
Molecular Epidemiological Investigation of a Nosocomial Cluster of C. auris: Evidence of Recent Emergence in Italy and Ease of Transmission during the COVID-19 Pandemic

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Candida auris Isolates
2.2. Antifungal Susceptibility Testing (AFST)Share and Cite
2.3. Whole-Genome Sequencing (WGS)
2.4. Phylogenomic Reconstruction and Comparative Analyses
3. Results
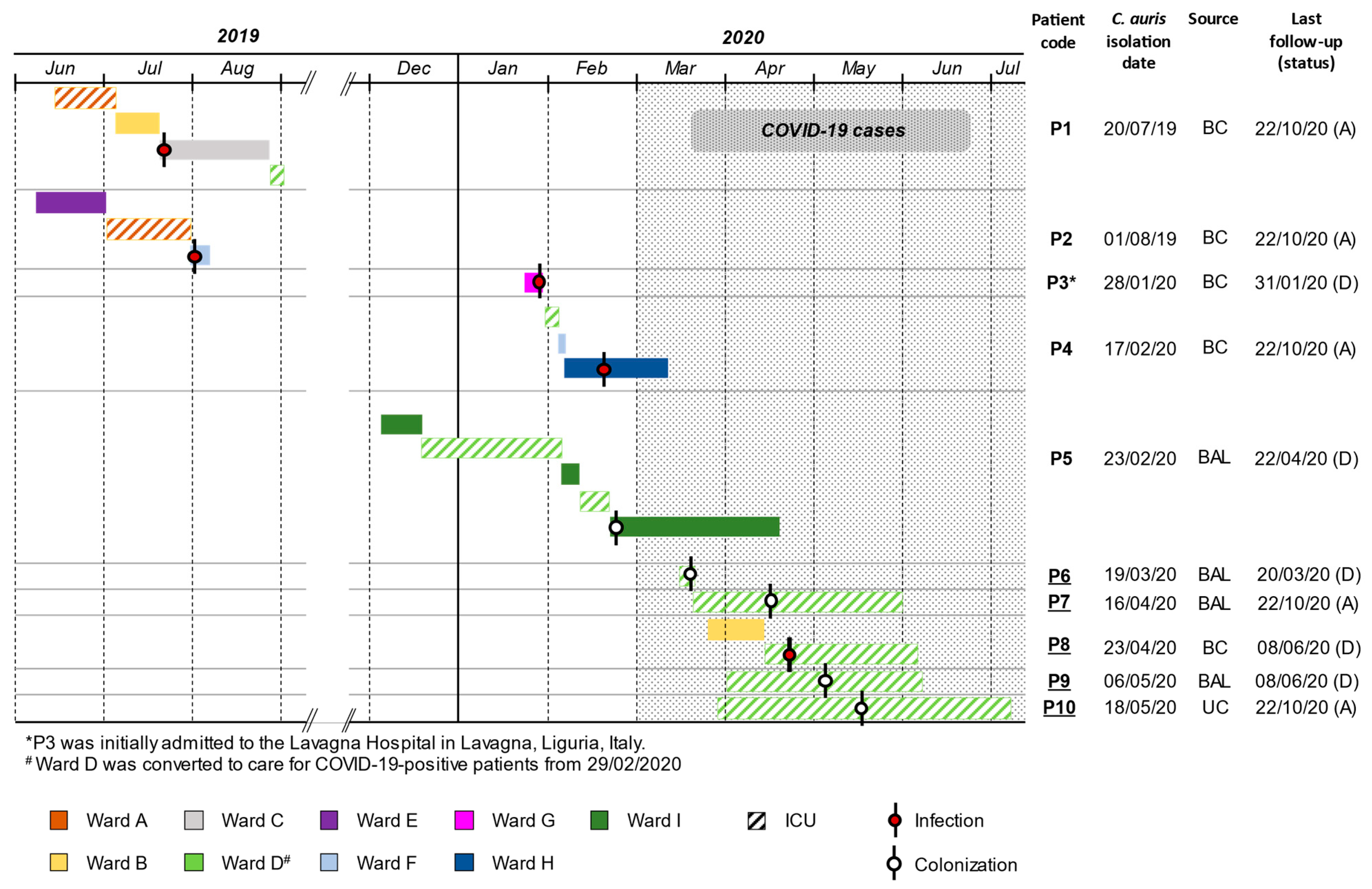
3.1. Case Series Description
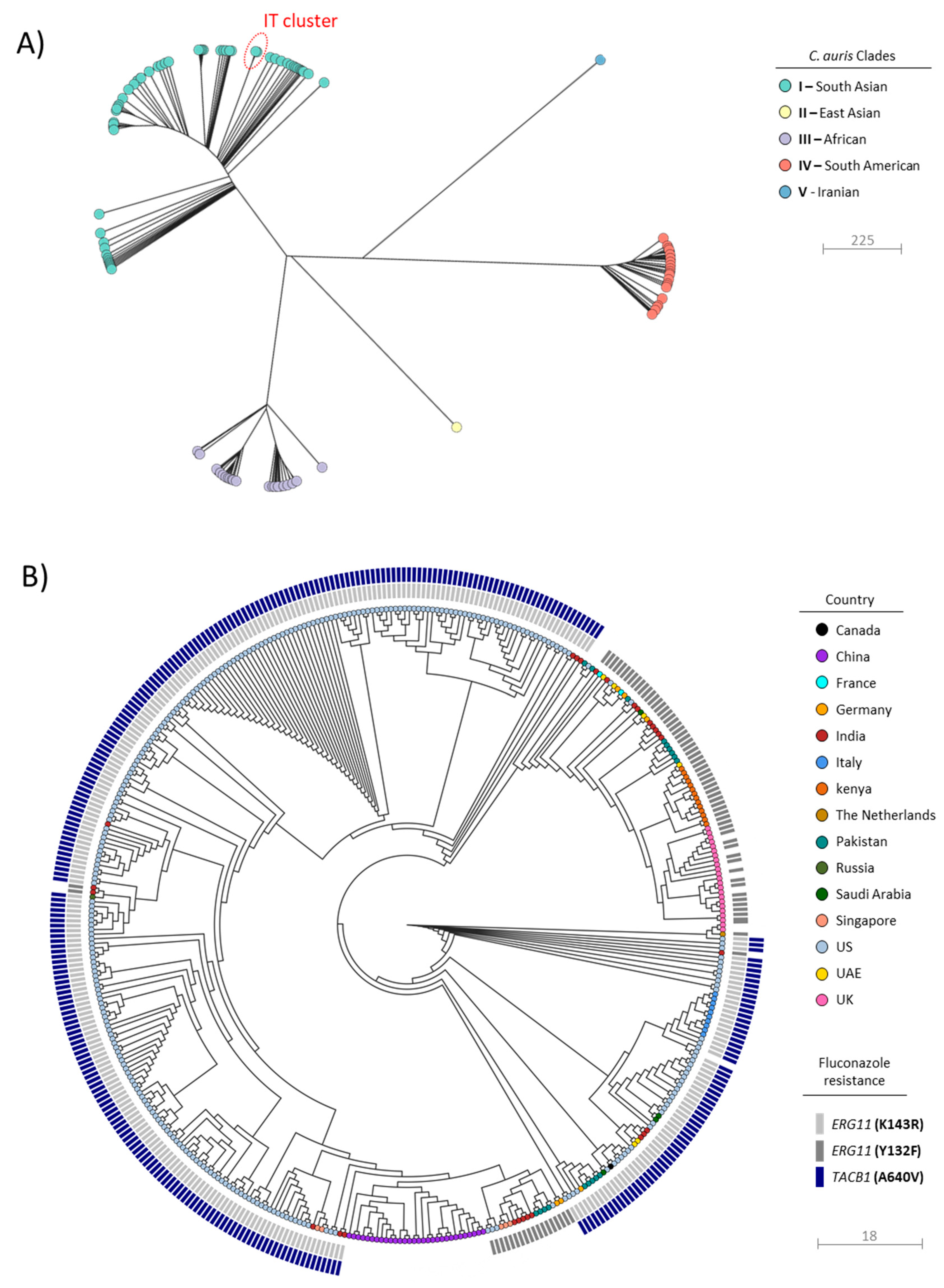
3.2. Inference of Putative Origin and Emergence of C. auris Isolates in Italy
3.3. Investigation of Clonal Relatedness and Putative Transmission Patterns of C. auris from HSM
3.4. Antifungal Susceptibility Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden killers: Human fungal infections. Sci. Transl. Med. 2012, 4, 165rv13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffery-Smith, A.; Taori, S.K.; Schelenz, S.; Jeffery, K.; Johnson, E.M.; Borman, A.; Manuel, R.; Brown, C.S.; Browna, C.S. Candida auris: A review of the literature. Clin. Microbiol. Rev. 2018, 31, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chybowska, A.D.; Childers, D.S.; Farrer, R.A. Nine Things Genomics Can Tell Us About Candida auris. Front. Genet. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Chow, N.A.; De Groot, T.; Badali, H.; Abastabar, M.; Chiller, T.M.; Meis, J.F. Potential fifth clade of Candida auris, Iran, 2018. Emerg. Infect. Dis. 2019, 25, 1780–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, N.A.; Muñoz, J.F.; Gade, L.; Berkow, E.L.; Li, X.; Welsh, R.M.; Forsberg, K.; Lockhart, S.R.; Adam, R.; Alanio, A.; et al. Tracing the Evolutionary History and Global Expansion of Candida auris Using Population Genomic Analyses. MBio 2020, 11. [Google Scholar] [CrossRef]
- Lone, S.A.; Ahmad, A. Candida auris-the growing menace to global health. Mycoses 2019, 62, 620–637. [Google Scholar] [CrossRef] [Green Version]
- Taori, S.K.; Khonyongwa, K.; Hayden, I.; Athukorala, G.D.A.; Letters, A.; Fife, A.; Desai, N.; Borman, A.M. Candida auris outbreak: Mortality, interventions and cost of sustaining control. J. Infect. 2019, 79, 601–611. [Google Scholar] [CrossRef]
- Rudramurthy, S.M.; Chakrabarti, A.; Paul, R.A.; Sood, P.; Kaur, H.; Capoor, M.R.; Kindo, A.J.; Marak, R.S.K.; Arora, A.; Sardana, R.; et al. Candida auris candidaemia in Indian ICUs: Analysis of risk factors. J. Antimicrob. Chemother. 2017, 72, 1794–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bustos, V.; Salavert, M.; Ruiz-Gaitán, A.C.; Cabañero-Navalon, M.D.; Sigona-Giangreco, I.A.; Pemán, J. A clinical predictive model of candidaemia by Candida auris in previously colonized critically ill patients. Clin. Microbiol. Infect. 2020, 26. [Google Scholar] [CrossRef]
- Yapar, N. Epidemiology and risk factors for invasive candidiasis. Clin. Risk Manag. 2014, 10, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohlenberg, A.; Struelens, M.J.; Monnet, D.L.; Plachouras, D. Candida auris: Epidemiological situation, laboratory capacity and preparedness in European Union and European Economic Area countries, 2013 to 2017. Eurosurveillance 2018, 23, 18-00136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crea, F.; Codda, G.; Orsi, A.; Battaglini, A.; Giacobbe, D.R.; Delfino, E.; Ungaro, R.; Marchese, A. Isolation of Candida auris from invasive and non-invasive samples of a patient suffering from vascular disease, Italy, July 2019. Euro Surveill. 2019, 24, 1900549. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Gaitán, A.C.; Fernández-Pereira, J.; Valentin, E.; Tormo-Mas, M.A.; Eraso, E.; Pemán, J.; de Groot, P.W.J. Molecular identification of Candida auris by PCR amplification of species-specific GPI protein-encoding genes. Int. J. Med. Microbiol. 2018, 308, 812–818. [Google Scholar] [CrossRef]
- CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeast, 4th ed.; CLSI standard M27; Clinical and Laboratory Standards Institute, Ed.; CLSI: Wayne, PA, USA, 2017; ISBN 1-56238-827-4. [Google Scholar]
- Lockhart, S.R.; Etienne, K.A.; Vallabhaneni, S.; Farooqi, J.; Chowdhary, A.; Govender, N.P.; Colombo, A.L.; Calvo, B.; Cuomo, C.A.; Desjardins, C.A.; et al. Simultaneous emergence of multidrug-resistant candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin. Infect. Dis. 2017, 64, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Moss, E.L.; Maghini, D.G.; Bhatt, A.S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 2020, 38, 701–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, E.G.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv 2012, arXiv:1207.3907. [Google Scholar]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhodes, J.; Abdolrasouli, A.; Farrer, R.A.; Cuomo, C.A.; Aanensen, D.M.; Armstrong-James, D.; Fisher, M.C.; Schelenz, S. Genomic epidemiology of the UK outbreak of the emerging human fungal pathogen Candida auris article. Emerg. Microbes Infect. 2018, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnasco, L.; Mikulska, M.; Giacobbe, D.R.; Taramasso, L.; Vena, A.; Dentone, C.; Dettori, S.; Tutino, S.; Labate, L.; Di Pilato, V.; et al. Spread of Carbapenem-Resistant Gram-Negatives and Candida auris during the COVID-19 Pandemic in Critically Ill Patients: One Step Back in Antimicrobial Stewardship? Microorganisms 2021, 9, 95. [Google Scholar] [CrossRef]
- Chow, N.A.; Gade, L.; Tsay, S.V.; Forsberg, K.; Greenko, J.A.; Southwick, K.L.; Barrett, P.M.; Kerins, J.L.; Lockhart, S.R.; Chiller, T.M.; et al. Multiple introductions and subsequent transmission of multidrug-resistant Candida auris in the USA: A molecular epidemiological survey. Lancet Infect. Dis. 2018, 18, 1377–1384. [Google Scholar] [CrossRef]
- Yadav, A.; Singh, A.; Wang, Y.; van Haren, M.H.; Singh, A.; de Groot, T.; Meis, J.F.; Xu, J.; Chowdhary, A. Colonisation and Transmission Dynamics of Candida auris among Chronic Respiratory Diseases Patients Hospitalised in a Chest Hospital, Delhi, India: A Comparative Analysis of Whole Genome Sequencing and Microsatellite Typing. J. Fungi (BaselSwitz.) 2021, 7, 81. [Google Scholar] [CrossRef]
- Carolus, H.; Pierson, S.; Muñoz, J.F.; Subotić, A.; Cruz, R.B.; Cuomo, C.A.; Van Dijck, P. Genome-wide analysis of experimentally evolved Candida auris reveals multiple novel mechanisms of multidrug-resistance. bioRxiv 2020. [Google Scholar] [CrossRef]
- Arendrup, M.C.; Patterson, T.F. Multidrug-Resistant Candida: Epidemiology, Molecular Mechanisms, and Treatment. J. Infect. Dis. 2017, 216, S445–S451. [Google Scholar] [CrossRef] [Green Version]
- Rybak, J.M.; Muñoz, J.F.; Barker, K.S.; Parker, J.E.; Esquivel, B.D.; Berkow, E.L.; Lockhart, S.R.; Gade, L.; Palmer, G.E.; White, T.C.; et al. Mutations in TAC1B: A Novel Genetic Determinant of Clinical Fluconazole Resistance in Candida auris. MBio 2020, 11. [Google Scholar] [CrossRef]
- Chowdhary, A.; Prakash, A.; Sharma, C.; Kordalewska, M.; Kumar, A.; Sarma, S.; Tarai, B.; Singh, A.; Upadhyaya, G.; Upadhyay, S.; et al. A multicentre study of antifungal susceptibility patterns among 350 Candida auris isolates (2009-17) in India: Role of the ERG11 and FKS1 genes in azole and echinocandin resistance. J. Antimicrob. Chemother. 2018, 73, 891–899. [Google Scholar] [CrossRef]
- Borman, A.M.; Johnson, E.M. Candida auris in the UK: Introduction, dissemination, and control. PLoS Pathog. 2020, 16, e1008563. [Google Scholar] [CrossRef] [PubMed]
- Prestel, C.; Anderson, E.; Forsberg, K.; Lyman, M.; de Perio, M.A.; Kuhar, D.; Edwards, K.; Rivera, M.; Shugart, A.; Walters, M.; et al. Candida auris Outbreak in a COVID-19 Specialty Care Unit—Florida, July–August 2020. Mmwr. Morb. Mortal Wkly Rep. 2021, 56–57. [Google Scholar] [CrossRef]
- Villanueva-Lozano, H.; Treviño-Rangel, R.d.J.; González, G.M.; Ramírez-Elizondo, M.T.; Lara-Medrano, R.; Aleman-Bocanegra, M.C.; Guajardo-Lara, C.E.; Gaona-Chávez, N.; Castilleja-Leal, F.; Torre-Amione, G.; et al. Outbreak of Candida auris infection in a COVID-19 hospital in Mexico. Clin. Microbiol. Infect. 2021, in press. [Google Scholar] [CrossRef]
- Chowdhary, A.; Tarai, B.; Singh, A.; Sharma, A. Multidrug-Resistant Candida auris Infections in Critically Ill Coronavirus Disease Patients, India, April-July 2020. Emerg. Infect. Dis. 2020, 26, 2694–2696. [Google Scholar] [CrossRef]
- Chowdhary, A.; Sharma, A. The lurking scourge of multidrug resistant Candida auris in times of COVID-19 pandemic. J. Glob. Antimicrob. Resist. 2020, 22, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, S.R. Candida auris and multidrug resistance: Defining the new normal. Fungal Genet. Biol. 2019, 131, 103243. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Antifungal Drugs | MIC Range | MIC50 | MIC90 |
|---|---|---|---|
| Fluconazole | >256 | >256 | >256 |
| Itraconazole | 0.5 | 0.5 | 0.5 |
| Voriconazole | 4 | 4 | 4 |
| Posaconazole | 0.25 | 0.25 | 0.25 |
| Caspofungin | 0.06–0.25 | 0.25 | 0.25 |
| Anidulafungin | 0.12–0.25 | 0.25 | 0.25 |
| Micafungin | 0.06–0.12 | 0.12 | 0.12 |
| Amphotericin B | 2–4 | 2 | 4 |
| Flucytosine | 0.12–0.5 | 0.25 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pilato, V.; Codda, G.; Ball, L.; Giacobbe, D.R.; Willison, E.; Mikulska, M.; Magnasco, L.; Crea, F.; Vena, A.; Pelosi, P.; et al. Molecular Epidemiological Investigation of a Nosocomial Cluster of C. auris: Evidence of Recent Emergence in Italy and Ease of Transmission during the COVID-19 Pandemic. J. Fungi 2021, 7, 140. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020140
Di Pilato V, Codda G, Ball L, Giacobbe DR, Willison E, Mikulska M, Magnasco L, Crea F, Vena A, Pelosi P, et al. Molecular Epidemiological Investigation of a Nosocomial Cluster of C. auris: Evidence of Recent Emergence in Italy and Ease of Transmission during the COVID-19 Pandemic. Journal of Fungi. 2021; 7(2):140. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020140
Chicago/Turabian StyleDi Pilato, Vincenzo, Giulia Codda, Lorenzo Ball, Daniele Roberto Giacobbe, Edward Willison, Malgorzata Mikulska, Laura Magnasco, Francesca Crea, Antonio Vena, Paolo Pelosi, and et al. 2021. "Molecular Epidemiological Investigation of a Nosocomial Cluster of C. auris: Evidence of Recent Emergence in Italy and Ease of Transmission during the COVID-19 Pandemic" Journal of Fungi 7, no. 2: 140. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020140