. Barriers to Clinical Translation of Cx43-Targeting Peptidic Therapeutics
There has been extensive research and development activity on peptidic therapeutics –recent reviews on the topic include [141,142]. Worldwide over 60 peptide drugs have passed regulatory approval for use in humans—with metabolic disorders and cancer being the most commonly targeted disease indications. Insulin was the first and probably longest-used therapy of this type [143,144], but the broad gamut now includes drugs such as liraglutide and glucagon-like peptide 1, which are being used for treatment of metabolic disorders, with annual sales currently exceeding two billion US dollars. No molecules based on or targeting Cx43 have as yet been approved for clinical use, though as discussed in preceding sections, some have advanced to late-stage clinical testing [4,26].
Whilst interest is burgeoning, it is recognized that significant challenges exist in clinical translation of peptidic therapeutics [145]. This includes the need to resolve difficulties associated with the administration of peptidic therapeutics, as well as a requirement to improve the stability, bioavailability, and tissue and cell penetration properties of these drug candidates. Issues such as these have likely tempered the enthusiasm of the pharmaceutical industry in pursuing development of peptide drugs, even when supporting preclinical data is strong. Short linear peptides such as those exemplified by Cx43 mimetics may present special challenges due to their relatively short life in fluids like blood—limiting the time window that such molecules achieve concentrations within the body sufficient for bioactivity. In one example, our own studies of the nine-mer αCT11 suggest that it is degraded within 30 min of incubation in mouse serum at 37 C. In this respect, it is also notable that much of the clinical development of alphaCT peptides have been as topical treatments for chronic skin wounds or injuries to cutaneous tissue [26] where degradative processes can be controlled to some extent by external application of the drug and controlled release from protective delivery vehicles. In the final section of the review, we survey strategies that have or have potential to mitigate drawbacks linked to peptidic drugs and how these lessons could be applied to Cx43-targeting therapeutics.
4.1. Stability of Peptidic Drugs In Vivo
The most common concern raised on peptide-based drugs are questions on their stability in vivo. Some naturally occurring peptides such as insulin have been designed by evolution to resist proteolysis and breakdown [146]. However, for novel and synthetic peptides such as most Cx43 mimetics this remains an issue. Various chemical strategies have been used to extend the half-life of peptides in vivo including addition of chemical groups to side chains and the NT and CT of the molecule, use of enantiomeric and other non-natural amino acids to assemble all or part of the sequence, and peptide stapling and cyclization modifications to increase stability of peptides [144,145,146]. For example, the Cx43-targeting peptide Danegaptide represents an enantiomeric form (i.e., composed of L- amino acids) of the naturally occurring peptide AAP10 [133]. However, modifying peptides chemically to achieve goals such as improving stability in vivo may not come without cost. For example, consideration as to whether such modifications increase toxicity, and in particular immunotoxicity, needs to be taken into account. Controlled release from a polymeric vehicle or capsule is another approach to the slow breakdown of peptides, as well as for sustaining effective systemic concentration of the drug, reducing the need for repeated dosing. This approach has been employed with αCT1, wherein the peptide has been encapsulated in alginate-poly-l-ornithine microcapsules (150 μm) and delivered in normal and diabetic corneal injury models in rabbits [147,148]. More recently, αCT1-containing nanoparticles (100 nm) were generated using double emulsion-solvent evaporation method from poly(lactic-co-glycolic acid) (PLGA). Encapsulation of αCT1 in PLGA nanoparticles enabled release of peptide over periods of three weeks in solution, characterized by an initial burst of approximately half of the encapsulated drug over the first three days, followed by sustained availability of active peptide over the remaining two and a half weeks [149].
4.2. Cell and Tissue Penetration of Peptidic Drugs
In general, peptide-based drugs have relatively poor penetration characteristics, restricting delivery to internal organs and tissues, as well as complicating administration. For example, oral administration is not an option for peptidic therapeutics, as most protein sequences, bar a few select di- and tri-peptides, are rapidly hydrolyzed following ingestion by digestive processes [150]. With a few important exceptions, peptides also do not efficiently cross tissue boundaries such as the blood–brain, blood eye, and dermal barriers. In a well-characterized example, peptides derived from the rabies virus glycoprotein have been shown to cross the blood–brain barrier and under study for their potential to deliver therapeutic cargos into brain tissues [151]. The most common employed strategy used to improve the delivery of Cx43-targeting peptides is the addition of a cell penetration sequence (CPP). A number of such constructs, including antennapedia, TAT, and XEntry, have already been discussed in this review. There are accounts that a TAT-conjugated version of Gap19 shows ability to penetrate into brain parenchyma from the circulation [109], though it remains unclear that this is a general property of TAT-conjugated Cx43 mimetic sequences. The principal rationale for addition of a CPP is to enable peptide sequences to cross cell membranes and engage cytoplasmic targets—and as such may not be a useful modification for extracellularly located connexin sequences such as Gap26 and Gap27. Nonetheless, it is notable that lipidation of EL-loop peptides increases potency by an order of magnitude or more [99]. There is also evidence that under some circumstances a CPP may not be required for cell penetration. Jiang et al. reported that αCT11, a nine amino acid peptide, was able to cross into the cytoplasm of myocytes with an efficiency similar to that of its CPP-containing parent peptide αCT1 [83]. At present, the mechanism for this is unclear, though as Cx43-formed channels have been shown to be permeated by linear peptides of up to 15 amino acids [152], open HCs thus may provide a route by which the αCT11 nine-mer is able to gain access to its cytoplasmic targets [83].
4.3. The Immune System and Peptidic Drugs
One aspect of peptide-based drugs that is not often well broached in literature is their potential interaction with the immune system. This consideration is significant in the bioavailability of peptides in vivo, particularly after a second or third administration, where a primed immune system may more efficiently clear the molecule. However, the safety implications of repeated exposure are also of interest. This concern may be further enhanced by strategies such as chemical modifications, use of non-natural amino acids, and peptide cyclization, which while improving stability in vivo, may augment the ability of the immune system to distinguish the molecule as nonself. CPPs such as TAT may also increase engagement by the immune system, promoting antigen uptake, processing, and presentation by antigen-presenting cells [153]. Unfortunately, there are few data from preclinical studies of Cx43-targeting peptides on this issue. There is some information from clinical studies on αCT1 in its Granexin formulation for topical use on skin wounds, wherein one Phase I and three Phase II clinical trials found no indication of antibodies against the peptide in patient sera [120,121,122]. The treatment regimens for chronic wounds (diabetic foot ulcers and venous leg ulcers) in these studies involved repeated weekly dosing with topically applied αCT1 over a 3 month study period, with the independent contract research organization carrying out the work finding no occurrences of severe adverse events, including no evidence of immune responses to the peptide drug. Such reports are encouraging, and the outcome in this case may point to the benefit of clinical testing strategies focused on external administration of a gel containing the peptidic drug to the skin, where exposure to the immune system may be reduced.
4.4. The Potential of Extracellular Vesicles as Vehicles for Mimetic Peptides
Topical application of Cx43-based peptides to skin has shown success in clinical testing. However, the short half-life, poor tissue penetration characteristics, and potential for interaction with the immune system of peptidic drugs remain drawbacks on extending their use to clinical indications within the body such as myocardial infarction. Fortunately, technologies are on the horizon that may enable some of these concerns to be surmounted. One avenue that our laboratory is putting emphasis on in ongoing work is the encapsulation of Cx43 therapeutic drug candidates such as αCT11 in exosomes. Mounting evidence suggests that this class of small extracellular vesicle may enable shielding and protection of fragile drug cargos during transport within body fluids [154,155]. The ability of exosomes to release their encapsulated cargo into the cytoplasm of target cells also obviates the need for CPP sequences. Exosomes demonstrate immune privilege and certain types of these nanovesicles are adept at crossing tissues boundaries, including the blood–brain-barrier [156,157,158,159,160]. Mammalian milk turns out to be a rich source of exosomes [161,162,163] opening up the possibility of production at industrial scale. Milk exosomes also show a unique propensity to be taken up from the gut and transferred to the circulation, a characteristic that may enable oral administration of peptide therapeutics [164]. Considerable work is still required, including development of cost-effective isolation techniques for producing pharmaceutical grade exosomes in large amounts, reliable approaches to drug loading, and extensive preclinical and clinical testing of these novel formulations.
5. Concluding Remarks
There is strong evidence from preclinical and clinical research that targeting gap junctional Cx43 may provide a path to amelioration of human pathologies, including diseases of the heart, the focus of this review. The last two decades have seen the emergence of drug-like peptidic molecules mimicking or interacting with Cx43 that have shown therapeutic potential. A number of questions and opportunities are presented for ongoing study. First, the potent bioactivity of synthetic Cx43 fragments presents the interesting question of whether these molecules are amplifying a naturally occurring injury response mechanism. It is well established that polypeptides corresponding to the Cx43 CT are generated by alternate translation [6]. This mechanism includes relatively short Cx43 CT sequences, including an 11 kDa peptide that has been reported to traffic to the nucleus, with associated effects on cell-cycle progression [165]. Ongoing research might include investigation of whether there are other small, bioactive Cx43 sequences present endogenously, generated by processes such as proteolytic cleavage. Second, techniques such as cryoelectron microscopy are enabling unprecedented views of connexon structure in the context of a lipid environment at 1.9 angstrom resolution [166]. This molecular-scale insight provides an opportunity for rationale design of connexin-selective ligands, including peptidic drugs, though the disordered nature of connexin CTs may hinder implementation of this approach for therapeutic candidates targeting this domain. This is a significant challenge to surmount given, as outlined in this review, a majority of currently available therapeutic molecules mimic or target the Cx43 CT. Third, a challenge to translating the clinical promise of existing Cx43 mimetic drug candidates has been to overcome the long-standing resistance to therapeutic peptides by the pharmaceutical industry. This includes addressing matters such as the limited options for administration of this class of drug molecules, as well as the short half-life and poor tissue penetration characteristics of peptide therapeutics. Nonetheless, with the advent of delivery modalities such as exosomes, this class of connexin targeting drugs could be on the cusp of a new era in which we finally see barriers to their clinical translation significantly lowered.
Funding
This work was supported by the U.S. National Institutes of Health R01 grants 2R01HL056728-18 and 5R01HL141855-03 to R.G.G.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
R.G.G. acknowledges the generous philanthropic grant of the Edward N. and Della L. Thome Memorial Foundation, Bank of America, N.A., Trustee, to support the work of his laboratory.
Conflicts of Interest
R.G.G. is a nonremunerated member of the Scientific Advisory Board of FirstString Research, which licensed α carboxyl terminus 1. R.G.G. has modest (<1% of total stock) ownership interests in the company. The remaining authors have no disclosures to report.
References
- Harris, A.L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys. 2001, 34, 325–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, E.C.; Berthoud, V.M. Gap junction gene and protein families: Connexins, innexins, and pannexins. Biochim. Biophys. Acta Biomembr. 2018, 1860, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Delmar, M.; Laird, D.W.; Naus, C.C.; Nielsen, M.S.; Verselis, V.K.; White, T.W. Connexins and Disease. Cold Spring Harb. Perspect. Biol. 2017, 10, a029348. [Google Scholar] [CrossRef]
- Laird, D.W.; Lampe, P.D. Therapeutic strategies targeting connexins. Nat. Rev. Drug Discov. 2018, 17, 905–921. [Google Scholar] [CrossRef]
- Harris, A.L. Electrical coupling and its channels. J. Gen. Physiol. 2018, 150, 1606–1639. [Google Scholar] [CrossRef] [Green Version]
- Gourdie, R.; Smyth, J.; Poelzing, S. Gap Junctional Connexin43: Novel Insights from the New Millennium and Their Clinical Implications. In Cardiac Electrophysiology: From Cell to Bedside, 8th ed.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Sorgen, P.L.; Trease, A.J.; Spagnol, G.; Delmar, M.; Nielsen, M.S. Protein(-)Protein Interactions with Connexin43: Regulation and Function. Int. J. Mol. Sci. 2018, 19, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoagland, D.T.; Santos, W.; Poelzing, S.; Gourdie, R.G. The role of the gap junction perinexus in cardiac conduction: Potential as a novel anti-arrhythmic drug target. Prog. Biophys. Mol. Biol. 2019, 144, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Martins-Marques, T.; Ribeiro-Rodrigues, T.; Batista-Almeida, D.; Aasen, T.; Kwak, B.R.; Girao, H. Biological Functions of Connexin43 Beyond Intercellular Communication. Trends Cell Biol. 2019, 29, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Rusiecka, O.M.; Montgomery, J.; Morel, S.; Batista-Almeida, D.; Van Campenhout, R.; Vinken, M.; Girao, H.; Kwak, B.R. Canonical and Non-Canonical Roles of Connexin43 in Cardioprotection. Biomolecules 2020, 10, 1225. [Google Scholar] [CrossRef]
- Laird, D.W.; Puranam, K.L.; Revel, J.P. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem. J. 1991, 273 Pt 1, 67–72. [Google Scholar] [CrossRef] [Green Version]
- Beardslee, M.A.; Laing, J.G.; Beyer, E.C.; Saffitz, J.E. Rapid turnover of connexin43 in the adult rat heart. Circ. Res. 1998, 83, 629–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyamada, M.; Takebe, K.; Oyamada, Y. Regulation of connexin expression by transcription factors and epigenetic mechanisms. Biochim. Biophys. Acta 2013, 1828, 118–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.; Yue, B.; Aoyama, H. Crucial motifs and residues in the extracellular loops influence the formation and specificity of connexin docking. Biochim. Biophys. Acta Biomembr. 2018, 1860, 9–21. [Google Scholar] [CrossRef]
- John, S.A.; Kondo, R.; Wang, S.-Y.; Goldhaber, J.I.; Weiss, J.N. Connexin-43 hemichannels opened by metabolic inhibition. J. Biol. Chem. 1999, 274, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.Y.; Kam, Y.; Koo, S.K.; Joe, C.O. Gating Connexin43 channels reconstituted in lipid vesicles by mitogen-activated protein kinase phosphorylation. J. Biol. Chem. 1999, 274, 5581–5587. [Google Scholar] [CrossRef] [Green Version]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [Green Version]
- Retamal, M.A.; Schalper, K.A.; Shoji, K.F.; Orellana, J.A.; Bennett, M.V.L.; Sáez, J.C. Possible involvement of different connexin43 domains in plasma membrane permeabilization induced by ischemia-reperfusion. J. Membr. Biol. 2007, 218, 49–63. [Google Scholar] [CrossRef]
- De Vuyst, E.; Wang, N.; Decrock, E.; De Bock, M.; Vinken, M.; Van Moorhem, M.; Lai, C.; Culot, M.; Rogiers, V.; Cecchelli, R.; et al. Ca (2+ ) regulation of Connexin43 hemichannels in C6 glioma and glial cells. Cell Calcium 2009, 46, 176–187. [Google Scholar] [CrossRef]
- Wang, N.; De Bock, M.; Decrock, E.; Bol, M.; Gadicherla, A.; Bultynck, G.; Leybaert, L. Connexin targeting peptides as inhibitors of voltage- and intracellular Ca-triggered Cx43 hemichannel opening. Neuropharmacology 2013, 75, 506–516. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17 (Suppl. 1), 11. [Google Scholar] [CrossRef] [Green Version]
- Ek-Vitorín, J.F.; Pontifex, T.K.; Burt, J.M. Cx43 Channel Gating and Permeation: Multiple Phosphorylation-Dependent Roles of the Carboxyl Terminus. Int. J. Mol. Sci. 2018, 19, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Bai, X.; Liu, Y.; Wang, K.; Shen, B.; Sun, X. Current Concepts and Perspectives on Connexin43: A Mini Review. Curr. Protein Pept. Sci. 2018, 19, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.E.; Gourdie, R.G. Cx43 and the Actin Cytoskeleton: Novel Roles and Implications for Cell-Cell Junction-Based Barrier Function Regulation. Biomolecules 2020, 10, 1656. [Google Scholar] [CrossRef] [PubMed]
- Grek, C.L.; Rhett, J.M.; Ghatnekar, G.S. Cardiac to cancer: Connecting connexins to clinical opportunity. FEBS Lett. 2014, 588, 1349–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugisho, O.O.; Green, C.R.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D. Connexin43 hemichannels: A potential drug target for the treatment of diabetic retinopathy. Drug Discov. Today 2019, 24, 1627–1636. [Google Scholar] [CrossRef]
- Mugisho, O.O.; Rupenthal, I.D.; Paquet-Durand, F.; Acosta, M.L.; Green, C.R. Targeting connexin hemichannels to control the inflammasome: The correlation between connexin43 and NLRP3 expression in chronic eye disease. Expert Opin. Ther. Targets 2019, 23, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Delvaeye, T.; Vandenabeele, P.; Bultynck, G.; Leybaert, L.; Krysko, D.V. Therapeutic Targeting of Connexin Channels: New Views and Challenges. Trends Mol. Med. 2018, 24, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Cocozzelli, A.G.; White, T.W. Connexin43 Mutations Lead to Increased Hemichannel Functionality in Skin Disease. Int. J. Mol. Sci. 2019, 20, 6186. [Google Scholar] [CrossRef] [Green Version]
- Varela-Vázquez, A.; Guitián-Caamaño, A.; Carpintero-Fernandez, P.; Fonseca, E.; Sayedyahossein, S.; Aasen, T.; Penuela, S.; Mayán, M.D. Emerging functions and clinical prospects of connexins and pannexins in melanoma. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188380. [Google Scholar] [CrossRef]
- Kieken, F.; Mutsaers, N.; Dolmatova, E.; Virgil, K.; Wit, A.L.; Kellezi, A.; Hirst-Jensen, B.J.; Duffy, H.S.; Sorgen, P.L. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ. Res. 2009, 104, 1103–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, J.H.; Green, C.R.; Peters, N.S.; Rothery, S.; Severs, N.J. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am. J. Pathol. 1991, 139, 801–821. [Google Scholar] [PubMed]
- Peters, N.S.; Coromilas, J.; Severs, N.J.; Wit, A.L. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation 1997, 95, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Ek-Vitorin, J.F.; King, T.J.; Heyman, N.S.; Lampe, P.D.; Burt, J.M. Selectivity of Connexin43 channels is regulated through protein kinase C-dependent phosphorylation. Circ. Res. 2006, 98, 1498–1505. [Google Scholar] [CrossRef] [Green Version]
- Lampe, P.D.; TenBroek, E.M.; Burt, J.M.; Kurata, W.E.; Johnson, R.G.; Lau, A.F. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J. Cell Biol. 2000, 149, 1503–1512. [Google Scholar] [CrossRef]
- Bao, X.; Altenberg, G.A.; Reuss, L. Mechanism of regulation of the gap junction protein Connexin43 by protein kinase C-mediated phosphorylation. Am. J. Physiol. Cell Physiol. 2004, 286, C647–C654. [Google Scholar] [CrossRef]
- Boengler, K.; Stahlhofen, S.; Van De Sand, A.; Gres, P.; Ruiz-Meana, M.; Garcia-Dorado, D.; Heusch, G.; Schulz, R. Presence of Connexin43 in subsarcolemmal, but not in interfibrillar cardiomyocyte mitochondria. Basic Res. Cardiol. 2009, 104, 141–147. [Google Scholar] [CrossRef]
- Solan, J.L.; Marquez-Rosado, L.; Sorgen, P.L.; Thornton, P.J.; Gafken, P.R.; Lampe, P.D. Phosphorylation at S365 is a gatekeeper event that changes the structure of Cx43 and prevents down-regulation by PKC. J. Cell Biol. 2007, 179, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Dunn, C.A.; Lampe, P.D. Injury-triggered Akt phosphorylation of Cx43: A ZO-1-driven molecular switch that regulates gap junction size. J. Cell Sci. 2014, 127, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Totland, M.Z.; Rasmussen, N.L.; Knudsen, L.M.; Leithe, E. Regulation of gap junction intercellular communication by connexin ubiquitination: Physiological and pathophysiological implications. Cell Mol Life Sci. 2020, 77, 573–591. [Google Scholar] [CrossRef] [Green Version]
- Laing, J.G.; Beyer, E.C. The gap junction protein connexin43 is degraded via the ubiquitin proteasome pathway. J. Biol. Chem. 1995, 270, 26399–26403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, E.; Girao, H.; Yuste, A.; Patel, B.; Marques, C.; Spray, D.C.; Pereira, P.; Cuervo, A.M. Autophagy modulates dynamics of connexins at the plasma membrane in a ubiquitin-dependent manner. Mol Biol Cell. 2012, 23, 2156–2169. [Google Scholar] [CrossRef]
- Leithe, E.; Kjenseth, A.; Sirnes, S.; Stenmark, H.; Brech, A.; Rivedal, E. Ubiquitylation of the gap junction protein connexin-43 signals its trafficking from early endosomes to lysosomes in a process mediated by Hrs and Tsg101. J Cell Sci. 2009, 122, 3883–3893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhett, J.M.; Ongstad, E.L.; Jourdan, J.; Gourdie, R.G. Cx43 associates with Na(v)1.5 in the cardiomyocyte perinexus. J. Membr. Biol. 2012, 245, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani-Ishida, K.; Uemura, K.; Yoshida, K. Hemichannels in cardiomyocytes open transiently during ischemia and contribute to reperfusion injury following brief ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1714–H1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, L.; Wen, N.; Liu, C.; Zhao, M.; Zheng, L. Connexin43 and Myocardial Ischemia-Reperfusion Injury. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 14–16. [Google Scholar] [CrossRef]
- Rodriguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; Garcia-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta Biomembr. 2018, 1860, 174–181. [Google Scholar] [CrossRef]
- Ramadan, R.; Baatout, S.; Aerts, A.; Leybaert, L. The role of connexin proteins and their channels in radiation-induced atherosclerosis. Cell Mol Life Sci. 2021, 78, 3087–3103. [Google Scholar] [CrossRef]
- Sinyuk, M.; Mulkearns-Hubert, E.E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front Oncol. 2018, 8, 646. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 12, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Farah, A.; Barbagelata, A. Unmet goals in the treatment of Acute Myocardial Infarction: Review. F1000Res 2017, 6, F1000 Faculty Rev-1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, T.C.; Williams, O.J.; Martin, P.E.; Evans, W.H. ATP release by cardiac myocytes in a simulated ischaemia model: Inhibition by a connexin mimetic and enhancement by an antiarrhythmic peptide. Eur. J. Pharmacol. 2009, 605, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Soleymani, S.; Madakshire, R.; Insel, P.A. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 2012, 26, 2580–2591. [Google Scholar] [CrossRef] [Green Version]
- Dosch, M.; Zindel, J.; Jebbawi, F.; Melin, N.; Sanchez-Taltavull, D.; Stroka, D.; Candinas, D.; Beldi, G. Connexin-43-dependent ATP release mediates macrophage activation during sepsis. Elife 2019, 8, 2670. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.M.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Gehmlich, K.; Syrris, P.; Reimann, M.; Asimaki, A.; Ehler, E.; Evans, A.; Quarta, G.; Pantazis, A.; Saffitz, J.E.; McKenna, W.J. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc. Pathol. 2012, 21, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Noorman, M.; Hakim, S.; Kessler, E.; Groeneweg, J.A.; Cox, M.G.; Asimaki, A.; van Rijen, H.V.; van Stuijvenberg, L.; Chkourko, H.; van der Heyden, M.A.; et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013, 10, 412–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.C.; Perez-Hernandez, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca(2+)i Homeostasis and Connexin43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef]
- Rampazzo, A.; Nava, A.; Malacrida, S.; Beffagna, G.; Bauce, B.; Rossi, V.; Zimbello, R.; Simionati, B.; Basso, C.; Thiene, G. Towbin JA and Danieli GA. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am. J. Hum. Genet. 2002, 71, 1200–1206. [Google Scholar] [CrossRef] [Green Version]
- Lyon, R.C.; Mezzano, V.; Wright, A.T.; Pfeiffer, E.; Chuang, J.; Banares, K.; Castaneda, A.; Ouyang, K.; Cui, L.; Contu, R.; et al. Connexin defects underlie arrhythmogenic right ventricular cardiomyopathy in a novel mouse model. Hum. Mol. Genet. 2014, 23, 1134–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ervasti, J.M.; Campbell, K.P. Dystrophin and the membrane skeleton. Curr. Opin. Cell Biol. 1993, 5, 82–87. [Google Scholar] [CrossRef]
- Shirokova, N.; Niggli, E. Cardiac phenotype of Duchenne Muscular Dystrophy: Insights from cellular studies. J. Mol. Cell Cardiol. 2013, 58, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushby, K.; Connor, E. Clinical outcome measures for trials in Duchenne muscular dystrophy: Report from International Working Group meetings. Clin. Investig. 2011, 1, 1217–1235. [Google Scholar] [CrossRef]
- Shaw, R.M.; Saffitz, J.E. A role for connexin-43 in Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1608–1610. [Google Scholar] [CrossRef]
- Lillo, M.A.; Himelman, E.; Shirokova, N.; Xie, L.-H.; Fraidenraich, D.; Contreras, J.E. S-nitrosylation of connexin43 hemichannels elicits cardiac stress-induced arrhythmias in Duchenne muscular dystrophy mice. JCI Insight 2019, 4, e130091. [Google Scholar] [CrossRef]
- Himelman, E.; Lillo, M.A.; Nouet, J.; Gonzalez, J.P.; Zhao, Q.; Xie, L.-H.; Li, H.; Liu, T.; Wehrens, X.H.; Lampe, P.D.; et al. Prevention of connexin-43 remodeling protects against Duchenne muscular dystrophy cardiomyopathy. J. Clin. Investig. 2020, 130, 1713–1727. [Google Scholar] [CrossRef]
- Vielma, A.Z.; Boric, M.P.; Gonzalez, D.R. Apocynin Treatment Prevents Cardiac Connexin43 Hemichannels Hyperactivity by Reducing Nitroso-Redox Stress in Mdx Mice. Int. J. Mol. Sci. 2020, 21, 5415. [Google Scholar] [CrossRef]
- Nouet, J.; Himelman, E.; Lahey, K.C.; Zhao, Q.; Fraidenraich, D. Connexin-43 reduction prevents muscle defects in a mouse model of manifesting Duchenne muscular dystrophy female carriers. Sci. Rep. 2020, 10, 5683. [Google Scholar] [CrossRef] [Green Version]
- Borin, D.; Peña, B.; Chen, S.N.; Long, C.S.; Taylor, M.R.; Mestroni, L.; Sbaizero, O. Altered microtubule structure, hemichannel localization and beating activity in cardiomyocytes expressing pathologic nuclear lamin A/C. Heliyon 2020, 6, e03175. [Google Scholar] [CrossRef]
- Iyyathurai, J.; D’Hondt, C.; Wang, N.; De Bock, M.; Himpens, B.; Retamal, M.A.; Stehberg, J.; Leybaert, L.; Bultynck, G. Peptides and peptide-derived molecules targeting the intracellular domains of Cx43: Gap junctions versus hemichannels. Neuropharmacology 2013, 75, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Faniku, C.; O’Shaughnessy, E.; Lorraine, C.; Johnstone, S.R.; Graham, A.; Greenhough, S.; Martin, P.E.M. The Connexin Mimetic Peptide Gap27 and Cx43-Knockdown Reveal Differential Roles for Connexin43 in Wound Closure Events in Skin Model Systems. Int. J. Mol. Sci. 2018, 19, 604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooreman, A.; Van Campenhout, R.; Ballet, S.; Annaert, P.; Bossche, B.V.D.; Colle, I.; Cogliati, B.; Vinken, M. Connexin and Pannexin (Hemi)Channels: Emerging Targets in the Treatment of Liver Disease. Hepatology 2019, 69, 1317–1323. [Google Scholar] [CrossRef]
- Hawat, G.; Benderdour, M.; Rousseau, G.; Baroudi, G. Connexin43 mimetic peptide Gap26 confers protection to intact heart against myocardial ischemia injury. Pflugers Arch. 2010, 460, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Johansen, D.; Cruciani, V.; Sundset, R.; Ytrehus, K.; Mikalsen, S.-O. Ischemia induces closure of gap junctional channels and opening of hemichannels in heart-derived cells and tissue. Cell Physiol. Biochem. 2011, 28, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawat, G.; Hélie, P.; Baroudi, G. Single intravenous low-dose injections of Connexin43 mimetic peptides protect ischemic heart in vivo against myocardial infarction. J. Mol. Cell Cardiol. 2012, 53, 559–566. [Google Scholar] [CrossRef]
- Behmenburg, F.; Pickert, E.; Mathes, A.; Heinen, A.; Hollmann, M.W.; Huhn, R.; Berger, M.M. The Cardioprotective Effect of Dexmedetomidine in Rats Is Dose-Dependent and Mediated by BKCa Channels. J. Cardiovasc. Pharmacol. 2017, 69, 228–235. [Google Scholar] [CrossRef]
- Lucero, C.M.; Andrade, D.C.; Toledo, C.; Díaz, H.S.; Pereyra, K.V.; Diaz-Jara, E.; Schwarz, K.G.; Marcus, N.J.; Retamal, M.A.; Quintanilla, R.A.; et al. Cardiac remodeling and arrhythmogenesis are ameliorated by administration of Cx43 mimetic peptide Gap27 in heart failure rats. Sci. Rep. 2020, 10, 6878. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of Cx43 hemichannels by Gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [Green Version]
- Boengler, K.; Ungefug, E.; Heusch, G.; Leybaert, L.; Schulz, R. Connexin43 impacts on mitochondrial potassium uptake. Front. Pharmacol. 2013, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, J.P.; Ramachandran, J.; Xie, L.H.; Contreras, J.E.; Fraidenraich, D. Selective Connexin43 Inhibition Prevents Isoproterenol-Induced Arrhythmias and Lethality in Muscular Dystrophy Mice. Sci. Rep. 2015, 5, 13490. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hoagland, D.; Palatinus, J.A.; He, H.; Iyyathurai, J.; Jourdan, L.J.; Bultynck, G.; Wang, Z.; Zhang, Z.; Schey, K.; et al. Interaction of alpha Carboxyl Terminus 1 Peptide with the Connexin43 Carboxyl Terminus Preserves Left Ventricular Function After Ischemia-Reperfusion Injury. J. Am. Heart Assoc. 2019, 8, e012385. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Jiang, J.; Abbate, A.; Jourdan, J.L.; Gourdie, R.G. Abstract 13803: A Short Connexin43 Carboxyl Terminal-Based Peptide Permeates Hemichannels and Provides Post-Infarction Cardioprotection in vivo. Circ. Res. 2019, 140, A13803. [Google Scholar]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell. 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.L.; Evans, W.H.; Green, C.R.; Warner, A. Functional analysis of amino acid sequences in connexin43 involved in intercellular communication through gap junctions. J. Cell Sci. 1995, 108 Pt 4, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Chaytor, A.T.; Evans, W.H.; Griffith, T.M. Peptides homologous to extracellular loop motifs of Connexin43 reversibly abolish rhythmic contractile activity in rabbit arteries. J. Physiol. 1997, 503 Pt 1, 99–110. [Google Scholar] [CrossRef]
- Desplantez, T.; Verma, V.; Leybaert, L.; Evans, W.H.; Weingart, R. Gap26, a connexin mimetic peptide, inhibits currents carried by connexin43 hemichannels and gap junction channels. Pharmacol. Res. 2012, 65, 546–552. [Google Scholar] [CrossRef]
- Warner, A.; Clements, D.K.; Parikh, S.; Evans, W.H.; DeHaan, R.L. Specific motifs in the external loops of connexin proteins can determine gap junction formation between chick heart myocytes. J. Physiol. 1995, 488 Pt 3, 721–728. [Google Scholar] [CrossRef]
- Berthoud, V.M.; Beyer, E.C.; Seul, K.H. Peptide inhibitors of intercellular communication. Am J Physiol Lung Cell Mol Physiol. 2000, 279, 619–622. [Google Scholar] [CrossRef]
- Boitano, S.; Evans, W.H. Connexin mimetic peptides reversibly inhibit Ca2+ signaling through gap junctions in airway cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L623–L630. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Arce, F.T.; Ramachandran, S.; Lal, R. Nanomechanics of hemichannel conformations: Connexin flexibility underlying channel opening and closing. J. Biol. Chem. 2006, 281, 23207–23217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Griffin, J.M.; Harris, P.W.; Chan, S.H.C.; Nicholson, L.F.; Brimble, M.A.; O’Carroll, S.J.; Green, C.R. Characterizing the mode of action of extracellular Connexin43 channel blocking mimetic peptides in an in vitro ischemia injury model. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 68–78. [Google Scholar] [CrossRef]
- Ujiie, H.; Chaytor, A.T.; Bakker, L.M.; Griffith, T.M. Essential role of Gap junctions in NO- and prostanoid-independent relaxations evoked by acetylcholine in rabbit intracerebral arteries. Stroke 2003, 34, 544–550. [Google Scholar] [CrossRef] [Green Version]
- Sandow, S.L.; Goto, K.; Rummery, N.M.; Hill, C.E. Developmental changes in myoendothelial gap junction mediated vasodilator activity in the rat saphenous artery. J. Physiol. 2004, 556, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Haddock, R.E.; Grayson, T.H.; Brackenbury, T.D.; Meaney, K.R.; Neylon, C.B.; Sandow, S.L.; Hill, C.E. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2047–H2056. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, T.; Inoue, T.; Kanno, Y.; Okada, H.; Hill, C.E.; Suzuki, H. Connexins 37 and 40 transduce purinergic signals mediating renal autoregulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1–R11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makino, A.; Platoshyn, O.; Suárez, J.; Yuan, J.X.-J.; Dillmann, W.H. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin-induced diabetic mice. Am. J. Physiol. Cell Physiol. 2008, 295, C221–C230. [Google Scholar] [CrossRef] [Green Version]
- Cotter, M.L.; Boitano, S.; Lampe, P.D.; Solan, J.L.; Vagner, J.; Ek-Vitorin, J.F.; Burt, J.M. The lipidated connexin mimetic peptide SRPTEKT-Hdc is a potent inhibitor of Cx43 channels with specificity for the pS368 phospho-isoform. Am. J. Physiol. Cell Physiol. 2019, 317, C825–C842. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Alkadhi, M.; Nicholson, L.F.B.; Green, C.R. Connexin43 mimetic peptides reduce swelling, astrogliosis, and neuronal cell death after spinal cord injury. Cell Commun. Adhes. 2008, 15, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Acosta, M.L.; Nor, M.N.M.; Guo, C.X.; Mugisho, O.O.; Coutinho, F.P.; Rupenthal, I.D.; Green, C.R. Connexin therapeutics: Blocking connexin hemichannel pores is distinct from blocking pannexin channels or gap junctions. Neural Regen. Res. 2021, 16, 482–488. [Google Scholar] [CrossRef]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Danesh-Meyer, H.V.; Kerr, N.M.; Zhang, J.; Eady, E.K.; O’Carroll, S.J.; Nicholson, L.F.; Johnson, C.S.; Green, C.R. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischaemia. Brain 2012, 135, 506–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonkin, R.S.; Bowles, C.; Perera, C.J.; Keating, B.A.; Makker, P.G.; Duffy, S.S.; Lees, J.G.; Tran, C.; Don, A.S.; Fath, T.; et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 2018, 300, 1–12. [Google Scholar] [CrossRef]
- Mugisho, O.O.; Green, C.R.; Squirrell, D.M.; Bould, S.; Danesh-Meyer, H.V.; Zhang, J.; Acosta, M.L.; Rupenthal, I.D. Connexin43 hemichannel block protects against the development of diabetic retinopathy signs in a mouse model of the disease. J. Mol. Med. 2019, 97, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.; Green, C.R.; Rupenthal, I.D.; Mugisho, O.O. Connexin43 hemichannel block protects against retinal pigment epithelial cell barrier breakdown. Acta Diabetol. 2020, 57, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Price, G.W.; Chadjichristos, C.E.; Kavvadas, P.; Tang, S.C.W.; Yiu, W.H.; Green, C.R.; Potter, J.A.; Siamantouras, E.; Squires, P.E.; Hills, C.E. Blocking Connexin-43 mediated hemichannel activity protects against early tubular injury in experimental chronic kidney disease. Cell Commun. Signal. 2020, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Cassara, C.; Lin, X.; Veenstra, R.D. Calcium-calmodulin gating of a pH-insensitive isoform of connexin43 gap junctions. Biochem. J. 2019, 476, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide Gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef] [Green Version]
- Lissoni, A.; Wang, N.; Nezlobinskii, T.; De Smet, M.; Panfilov, A.V.; Vandersickel, N.; Leybaert, L.; Witschas, K. Gap19, a Cx43 Hemichannel Inhibitor, Acts as a Gating Modifier That Decreases Main State Opening While Increasing Substate Gating. Int. J. Mol. Sci. 2020, 21, 7340. [Google Scholar] [CrossRef]
- Freitas-Andrade, M.; Wang, N.; Bechberger, J.F.; De Bock, M.; Lampe, P.D.; Leybaert, L.; Naus, C.C. Targeting MAPK phosphorylation of Connexin43 provides neuroprotection in stroke. J. Exp. Med. 2019, 216, 916–935. [Google Scholar] [CrossRef]
- Coutinho, F.P.; Green, C.R.; Acosta, M.L.; Rupenthal, I.D. Xentry-Gap19 inhibits Connexin43 hemichannel opening especially during hypoxic injury. Drug Deliv. Transl. Res. 2020, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, R.; Vromans, E.; Anang, D.C.; Goetschalckx, I.; Hoorelbeke, D.; Decrock, E.; Baatout, S.; Leybaert, L.; Aerts, A. Connexin43 Hemichannel Targeting With TAT-Gap19 Alleviates Radiation-Induced Endothelial Cell Damage. Front. Pharmacol. 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef]
- D’Hondt, C.; Iyyathurai, J.; Vinken, M.; Rogiers, V.; Leybaert, L.; Himpens, B.; Bultynck, G. Regulation of connexin- and pannexin-based channels by post-translational modifications. Biol. Cell 2013, 105, 373–398. [Google Scholar] [CrossRef] [Green Version]
- Spagnol, G.; Al-Mugotir, M.; Kopanic, J.L.; Zach, S.; Li, H.; Trease, A.J.; Stauch, K.L.; Grosely, R.; Cervantes, M.; Sorgen, P.L.; et al. Secondary structural analysis of the carboxyl-terminal domain from different connexin isoforms. Biopolymers 2016, 105, 143–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Quinn, M.P.; Palatinus, J.A.; Harris, B.S.; Hewett, K.W.; Gourdie, R.G. A Peptide Mimetic of the Connexin43 Carboxyl Terminus Reduces Gap Junction Remodeling and Induced Arrhythmia Following Ventricular Injury. Circ. Res. 2011, 108, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Bryant, Z.J.; Ghatnekar, G.; Singh, U.P.; Gourdie, R.G.; Potts, J.D. A synthetic Connexin43 mimetic peptide augments corneal wound healing. Exp. Eye Res. 2013, 115, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Prasad, G.M.; Viswanathan, V.; Armstrong, D.G.; Gourdie, R.G.; Ghatnekar, G.S. Topical administration of a connexin43-based peptide augments healing of chronic neuropathic diabetic foot ulcers: A multicenter, randomized trial. Wound Repair Regen. 2015, 23, 203–212. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; Grek, C.L.; Armstrong, D.G.; Desai, S.C.; Gourdie, R.G. The effect of a connexin43-based Peptide on the healing of chronic venous leg ulcers: A multicenter, randomized trial. J. Investig. Dermatol. 2015, 135, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.S.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A Multicenter Randomized Controlled Trial Evaluating a Cx43-Mimetic Peptide in Cutaneous Scarring. J. Investig. Dermatol. 2017, 137, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basheer, W.A.; Fu, Y.; Shimura, D.; Xiao, S.; Agvanian, S.; Hernandez, D.M.; Hitzeman, T.C.; Hong, T.; Shaw, R.M. Stress response protein GJA1-20k promotes mitochondrial biogenesis, metabolic quiescence, and cardioprotection against ischemia/reperfusion injury. JCI Insight 2018, 20, e121900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponsaerts, R.; De Vuyst, E.; Retamal, M.; D’Hondt, C.; Vermeire, D.; Wang, N.; De Smedt, H.; Zimmermann, P.; Himpens, B.; Vereecke, J.; et al. Intramolecular loop/tail interactions are essential for Connexin43-hemichannel activity. FASEB J. 2010, 24, 4378–4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delvaeye, T.; De Smet, M.A.J.; Verwaerde, S.; Decrock, E.; Czekaj, A.; Vandenbroucke, R.E.; Lemeire, K.; Goncalves, A.; Declercq, W.; Vandenabeele, P.; et al. Blocking connexin43 hemichannels protects mice against tumour necrosis factor-induced inflammatory shock. Sci. Rep. 2019, 9, 16623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palatinus, J.A.; Rhett, J.M.; Gourdie, R.G. Enhanced PKCepsilon mediated phosphorylation of connexin43 at serine 368 by a carboxyl-terminal mimetic peptide is dependent on injury. Channels 2011, 5, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Calder, B.W.; Matthew Rhett, J.; Bainbridge, H.; Fann, S.A.; Gourdie, R.G.; Yost, M.J. Inhibition of Connexin43 hemichannel-mediated ATP release attenuates early inflammation during the foreign body response. Tissue Eng. Part A 2015, 21, 1752–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giepmans, B.N.; Verlaan, I.; Hengeveld, T.; Janssen, H.; Calafat, J.; Falk, M.M.; Moolenaar, W.H. Gap junction protein connexin-43 interacts directly with microtubules. Curr. Biol. 2001, 11, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Lamouille, S.; Smyth, J.W.; O’Rourke, L.; Kanabur, P.; Guo, S.; Jourdan, L.J.; Sheng, Z.; Gourdie, R.G. Targeting glioblastoma cancer stem cells with a novel Connexin43 mimetic peptide. Cancer Res. 2020, 77, 4765. [Google Scholar]
- Pelaz, S.G.; Jaraíz-Rodríguez, M.; Álvarez-Vázquez, A.; Talaverón, R.; García-Vicente, L.; Flores-Hernández, R.; De Cedrón, M.G.; Tabernero, M.; De Molina, A.R.; Lillo, C.; et al. Targeting metabolic plasticity in glioma stem cells in vitro and in vivo through specific inhibition of c-Src by TAT-Cx43266-283. EBioMedicine 2020, 62, 103134. [Google Scholar] [CrossRef]
- Shibayama, J.; Lewandowski, R.; Kieken, F.; Coombs, W.; Shah, S.; Sorgen, P.L.; Taffet, S.M.; Delmar, M. Identification of a novel peptide that interferes with the chemical regulation of connexin43. Circ. Res. 2006, 98, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Verma, V.; Larsen, B.D.; Coombs, W.; Lin, X.; Spagnol, G.; Sorgen, P.L.; Taffet, S.M.; Delmar, M. Novel Pharmacophores of Connexin43 Based on the “RXP” Series of Cx43-Binding Peptides. Circ. Res. 2009, 105, 176–184. [Google Scholar] [CrossRef] [Green Version]
- Dhein, S.; Hagen, A.; Jozwiak, J.; Dietze, A.; Garbade, J.; Barten, M.; Kostelka, M.; Mohr, F.-W. Improving cardiac gap junction communication as a new antiarrhythmic mechanism: The action of antiarrhythmic peptides. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 381, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Gottwald, M.; Tudyka, T.; Linke, W.; Klaus, W.; Dhein, S. Increase in gap junction conductance by an antiarrhythmic peptide. Eur. J. Pharmacol. 1997, 327, 65–72. [Google Scholar] [CrossRef]
- Xing, D.; Kjolbye, A.L.; Nielsen, M.S.; Petersen, J.S.; Harlow, K.W.; Holstein-Rathlou, N.H.; Martins, J.B. ZP123 increases gap junctional conductance and prevents reentrant ventricular tachycardia during myocardial ischemia in open chest dogs. J. Cardiovasc. Electrophysiol. 2003, 14, 510–520. [Google Scholar] [CrossRef] [PubMed]
- Hennan, J.K.; Swillo, R.E.; Morgan, G.A.; Keith, J.C.; Schaub, R.G.; Smith, R.P.; Feldman, H.S.; Haugan, K.; Kantrowitz, J.; Wang, P.J.; et al. Rotigaptide (ZP123) prevents spontaneous ventricular arrhythmias and reduces infarct size during myocardial ischemia/reperfusion injury in open-chest dogs. J. Pharmacol. Exp. Ther. 2006, 317, 236–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-S.; Zhong, J.-Q.; Zeng, Q.-X.; Liu, H.-Z.; Su, G.-Y.; Zhang, Y. Effect of ZP123, a gap junction modifier, on prolonged ventricular fibrillation in swine. Cardiology 2011, 118, 147–152. [Google Scholar] [CrossRef]
- Skyschally, A.; Walter, B.; Hansen, R.S.; Heusch, G. The antiarrhythmic dipeptide ZP1609 (danegaptide) when given at reperfusion reduces myocardial infarct size in pigs. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 383–391. [Google Scholar] [CrossRef]
- Reynolds, J. Zealand announces results of a Phase II Proof-of-Concept trial with danegaptide for cardiac reperfusion injuries. Fierce Biotech 2016. Available online: https://www.fiercebiotech.com/biotech/zealand-announces-results-of-a-phase-ii-proof-of-concept-trial-danegaptide-for-cardiac (accessed on 1 March 2021).
- ClinicalTrials.gov. Gap Junction Potentiation of Endothelial Function with Rotigaptide in the Human Forearm Arterial Circulation—Effects of Ischaemia Induced Endothelial Dysfunction. 2010. Available online: https://clinicaltrials.gov/ct2/show/NCT00901563?term=rotigaptide&draw=2&rank=1 (accessed on 1 March 2021).
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Bliss, M. Banting’s, Best’s, and Collip’s accounts of the discovery of insulin. Bull. Hist. Med. 1982, 56, 554–568. [Google Scholar]
- Chang, S.-G.; Choi, K.-D.; Jang, S.-H.; Shin, H.-C. Role of disulfide bonds in the structure and activity of human insulin. Mol. Cells 2003, 16, 323–330. [Google Scholar] [PubMed]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.A.; Deber, C.M. Therapeutic design of peptide modulators of protein-protein interactions in membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2017; 1859, 577–585. [Google Scholar]
- Moore, K.; Ghatnekar, G.; Gourdie, R.G.; Potts, J.D. Impact of the controlled release of a Connexin43 peptide on corneal wound closure in an STZ model of type I diabetes. PLoS ONE 2014, 9, e86570. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Amos, J.; Davis, J.; Gourdie, R.; Potts, J.D. Characterization of polymeric microcapsules containing a low molecular weight peptide for controlled release. Microsc. Microanal. 2013, 19, 213–226. [Google Scholar] [CrossRef]
- Roberts, R.; Smyth, J.W.; Will, J.; Roberts, P.; Grek, C.L.; Ghatnekar, G.S.; Sheng, Z.; Gourdie, R.G.; Lamouille, S.; Foster, E.J. Development of PLGA nanoparticles for sustained release of a connexin43 mimetic peptide to target glioblastoma cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 108, 110191. [Google Scholar] [CrossRef]
- Miner-Williams, W.M.; Stevens, B.R.; Moughan, P.J. Are intact peptides absorbed from the healthy gut in the adult human? Nutr. Res. Rev. 2014, 27, 308–329. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Xiang, Y.; Li, X.; Fu, A. Targeted transport of nanocarriers into brain for theranosis with rabies virus glycoprotein-derived peptide. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 87, 155–166. [Google Scholar] [CrossRef]
- Neijssen, J.; Herberts, C.; Drijfhout, J.W.; Reits, E.A.J.; Janssen, L.; Neefjes, J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature 2005, 434, 83–88. [Google Scholar] [CrossRef]
- Yang, J.; Luo, Y.; Shibu, M.A.; Toth, I.; Skwarczynskia, M.; Skwarczynski, M.; Skwarczyski, M. Cell-penetrating Peptides: Efficient Vectors for Vaccine Delivery. Curr. Drug Deliv. 2019, 16, 430–443. [Google Scholar] [CrossRef]
- Pinheiro, A.; Silva, A.M.; Teixeira, J.H.; Goncalves, R.M.; Almeida, M.I.; Barbosa, M.A.; Santos, S.G. Extracellular vesicles: Intelligent delivery strategies for therapeutic applications. J. Control. Release 2018, 289, 56–69. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, J.; Ma, J.; Sun, T.; Zhou, Q.; Wang, W.; Wang, G.; Wu, P.; Wang, H.; Jiang, L.; et al. Exosomal circRNAs: Biogenesis, effect and application in human diseases. Mol. Cancer 2019, 18, 116. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Gould, S.J. Exosomes. Annu. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Antes, T.J.; Middleton, R.C.; Luther, K.M.; Ijichi, T.; Peck, K.A.; Liu, W.J.; Valle, J.; Echavez, A.K.; Marbán, E. Targeting extracellular vesicles to injured tissue using membrane cloaking and surface display. J. Nanobiotechnol. 2018, 16, 61. [Google Scholar] [CrossRef]
- Sarko, D.K.; McKinney, C.E. Exosomes: Origins and Therapeutic Potential for Neurodegenerative Disease. Front. Neurosci. 2017, 11, 82. [Google Scholar] [CrossRef] [Green Version]
- Kishore, R.; Khan, M. More Than Tiny Sacks: Stem Cell Exosomes as Cell-Free Modality for Cardiac Repair. Circ. Res. 2016, 118, 330–343. [Google Scholar] [CrossRef] [Green Version]
- Sanwlani, R.; Fonseka, P.; Chitti, S.V.; Mathivanan, S. Milk-Derived Extracellular Vesicles in Inter-Organism, Cross-Species Communication and Drug Delivery. Proteomes 2020, 8, 11. [Google Scholar] [CrossRef]
- Galley, J.D.; Besner, G.E. The Therapeutic Potential of Breast Milk-Derived Extracellular Vesicles. Nutrients 2020, 12, 745. [Google Scholar] [CrossRef] [Green Version]
- Zempleni, J.; Sukreet, S.; Zhou, F.; Wu, D.; Mutai, E. Milk-Derived Exosomes and Metabolic Regulation. Annu. Rev. Anim. Biosci. 2019, 7, 245–262. [Google Scholar] [CrossRef]
- Manca, S.; Upadhyaya, B.; Mutai, E.; Desaulniers, A.T.; Cederberg, R.A.; White, B.R.; Zempleni, J. Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci. Rep. 2018, 8, 11321. [Google Scholar] [CrossRef] [Green Version]
- Epifantseva, I.; Xiao, S.; Baum, R.E.; Kléber, A.G.; Hong, T.; Shaw, R.M. An Alternatively Translated Connexin43 Isoform, GJA1-11k, Localizes to the Nucleus and Can Inhibit Cell Cycle Progression. Biomolecules 2020, 10, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, J.A.; Haddad, B.G.; Dolan, K.A.; Myers, J.B.; Yoshioka, C.C.; Copperman, J.; Zuckerman, D.M.; Reichow, S.L. Connexin-46/50 in a dynamic lipid environment resolved by CryoEM at 1.9 Å. Nat. Commun. 2020, 11, 4331. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Model of gap junction between two cells. Connexon hemichannels dock to form intercellular channels coupling the cells, enabling cytoplasmic exchange of ions and small molecules typically less than 1000 Da in molecular weight. Undocked hemichannels, concentrated in the perinexus surrounding the gap junction [8], are typically closed but can open in response to prompts like ischemic stress. Open hemichannels thereby underpin flow of channel-permeant molecules from the cytoplasm into the extracellular space.
Figure 1.
Model of gap junction between two cells. Connexon hemichannels dock to form intercellular channels coupling the cells, enabling cytoplasmic exchange of ions and small molecules typically less than 1000 Da in molecular weight. Undocked hemichannels, concentrated in the perinexus surrounding the gap junction [8], are typically closed but can open in response to prompts like ischemic stress. Open hemichannels thereby underpin flow of channel-permeant molecules from the cytoplasm into the extracellular space.

Figure 2.
Connexin-43 mimetic peptides discussed in this review.


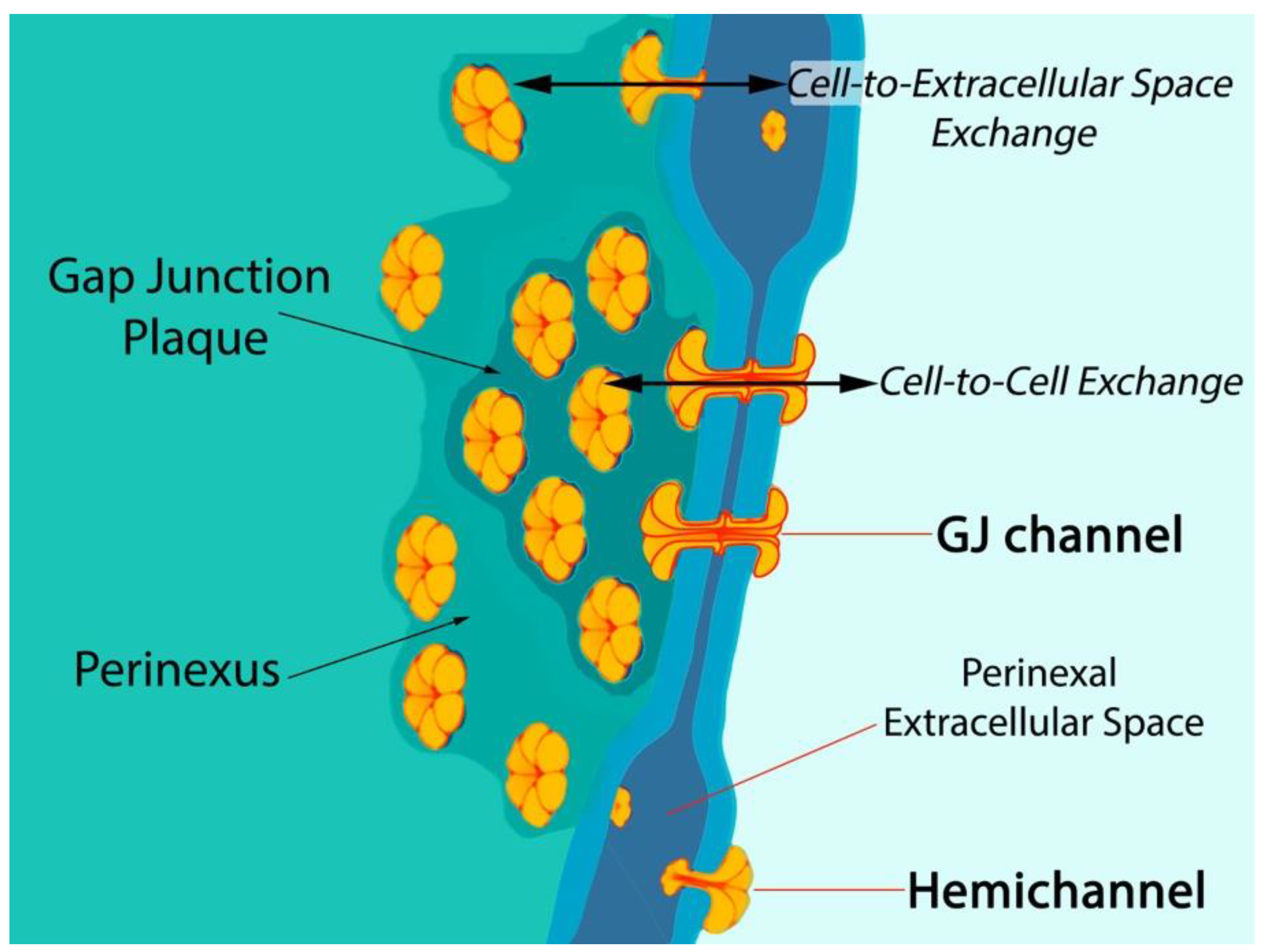{kind=link}
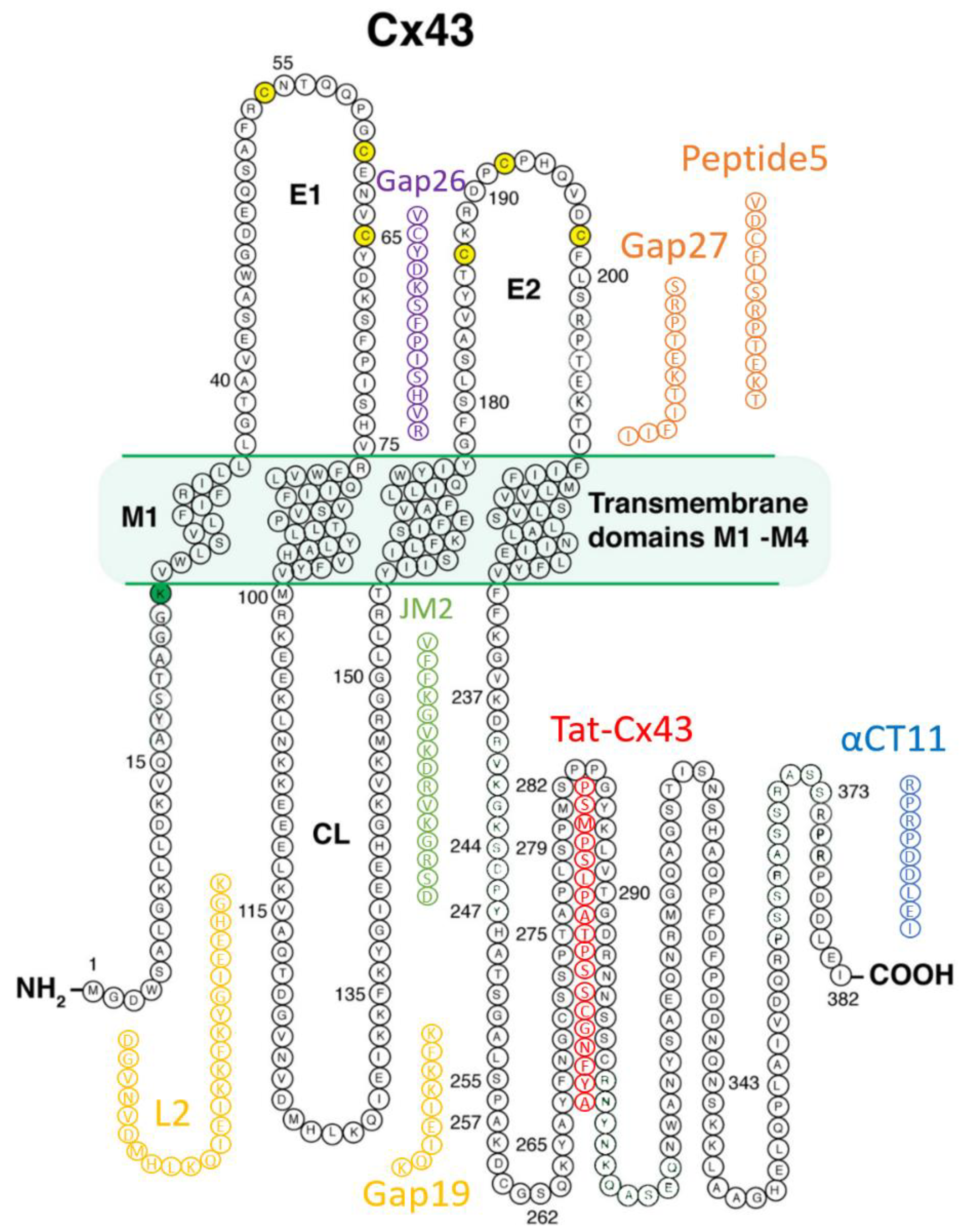{kind=link}
Table 1.
Overview of Cx43 mimetic peptides and their currently known effects.
| Peptide Name | Type | Sequence | Effects in Disease Models Pertinent to Heart |
|---|---|---|---|
| Gap26 | Extracellular Loop | VCYDKSFPISJVR | Improves myocyte viability postischemia in vitro [46,75,76,77] |
| Gap27 | Extracellular Loop | SRPTEKTIFII | Reduces cardiac I/R injury severity ex vivo and in vivo [77,78,79] |
| L2 | Cytoplasmic Loop | DGVNVDMHLKQIEIKKFKYGIEEHGK | Protects myocytes against I/R injury [80] |
| Gap19 | Cytoplasmic Loop | KQIEIKKFK | Reduces cardiac I/R injury severity in vivo [80,81,82] |
| αCT1 | Cytoplasmic Terminus | Ant-RPRPDDLEI | Preischemic treatment decreases cardiac I/R injury severity ex vivo. In clinical testing in humans [26,83] |
| αCT11 | Cytoplasmic Terminus | RPRPDDLEI | Reduces cardiac I/R injury ex vivo and in vivo [83,84,85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Marsh, S.R.; Williams, Z.J.; Pridham, K.J.; Gourdie, R.G. Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine. J. Cardiovasc. Dev. Dis. 2021, 8, 52. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050052
AMA Style
Marsh SR, Williams ZJ, Pridham KJ, Gourdie RG. Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine. Journal of Cardiovascular Development and Disease. 2021; 8(5):52. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050052
Chicago/Turabian StyleMarsh, Spencer R., Zachary J. Williams, Kevin J. Pridham, and Robert G. Gourdie. 2021. "Peptidic Connexin43 Therapeutics in Cardiac Reparative Medicine" Journal of Cardiovascular Development and Disease 8, no. 5: 52. https://0-doi-org.brum.beds.ac.uk/10.3390/jcdd8050052
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.