Mass Spectrometry-Based Proteomics of Fungal Pathogenesis, Host–Fungal Interactions, and Antifungal Development


{kind=link}
{kind=link}
{kind=link}
Abstract
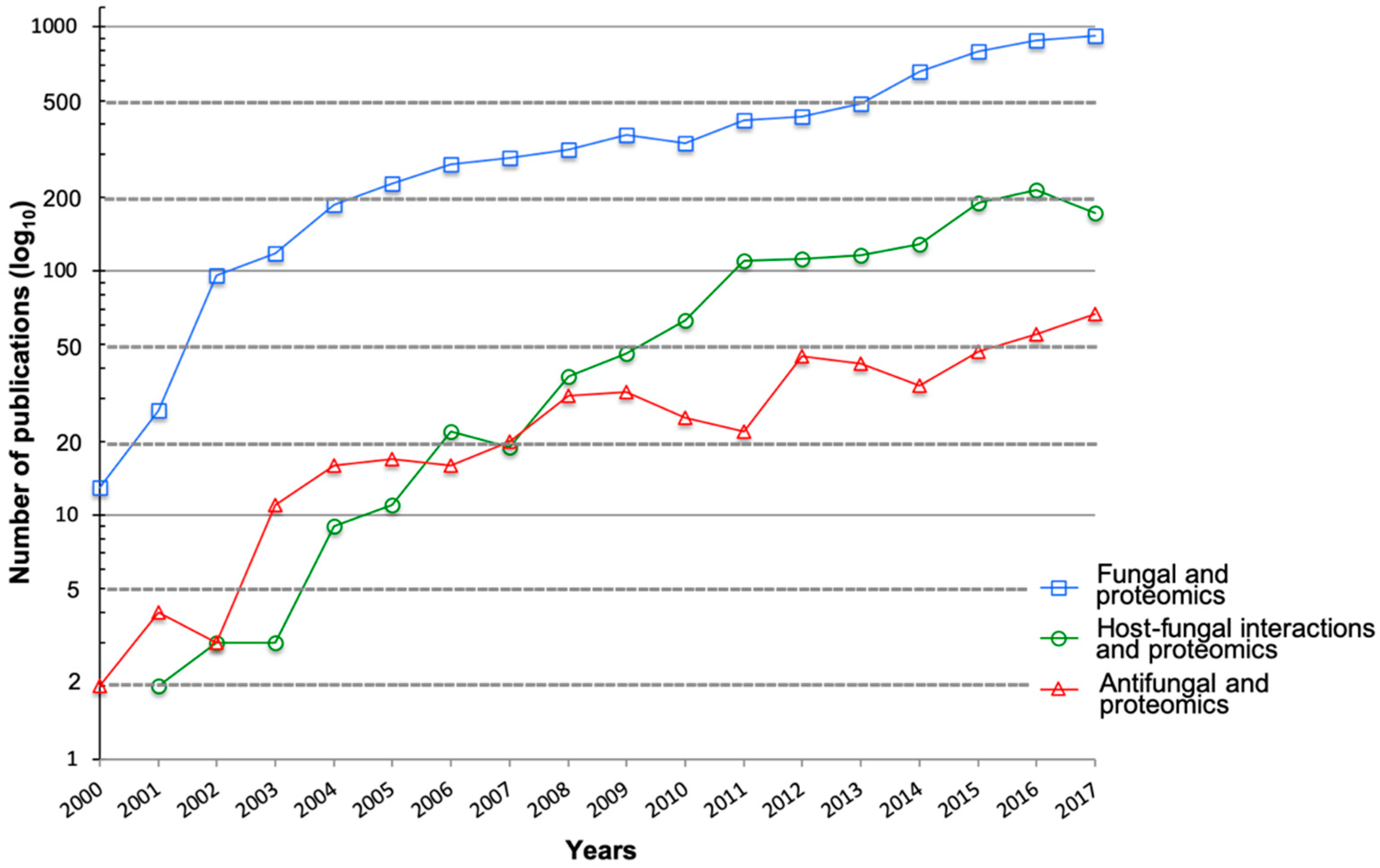
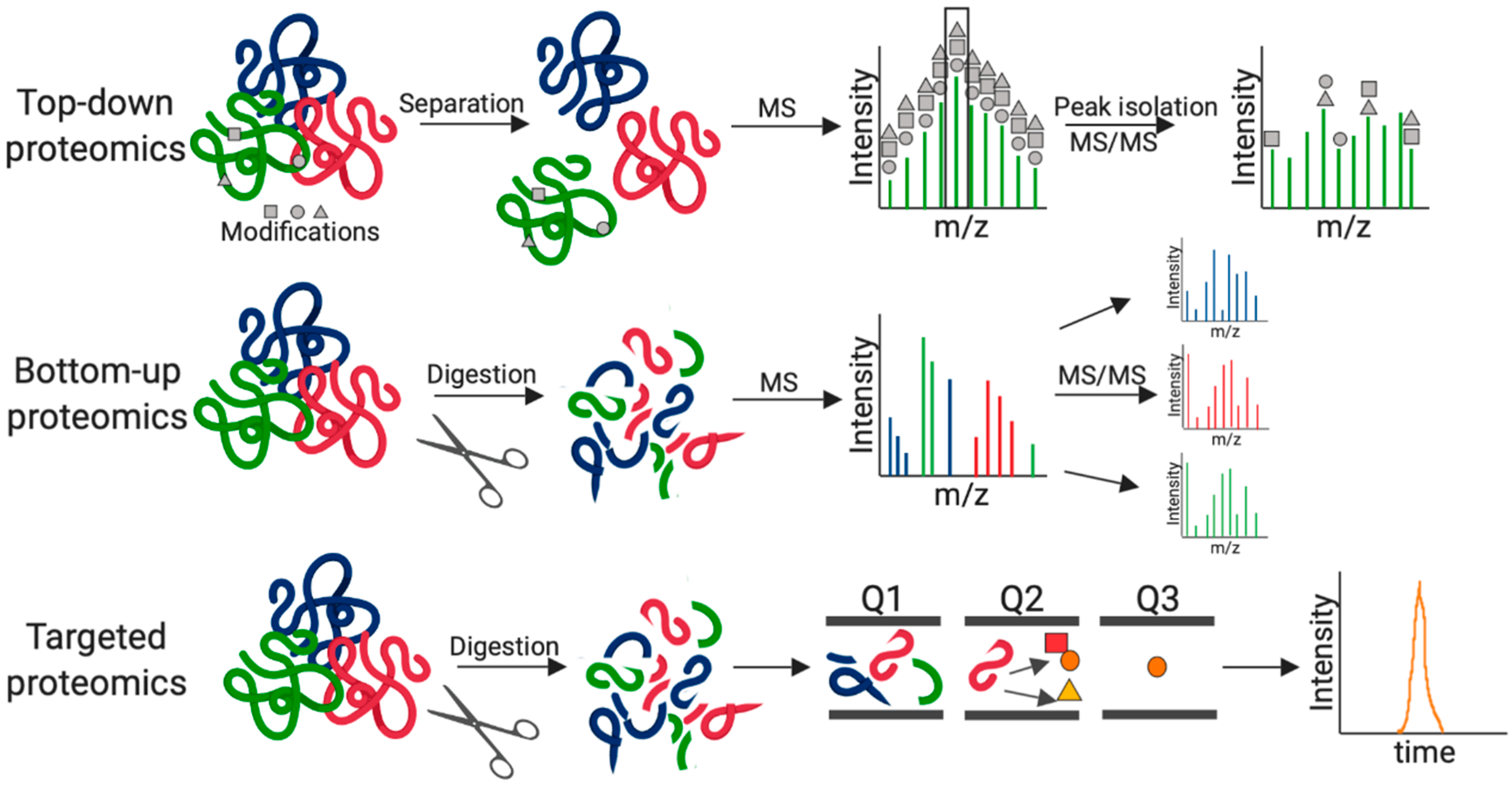
:1. Introduction
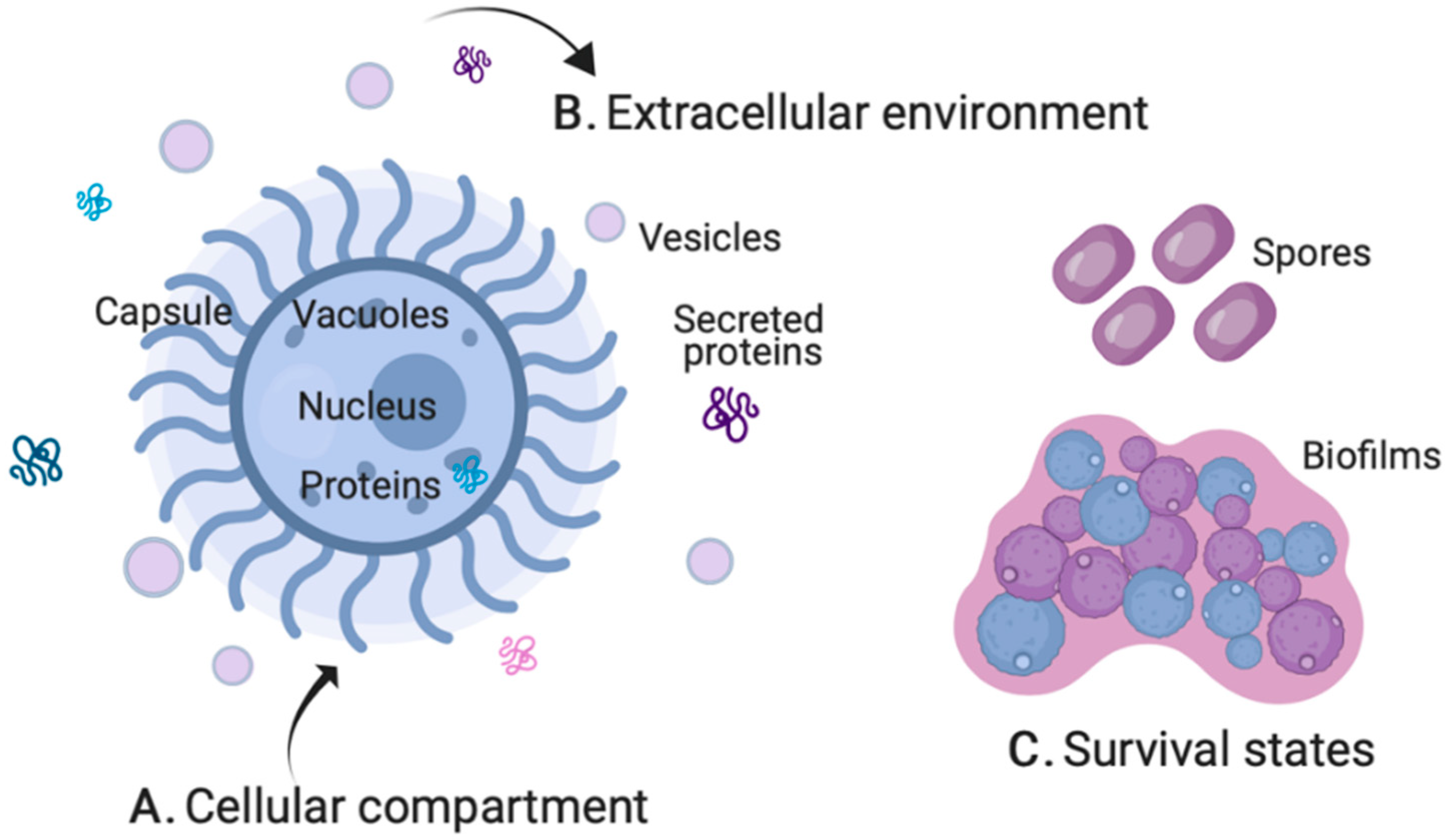
2. Fungal Pathogenesis by MS-Based Proteomics
2.1. Cellular Compartment
2.2. Extracellular Environment
2.3. Survival States
2.4. Modifications and Interactions
3. MS-Based Proteomics of Host–Fungal Interactions
3.1. Host Perspective
3.2. Pathogen Perspective
3.3. Dual Perspective
4. MS-Based Proteomics for the Development of Novel Antifungals
4.1. Antifungal Resistance Mechanisms
4.2. Microbial Competition Showcasing Antifungal Properties
4.3. Drug Repurposing, Vaccine Design, and New Antifungal Development
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Calderone, R. Candida and Candidiasis. Clin. Infect. Dis. 2002. [Google Scholar] [CrossRef]
- Calderone, R.A.; Fonzi, W.A. Virulence factors of Candida albicans. Trends Microbiol. 2001. [Google Scholar] [CrossRef]
- Barelle, C.J.; Priest, C.L.; MacCallum, D.M.; Gow, N.A.R.; Odds, F.C.; Brown, A.J.P. Niche-specific regulation of central metabolic pathways in a fungal pathogen. Cell. Microbiol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017. [Google Scholar] [CrossRef]
- Dagenais, T.R.T.; Keller, N.P. Pathogenesis of Aspergillus fumigatus in invasive aspergillosis. Clin. Microbiol. Rev. 2009. [Google Scholar] [CrossRef] [PubMed]
- Mittal, J.; Ponce, M.G.; Gendlina, I.; Nosanchuk, J.D. Histoplasma capsulatum: Mechanisms for Pathogenesis. Curr. Top. Microbiol. Immunol. 2018. [Google Scholar] [CrossRef]
- Chen, S.C.A.; Meyer, W.; Sorrell, T.C. Cryptococcus gattii infections. Clin. Microbiol. Rev. 2014. [Google Scholar] [CrossRef]
- Hope, W.W.; Walsh, T.J.; Denning, D.W. The invasive and saprophytic syndromes due to Aspergillus spp. Med. Mycol. 2005. [Google Scholar] [CrossRef]
- Perfect, J.R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 2017. [Google Scholar] [CrossRef]
- Geddes-Mcalister, J.; Shapiro, R.S. New pathogens, new tricks: Emerging, drug-resistant fungal pathogens and future prospects for antifungal therapeutics. Ann. N. Y. Acad. Sci. 2018. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The Coming Age of Complete, Accurate, and Ubiquitous Proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappsilber, J.; Mann, M.; Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2007, 2, 1896–1906. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.-E.; Schenone, M.; Margolin, A.A.; Li, X.; Do, K.; Doud, M.K.; Mani, D.R.; Kuai, L.; Wang, X.; Wood, J.L.; et al. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc. Natl. Acad. Sci. USA 2009, 106, 4617–4622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouw, J.W.; Krijgsveld, J.; Heck, A.J.R. Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteom. 2010, 9, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed Protein Quantitation in Saccharomyces cerevisiae Using Amine-reactive Isobaric Tagging Reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Dayon, L.; Hainard, A.; Licker, V.; Turck, N.; Kuhn, K.; Hochstrasser, D.F.; Burkhard, P.R.; Sanchez, J.C. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 2008, 80, 2921–2931. [Google Scholar] [CrossRef]
- Cox, J.; Hein, M.Y.; Luber, C.A.; Paron, I.; Nagaraj, N.; Mann, M. Accurate Proteome-wide Label-free Quantification by Delayed Normalization and Maximal Peptide Ratio Extraction, Termed MaxLFQ. Mol. Cell. Proteom. 2014, 13, 2513–2526. [Google Scholar] [CrossRef] [Green Version]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.-Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; et al. Extending the Limits of Quantitative Proteome Profiling with Data-Independent Acquisition and Application to Acetaminophen-Treated Three-Dimensional Liver Microtissues. Mol. Cell. Proteom. 2015, 14, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized P.P.B.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: Comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Röst, H.L.; Rosenberger, G.; Navarro, P.; Gillet, L.; Miladinoviä, S.M.; Schubert, O.T.; Wolski, W.; Collins, B.C.; Malmström, J.; Malmström, L.; et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat. Biotechnol. 2014, 32, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, K.J.; Lechner, B.E.; Keeffe, G.O.; Keller, M.A.; Werner, E.R.; Lindner, H.; Jones, G.W.; Haas, H.; Doyle, S. Ergothioneine Biosynthesis and Functionality in the Opportunistic Fungal Pathogen, Aspergillus fumigatus. Sci. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, M.D.; Beynon, R.J.; Gethings, L.A.; Claydon, A.J.; Langridge, J.I.; Vissers, J.P.C.; Brown, A.J.P.; Hammond, D.E. Specificity of the osmotic stress response in Candida albicans highlighted by quantitative proteomics. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Soares, C.M.; Araújo, D.S.; Pereira, M.; Portis, I.G.; dos Santos Junior, A.D.; Fontes, W.; de Sousa, M.V.; Assunção, L.D.; Baeza, L.C.; Bailão, A.M.; et al. Metabolic Peculiarities of Paracoccidioides brasiliensis Dimorphism as Demonstrated by iTRAQ Labeling Proteomics. Front. Microbiol. 2019. [Google Scholar] [CrossRef]
- Kaneva, I.N.; Longworth, J.; Sudbery, P.E.; Dickman, M.J. Quantitative Proteomic Analysis in Candida albicans Using SILAC-Based Mass Spectrometry. Proteomics 2018. [Google Scholar] [CrossRef]
- Fröhlich, F.; Christiano, R.; Walther, T.C. Native SILAC: Metabolic Labeling of Proteins in Prototroph Microorganisms Based on Lysine Synthesis Regulation. Mol. Cell. Proteom. 2013. [Google Scholar] [CrossRef]
- Kronstad, J.W.; Hu, G.; Choi, J. The cAMP/protein kinase A pathway and virulence in Cryptococcus neoformans. Mycobiology 2011. [Google Scholar] [CrossRef]
- Geddes, J.M.H.; Croll, D.; Caza, M.; Stoynov, N.; Foster, L.J.; Kronstad, J.W. Secretome profiling of Cryptococcus neoformans reveals regulation of a subset of virulence-associated proteins and potential biomarkers by protein kinase A. BMC Microbiol. 2015, 15, 1–26. [Google Scholar] [CrossRef]
- Clarke, S.C.; Dumesic, P.A.; Homer, C.M.; O’Donoghue, A.J.; La Greca, F.; Pallova, L.; Majer, P.; Madhani, H.D.; Craik, C.S. Integrated Activity and Genetic Profiling of Secreted Peptidases in Cryptococcus neoformans Reveals an Aspartyl Peptidase Required for Low pH Survival and Virulence. PLoS Pathog. 2016. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, A.J.; Alegra Eroy-Reveles, A.A.; Knudsen, G.M.; Ingram, J.; Zhou, M.; Statnekov, J.B.; Greninger, A.L.; Hostetter, D.R.; Qu, G.; Maltby, D.A.; et al. Global identification of peptidase specificity by multiplex substrate profiling. Nat. Methods 2012. [Google Scholar] [CrossRef] [PubMed]
- Gil-Bona, A.; Llama-Palacios, A.; Parra, C.M.; Vivanco, F.; Nombela, C.; Monteoliva, L.; Gil, C. Proteomics unravels extracellular vesicles as carriers of classical cytoplasmic proteins in Candida albicans. J. Proteome Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.M.; Espadas-Moreno, J.; Luque-Garcia, J.L.; Casadevall, A. Interaction of Cryptococcus neoformans Extracellular Vesicles with the Cell Wall. Eukaryot. Cell 2014. [Google Scholar] [CrossRef] [PubMed]
- Fanning, S.; Mitchell, A.P. Fungal Biofilms. PLoS Pathog. 2012. [Google Scholar] [CrossRef] [PubMed]
- Santi, L.; Beys-Da-Silva, W.O.; Berger, M.; Calzolari, D.; Guimarães, J.A.; Moresco, J.J.; Yates, J.R. Proteomic profile of Cryptococcus neoformans biofilm reveals changes in metabolic processes. J. Proteome Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Brakhage, A.A.; Langfelder, K. Menacing Mold: The Molecular Biology of Aspergillus fumigatus. Annu. Rev. Microbiol. 2002. [Google Scholar] [CrossRef] [PubMed]
- Voltersen, V.; Blango, M.G.; Herrmann, S.; Schmidt, F.; Heinekamp, T.; Strassburger, M.; Krüger, T.; Bacher, P.; Lother, J.; Weiss, E.; et al. Proteome Analysis Reveals the Conidial Surface Protein CcpA Essential for Virulence of the Pathogenic Fungus Aspergillus fumigatus. mBio 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Hebert, A.S.; Coon, J.J.; Hull, C.M. Protein Composition of Infectious Spores Reveals Novel Sexual Development and Germination Factors in Cryptococcus. PLoS Genet. 2015. [Google Scholar] [CrossRef]
- Juvvadi, P.R.; Cole, D.C.; Falloon, K.; Waitt, G.; Soderblom, E.J.; Moseley, M.A.; Steinbach, W.J. Kin1 kinase localizes at the hyphal septum and is dephosphorylated by calcineurin but is dispensable for septation and virulence in the human pathogen Aspergillus fumigatus. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef]
- Park, H.S.; Chow, E.W.L.; Fu, C.; Soderblom, E.J.; Moseley, M.A.; Heitman, J.; Cardenas, M.E. Calcineurin Targets Involved in Stress Survival and Fungal Virulence. PLoS Pathog. 2016. [Google Scholar] [CrossRef]
- Xie, L.; Li, J.; Deng, W.; Yu, Z.; Fang, W.; Chen, M.; Liao, W.; Xie, J.; Pan, W. Proteomic analysis of lysine succinylation of the human pathogen Histoplasma capsulatum. J. Proteomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Fang, W.; Deng, W.; Yu, Z.; Li, J.; Chen, M.; Liao, W.; Xie, J.; Pan, W. Global profiling of lysine acetylation in human histoplasmosis pathogen Histoplasma capsulatum. Int. J. Biochem. Cell Biol. 2016. [Google Scholar] [CrossRef]
- Kaneva, I.N.; Sudbery, I.M.; Dickman, M.J.; Sudbery, P.E. Proteins that physically interact with the phosphatase Cdc14 in Candida albicans have diverse roles in the cell cycle. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Selvan, L.D.N.; Sreenivasamurthy, S.K.; Kumar, S.; Yelamanchi, S.D.; Madugundu, A.K.; Anil, A.K.; Renuse, S.; Nair, B.G.; Gowda, H.; Mathur, P.P.; et al. Characterization of host response to Cryptococcus neoformans through quantitative proteomic analysis of cryptococcal meningitis co-infected with HIV. Mol. Biosyst. 2015. [Google Scholar] [CrossRef] [PubMed]
- Toor, A.; Culibrk, L.; Singhera, G.K.; Moon, K.M.; Prudova, A.; Foster, L.J.; Moore, M.M.; Dorscheid, D.R.; Tebbutt, S.J. Transcriptomic and proteomic host response to Aspergillus fumigatus conidia in an air-liquid interface model of human bronchial epithelium. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Lee, M.V.; Topper, S.E.; Hubler, S.L.; Hose, J.; Wenger, C.D.; Coon, J.J.; Gasch, A.P. A dynamic model of proteome changes reveals new roles for transcript alteration in yeast. Mol. Syst. Biol. 2011. [Google Scholar] [CrossRef]
- Selbach, M.; Schwanhäusser, B.; Thierfelder, N.; Fang, Z.; Khanin, R.; Rajewsky, N. Widespread changes in protein synthesis induced by microRNAs. Nature 2008. [Google Scholar] [CrossRef]
- Schwanhüusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011. [Google Scholar] [CrossRef]
- Curty, N.; Kubitschek-Barreira, P.H.; Neves, G.W.; Gomes, D.; Pizzatti, L.; Abdelhay, E.; Souza, G.H.M.F.; Lopes-Bezerra, L.M. Discovering the infectome of human endothelial cells challenged with Aspergillus fumigatus applying a mass spectrometry label-free approach. J. Proteom. 2014. [Google Scholar] [CrossRef]
- Pandey, A.; Ding, S.L.; Qin, Q.M.; Gupta, R.; Gomez, G.; Lin, F.; Feng, X.; Fachini da Costa, L.; Chaki, S.P.; Katepalli, M.; et al. Global Reprogramming of Host Kinase Signaling in Response to Fungal Infection. Cell Host Microbe 2017, 21, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Tirado, F.H.; Peng, T.; Yang, M.; Hang, H.C.; Doering, T.L. A Single Protein S-acyl Transferase Acts through Diverse Substrates to Determine Cryptococcal Morphology, Stress Tolerance, and Pathogenic Outcome. PLoS Pathog. 2015. [Google Scholar] [CrossRef] [PubMed]
- Tomazett, M.V.; Baeza, L.C.; Paccez, J.D.; Parente-Rocha, J.A.; Ribeiro-Dias, F.; de A. Soares, C.M. Identification and characterization of Paracoccidioides lutzii proteins interacting with macrophages. Microbes Infect. 2019. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.J.; Vallhov, H.; Holm, T.; Gehrmann, U.; Andersson, A.; Johansson, C.; Blom, H.; Carroni, M.; Lehtiö, J.; Scheynius, A. Extracellular nanovesicles released from the commensal yeast Malassezia sympodialis are enriched in allergens and interact with cells in human skin. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Vlaic, S.; Krüger, T.; Schmidt, F.; Balkenhol, J.; Dandekar, T.; Guthke, R.; Kniemeyer, O.; Heinekamp, T.; Brakhage, A.A. Proteomics of Aspergillus fumigatus Conidia-containing Phagolysosomes Identifies Processes Governing Immune Evasion. Mol. Cell. Proteom. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kitahara, N.; Morisaka, H.; Aoki, W.; Takeda, Y.; Shibasaki, S.; Kuroda, K.; Ueda, M. Description of the interaction between Candida albicans and macrophages by mixed and quantitative proteome analysis without isolation. AMB Express 2015. [Google Scholar] [CrossRef]
- Truong, T.; Zeng, G.; Qingsong, L.; Kwang, L.T.; Tong, C.; Chan, F.Y.; Wang, Y.; Seneviratne, C.J. Comparative Ploidy Proteomics of Candida albicans Biofilms Unraveled the Role of the AHP1 Gene in the Biofilm Persistence Against Amphotericin B. Mol. Cell. Proteom. 2016. [Google Scholar] [CrossRef]
- Pais, P.; Costa, C.; Pires, C.; Shimizu, K.; Chibana, H.; Teixeira, M.C. Membrane Proteome-Wide Response to the Antifungal Drug Clotrimazole in Candida glabrata: Role of the Transcription Factor CgPdr1 and the Drug:H+ Antiporters CgTpo1_1 and CgTpo1_2. Mol. Cell. Proteom. 2016. [Google Scholar] [CrossRef]
- Zipperer, A.; Konnerth, M.C.; Laux, C.; Berscheid, A.; Janek, D.; Weidenmaier, C.; Burian, M.; Schilling, N.A.; Slavetinsky, C.; Marschal, M.; et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 2016, 535, 511–516. [Google Scholar] [CrossRef]
- Mayer, F.L.; Kronstad, J.W. Discovery of a Novel Antifungal Agent in the Pathogen Box. mSphere 2017. [Google Scholar] [CrossRef]
- Mayer, F.L.; Kronstad, J.W. Disarming Fungal Pathogens: Bacillus safensis Inhibits Virulence Factor Production and Biofilm Formation by Cryptococcus neoformans and Candida albicans. mBio 2017. [Google Scholar] [CrossRef] [PubMed]
- Junker, K.; Bravo Ruiz, G.; Lorenz, A.; Walker, L.; Gow, N.A.R.; Wendland, J. The mycoparasitic yeast Saccharomycopsis schoenii predates and kills multi-drug resistant Candida auris. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Junker, K.; Chailyan, A.; Hesselbart, A.; Forster, J.; Wendland, J. Multi-omics characterization of the necrotrophic mycoparasite Saccharomycopsis schoenii. PLoS Pathog. 2019. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.M.H.; Caza, M.; Croll, D.; Stoynov, N.; Foster, L.J.; Kronstad, J.W. Analysis of the protein kinase a-regulated proteome of Cryptococcus neoformans identifies a role for the ubiquitin-proteasome pathway in capsule formation. mBio 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Figueiredo-Pereira, M.E. Ubiquitin/proteasome pathway impairment in neurodegeneration: Therapeutic implications. Apoptosis 2010. [Google Scholar] [CrossRef] [PubMed]
- Huffnagle, G.B.; Lipscomb, M.F. Cells and cytokines in pulmonary cryptococcosis. Res. Immunol. 1998. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Weintraub, S.T.; Lopez-Ribot, J.L.; Wormley, F.L. Identification and characterization of Cryptococcus neoformans protein fractions that induce protective immune responses. Proteomics 2013. [Google Scholar] [CrossRef] [PubMed]
- Champer, J.; Ito, J.; Clemons, K.; Stevens, D.; Kalkum, M. Proteomic Analysis of Pathogenic Fungi Reveals Highly Expressed Conserved Cell Wall Proteins. J. Fungi 2016, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo Silva, L.; Ossa, D.P.; da Conceição Castro, S.V.; Pires, L.B.; de Oliveira, C.M.; da Silva, C.C.; Coelho, N.P.; Bailão, A.M.; Parente-Rocha, J.A.; de Almeida Soares, C.M.; et al. Transcriptome Profile of the Response of Paracoccidioides spp. to a Camphene Thiosemicarbazide Derivative. PLoS ONE 2015. [Google Scholar] [CrossRef]
- Borba, J.V.V.B.; Tauhata, S.B.F.; De Oliveira, C.M.A.; Marques, M.F.; Bailão, A.M.; De Almeida Soares, C.M.; Pereira, M. Chemoproteomic identification of molecular targets of antifungal prototypes, thiosemicarbazide and a camphene derivative of thiosemicarbazide, in Paracoccidioides brasiliensis. PLoS ONE 2018. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ball, B.; Bermas, A.; Carruthers-Lay, D.; Geddes-McAlister, J. Mass Spectrometry-Based Proteomics of Fungal Pathogenesis, Host–Fungal Interactions, and Antifungal Development. J. Fungi 2019, 5, 52. https://0-doi-org.brum.beds.ac.uk/10.3390/jof5020052
Ball B, Bermas A, Carruthers-Lay D, Geddes-McAlister J. Mass Spectrometry-Based Proteomics of Fungal Pathogenesis, Host–Fungal Interactions, and Antifungal Development. Journal of Fungi. 2019; 5(2):52. https://0-doi-org.brum.beds.ac.uk/10.3390/jof5020052
Chicago/Turabian StyleBall, Brianna, Arianne Bermas, Duncan Carruthers-Lay, and Jennifer Geddes-McAlister. 2019. "Mass Spectrometry-Based Proteomics of Fungal Pathogenesis, Host–Fungal Interactions, and Antifungal Development" Journal of Fungi 5, no. 2: 52. https://0-doi-org.brum.beds.ac.uk/10.3390/jof5020052