
Invasive Fungal Infections after Anti-CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy: State of the Evidence and Future Directions

Abstract
:1. Introduction
2. Epidemiology of Fungal Infections after CAR-T-Cell Therapy
2.1. Yeast Infections
2.2. Mold Infections
2.3. Other Fungal Infections
3. Risk Factors for Fungal Infections after CAR-T-Cell Therapy
4. Anti-Fungal Prophylaxis Following CAR-T-Cell Therapy
4.1. Yeast Versus Mold-Active Prophylaxis
4.2. Prophylaxis Against PCP
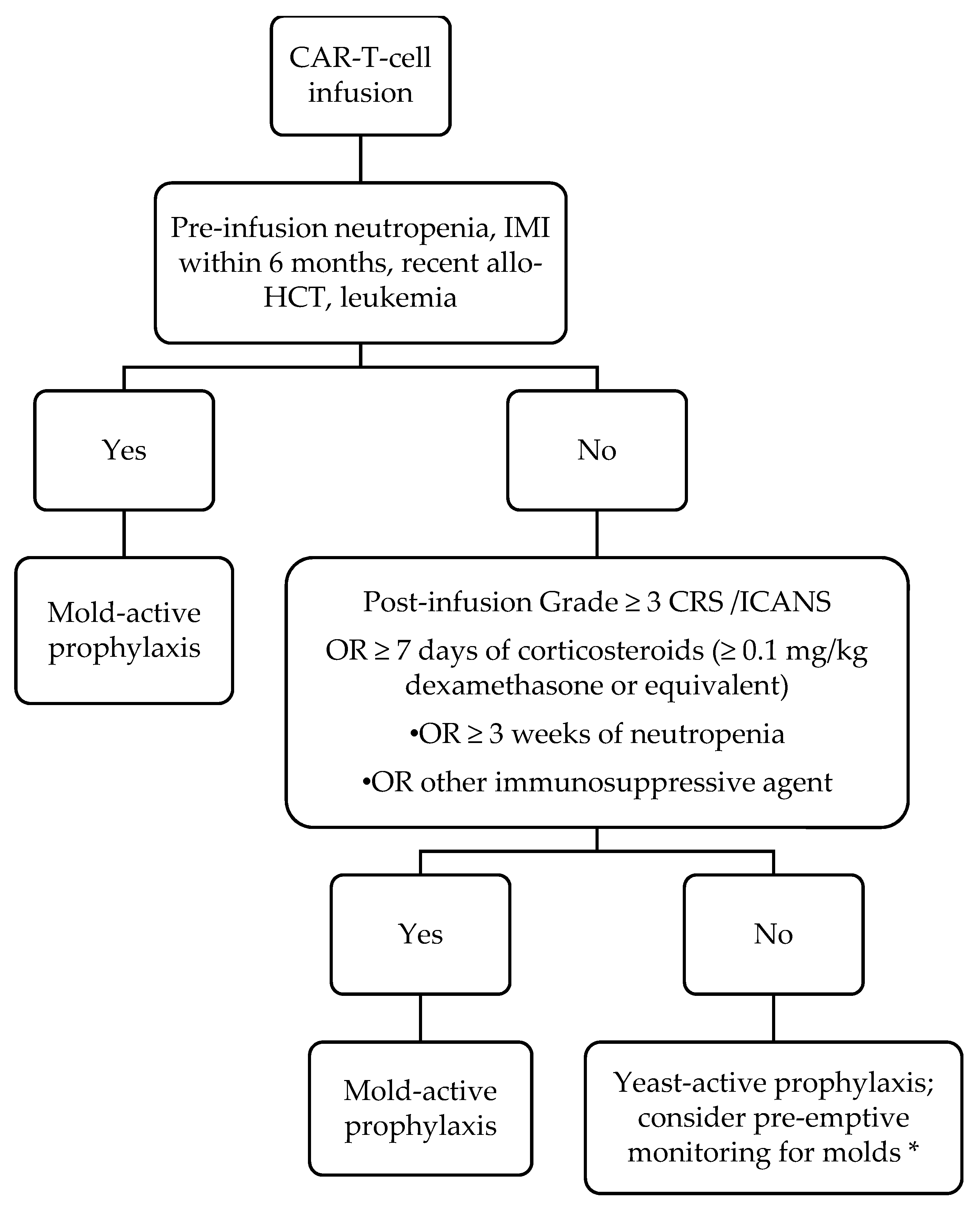
5. Our Approach to Work-Up and Management of Fungal Infections after CAR-T-Cell Therapy
6. Conclusions
Funding
Conflicts of Interest
References
- Kansagra, A.J.; Frey, N.V.; Bar, M.; Laetsch, T.W.; Carpenter, P.A.; Savani, B.N.; Heslop, H.E.; Bollard, C.M.; Komanduri, K.V.; Gastineau, D.A.; et al. Clinical Utilization of Chimeric Antigen Receptor T-Cells (Car-T) in B-Cell Acute Lymphoblastic Leukemia (All)—An Expert Opinion from the European Society for Blood and Marrow Transplantation (Ebmt) and the American Society for Blood and Marrow Transplantation (Asbmt). Bone Marrow Transplant. 2019, 54, 1868–1880. [Google Scholar] [PubMed]
- Mahadeo, K.M.; Khazal, S.J.; Abdel-Azim, H.; Fitzgerald, J.C.; Taraseviciute, A.; Bollard, C.M.; Tewari, P.; Duncan, C.; Traube, C.; McCall, D.; et al. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat. Rev. Clin. Oncol. 2019, 16, 45–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Haidar, G.; Garner, W.; Hill, J.A. Infections after Anti-Cd19 Chimeric Antigen Receptor T-Cell Therapy for Hematologic Malignancies: Timeline, Prevention, and Uncertainties. Curr. Opin. Infect. Dis. 2020, 33, 449–457. [Google Scholar] [CrossRef]
- Fried, S.; Avigdor, A.; Bielorai, B.; Meir, A.; Besser, M.J.; Schachter, J.; Shimoni, A.; Nagler, A.; Toren, A.; Jacoby, E. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transplant. 2019, 54, 1643–1650. [Google Scholar] [CrossRef]
- Garner, W.; Samanta, P.; Dorritie, K.; Sehgal, A.; Winfield, D.; Agha, M.; Boudreau, R.; Nguyen, M.H.T.; Haidar, G. 1105. The Burden of Infections Prior to Chimeric Antigen Receptor (CAR) Modified T-cell Therapy Predicts Post-CAR T-cell Infectious Complications. Open Forum Infect. Dis. 2020, 7, S583. [Google Scholar] [CrossRef]
- Hill, J.A.; Li, D.; Hay, K.A.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious complications of CD19-targeted chimeric antigen receptor–modified T-cell immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef]
- Cordeiro, A.; Bezerra, E.D.; Hirayama, A.V.; Hill, J.A.; Wu, Q.V.; Voutsinas, J.; Sorror, M.L.; Turtle, C.J.; Maloney, D.G.; Bar, M. Late Events after Treatment with CD19-Targeted Chimeric Antigen Receptor Modified T Cells. Biol. Blood Marrow Transplant. 2020, 26, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.H.; Romero, F.A.; Taur, Y.; Sadelain, M.; Brentjens, R.J.; Hohl, T.M.; Seo, S.K. Cytokine Release Syndrome Grade as a Predictive Marker for Infections in Patients With Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia Treated With Chimeric Antigen Receptor T Cells. Clin. Infect. Dis. 2018, 67, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.B.; Waghmare, A.; A Englund, J.; Qu, P.; A Gardner, R.; A Hill, J. Infectious Complications Following CD19 Chimeric Antigen Receptor T-cell Therapy for Children, Adolescents, and Young Adults. Open Forum Infect. Dis. 2020, 7, ofaa121. [Google Scholar] [CrossRef] [Green Version]
- Wudhikarn, K.; Palomba, M.L.; Pennisi, M.; Garcia-Recio, M.; Flynn, J.R.; Devlin, S.M.; Afuye, A.; Silverberg, M.L.; Maloy, M.A.; Shah, G.L.; et al. Infection during the first year in patients treated with CD19 CAR T cells for diffuse large B cell lymphoma. Blood Cancer J. 2020, 10, 79. [Google Scholar] [CrossRef]
- Logue, J.M.; Zucchetti, E.; Bachmeier, C.A.; Krivenko, G.S.; Larson, V.; Ninh, D.; Grillo, G.; Cao, B.; Kim, J.; Chavez, J.C.; et al. Immune reconstitution and associated infections following axicabtagene ciloleucel in relapsed or refractory large B-cell lymphoma. Haematologica 2020. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.; Eschenauer, G.; Scappaticci, G.; Frame, D.; Miceli, M.; Patel, T. Infections in Patients Treated with Chimeric Antigen Receptor T-Cells (Car-T) Therapy. Open Forum Infect. Dis. 2020, 7, S354. [Google Scholar] [CrossRef]
- Wingard, J.R.; Leather, H.L. Empiric antifungal therapy for the neutropenic patient. Oncology (Williston Park) 2001, 15, 351–363. [Google Scholar]
- Lortholary, O.; Gangneux, J.-P.; Sitbon, K.; Lebeau, B.; De Monbrison, F.; Le Strat, Y.; Coignard, B.; Dromer, F.; Bretagne, S. Epidemiological trends in invasive aspergillosis in France: The SAIF network (2005–2007). Clin. Microbiol. Infect. 2011, 17, 1882–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, L.; Akova, M.; Dimopoulos, G.; Herbrecht, R.; Drgona, L.; Blijlevens, N. Risk assessment and prognostic factors for mould-related diseases in immunocompromised patients. J. Antimicrob. Chemother. 2010, 66, i5–i14. [Google Scholar] [CrossRef] [Green Version]
- Wingard, J.R. The changing face of invasive fungal infections in hematopoietic cell transplant recipients. Curr. Opin. Oncol. 2005, 17, 89–92. [Google Scholar] [CrossRef]
- Wingard, J. Fungal infections after bone marrow transplant. Biol. Blood Marrow Transplant. 1999, 5, 55–68. [Google Scholar] [CrossRef] [Green Version]
- Tisi, M.C.; Hohaus, S.; Cuccaro, A.; Innocenti, I.; De Carolis, E.; Za, T.; D’Alò, F.; Laurenti, L.; Fianchi, L.; Sica, S.; et al. Invasive fungal infections in chronic lymphoproliferative disorders: A monocentric retrospective study. Haematologica 2016, 102, e108–e111. [Google Scholar] [CrossRef]
- Garcia-Vidal, C.; Upton, A.; Kirby, K.A.; Marr, K.A. Epidemiology of Invasive Mold Infections in Allogeneic Stem Cell Transplant Recipients: Biological Risk Factors for Infection According to Time after Transplantation. Clin. Infect. Dis. 2008, 47, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; Van Vollenhoven, R.F. Integrated safety in tocilizumab clinical trials. Arthritis Res. Ther. 2011, 13, R141. [Google Scholar] [CrossRef] [Green Version]
- Pagano, L.; Caira, M.; Candoni, A.; Offidani, M.; Fianchi, L.; Martino, B.; Pastore, D.; Picardi, M.; Bonini, A.; Chierichini, A.; et al. The epidemiology of fungal infections in patients with hematologic malignancies: The SEIFEM-2004 study. Haematologica 2006, 91, 1068–1075. [Google Scholar]
- Haidar, G.; Dorritie, K.; Farah, R.; Bogdanovich, T.; Nguyen, M.H.; Samanta, P. Invasive Mold Infections After Chimeric Antigen Receptor–Modified T-Cell Therapy: A Case Series, Review of the Literature, and Implications for Prophylaxis. Clin. Infect. Dis. 2019, 71, 672–676. [Google Scholar] [CrossRef] [PubMed]
- E Lewis, R.; Kontoyiannis, D.P. Chimeric Antigen Receptor T-cell Immunotherapy and Need for Prophylaxis for Invasive Mold Infections. Clin. Infect. Dis. 2020, 71, 1802–1803. [Google Scholar] [CrossRef]
- Haidar, G.; Nguyen, M.H.; Samanta, P. Reply to Lewis and Kontoyiannis. Clin. Infect. Dis. 2020, 71, 1803–1804. [Google Scholar] [CrossRef]
- A Maertens, J.; Girmenia, C.; Brüggemann, R.J.; Duarte, R.F.; Kibbler, C.C.; Ljungman, P.; Racil, Z.; Ribaud, P.; A Slavin, M.; A Cornely, O.; et al. European guidelines for primary antifungal prophylaxis in adult haematology patients: Summary of the updated recommendations from the European Conference on Infections in Leukaemia. J. Antimicrob. Chemother. 2018, 73, 3221–3230. [Google Scholar] [CrossRef] [PubMed]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of adults and children undergoing chimeric antigen receptor T-cell therapy: Best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2019, 105, 297–316. [Google Scholar] [CrossRef]
- Los-Arcos, I.; Iacoboni, G.; Aguilar-Guisado, M.; Alsina-Manrique, L.; De Heredia, C.D.; Fortuny-Guasch, C.; García-Cadenas, I.; García-Vidal, C.; González-Vicent, M.; Hernani, R.; et al. Recommendations for screening, monitoring, prevention, and prophylaxis of infections in adult and pediatric patients receiving CAR T-cell therapy: A position paper. Infection 2020, 1–17. [Google Scholar] [CrossRef]
- Hill, J.A.; Seo, S.K. How I prevent infections in patients receiving CD19-targeted chimeric antigen receptor T cells for B-cell malignancies. Blood 2020, 136, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Ullmann, A.J.; Lipton, J.H.; Vesole, D.H.; Chandrasekar, P.; Langston, A.; Tarantolo, S.R.; Greinix, H.; De Azevedo, W.M.; Reddy, V.; Boparai, N.; et al. Posaconazole or Fluconazole for Prophylaxis in Severe Graft-versus-Host Disease. N. Engl. J. Med. 2007, 356, 335–347. [Google Scholar] [CrossRef]
- Cornely, O.A.; Maertens, J.; Winston, D.J.; Perfect, J.; Ullmann, A.J.; Walsh, T.J.; Helfgott, D.; Holowiecki, J.; Stockelberg, D.; Goh, Y.-T.; et al. Posaconazole Vs. Flu-conazole or Itraconazole Prophylaxis in Patients with Neutropenia. N. Engl. J. Med. 2007, 356, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maertens, J.; Theunissen, K.; Verhoef, G.; Verschakelen, J.; Lagrou, K.; Verbeken, E.; Wilmer, A.; Verhaegen, J.; Boogaerts, M.; Van Eldere, J. Galactomannan and computed tomography-based preemptive antifungal therapy in neutropenic patients at high risk for invasive fungal infection: A prospective feasibility study. Clin. Infect. Dis. 2005, 41, 1242–1250. [Google Scholar] [CrossRef] [Green Version]
- Freifeld, A.G.; Bow, E.J.; Sepkowitz, K.A.; Boeckh, M.J.; Ito, J.I.; Mullen, C.A.; Raad, I.I.; Rolston, K.V.; Young, J.H.; Wingard, J.H.; et al. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the infectious diseases society of america. Clin. Infect. Dis. 2011, 52, e56–e93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Wang, N.; Huang, L.; Zhou, X.; Jin, J.; Li, C.; Wang, D.; Xu, B.; Xu, J.; Jiang, L.; et al. Inflammatory signatures for quick diagnosis of life-threatening infection during the CAR T-cell therapy. J. Immunother. Cancer 2019, 7, 271. [Google Scholar] [CrossRef]


{kind=link}
| Ref. | Fungal Infection | Cancer | Prophylaxis | Neutropenia | Lymphopenia | Time of Onset of Infection | CRS | Steroids | Tocilizumab Given? | Previous Transplant | Died of Fungal Infection? |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Park et al. [11] | Saccharomyces cerevisiae: fungemia | ALL | Micafungin | Yes | – | Day 0–30 | Grade 3 | – | – | – | No |
| Garner et al. [8] | Candida tropicalis: fungemia | DLBCL | Fluconazole | Yes | Yes | Day 0–30 | Grade 2 | Yes | Yes (2 doses) | No | Yes |
| Candida glabrata: intra-abdominal infection | DLBCL | Fluconazole | No | Yes | Day 0–30 | Grade 1 | Yes | Yes (1 dose) | Yes (autologous) | No | |
| Candida esophagitis | DLBCL | Fluconazole | No | Yes | Day 0–30 | Grade 2 | Yes | Yes (1 dose) | Yes (autologous) | No | |
| Candida albicans: fungemia | DLBCL | None | No | Yes | Day 30+ | Grade 2 | No | Yes (1 dose) | Yes (autologous) | No | |
| Candida albicans: vertebral osteomyelitis | DLBCL | None | – | – | Day 30+ | Grade 2 | No | Yes (1 dose) | Yes (autologous) | No | |
| Candida esophagitis | DLBCL | None | No | Yes | Day 30+ | No | No | No | No | No | |
| Hill et al. [9] | Candida glabrata: fungemia | – | Fluconazole | – | – | Day 0–30 | – | – | – | – | No |
| Candida glabrata: fungemia | – | Fluconazole | – | – | Day 0–30 | – | – | – | – | No | |
| Candida glabrata: lungs a | – | Fluconazole | – | – | Day 0–30 | – | – | – | – | No | |
| Candida bracarensis: lungs a | – | Fluconazole | – | – | Day 0–30 | – | – | – | – | No | |
| Tran et al. [15] | Candida glabrata: fungemia | – | – | – | – | Day 30+ | – | – | – | – | – |
| Cordeiro et al. [10] | Oral candidiasis | – | – | – | – | Day 30+ | – | – | – | – | No |
| Louge et al. [14] | Candida krusei fungemia | DLBCL | Fluconazole | – | – | 38 days | – | Yes, for ICANS | – | No | Yes |
| Ref. | Fungal Infection | Cancer | Prophylaxis | Neutropenia | Lymphopenia | Time of Onset of Infection | CRS | Steroids | Tocilizumab Given? | Previous Transplant | Died of Fungal Infection? |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Park et al. [11] | Aspergillus fumigatus: pulmonary | ALL | Micafungin | Yes | – | Day 0–30 | Grade 3 | – | – | – | Yes |
| Probable pulmonary aspergillosis (+BAL galactomannan) | ALL | Micafungin | Yes | – | Day 0–30 | Grade 1 | – | – | – | No | |
| Mucormycosis: lung | ALL | Micafungin | Yes | – | Day 0–30 | No | – | – | – | No | |
| Probable pulmonary aspergillosis (+serum galactomannan) | ALL | None | Yes | – | Day 30+ | – | – | – | – | No | |
| Garner et al. [8] | Fusarium solani; skin and sinuses | ALL | Fluconazole → posaconazole | Yes | Yes | Day 0–30 possibly pre-infusion | Grade 1 | No | Yes (3 doses) | No | No |
| Mucorales; sinuses | Hairy cell leukemia | Voriconazole | Yes | Yes | Day 30+ | Grade 1 | No | Yes (1 dose) | Yes (allogeneic) | Yes | |
| Possible pulmonary aspergillosis | CLL | None | Yes | No | Day 30+ | None | No | No | No | No | |
| Hill et al. [9] | Aspergillus ustus; lungs | CLL | Fluconazole | No | – | Day 0–30 | ≥Grade 3 | – | – | – | Yes |
| Unknown mold; sinuses | ALL | Fluconazole | – | – | Day 0–30; possibly pre-infusion | ≥Grade 3 | – | – | – | No | |
| Aspergillus fumigatus; sinuses | CLL | Fluconazole | Yes | – | Day 30+ | ≥Grade 3 | – | – | Yes (allogeneic) | No | |
| Tran et al. [15] | Aspergillus + Rhizopus species: skin and soft tissue | – | – | – | – | Day 30+ | – | -– | – | – | – |
| Cordeiro et al. [10] | Aspergillosis (n = 2) | – | – | – | – | Day 30+ | – | – | – | – | No |
| Logue et al. [14] | Disseminated fusariosis | DLBCL | Fluconazole → micafungin | – | – | Day 0–30 | – | Yes, for ICANS | – | No | Yes |
| Vora et al. [12] | Cunninghamella species: lung | ALL | Voriconazole | Yes | – | Day 0–30; possibly pre-infusion | Grade 1 | No | Yes | No | Yes |
| Ref. | Fungal Infection | Cancer | Prophylaxis | Neutropenia | Lymphopenia | Time of Onset of Infection | CRS | Steroids | Tocilizumab Given? | Previous Transplant | Died of Fungal Infection? |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Garner et al. [8] | PCP | DLBCL | None (completed TMP/SMX) | No | Yes | Day 30+ | Grade 2 | Yes | Yes (2 doses) | No | No |
| Hill et al. [9] | PCP | ALL | None (TMP/SMX non-compliance) | – | – | Day 30+ | – | – | – | – | No |
| Wudhikarn et al. [13] | PCP | – | None (completed pentamidine) | – | Yes | Day 30+ | – | – | – | – | No |
| Cordeiro et al. [10] | Coccidioides infection | – | – | – | – | Day 30+ | – | – | – | – | No |
| Risk Factors for Any Infection after CAR-T-Cell Therapy | Other Potential Risk Factors for IFI after CAR-T-Cell Therapy a | |
|---|---|---|
| Pre-Infusion Factors | Prior history HCT # of prior lines of chemotherapy CAR-T-cell dose ALL History of infection prior to CAR-T-cell therapy | Refractory disease/# of prior lines of chemotherapy Type of underlying malignancy Prior history of HCT Previous history of IFI Indwelling CVCs |
| Post-Infusion Factors | Higher CRS grade (≥3) | Neutropenia Lymphopenia Steroids (dose/duration) ICU admission Indwelling CVCs |
| Potential Additional Post-Infusions Factors | Neutropenia Lymphopenia Tocilizumab (# of doses) Steroids (dose/duration) Use of alternative immunosuppressing agents in treatment of CRS or ICANS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garner, W.; Samanta, P.; Haidar, G. Invasive Fungal Infections after Anti-CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy: State of the Evidence and Future Directions. J. Fungi 2021, 7, 156. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020156
Garner W, Samanta P, Haidar G. Invasive Fungal Infections after Anti-CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy: State of the Evidence and Future Directions. Journal of Fungi. 2021; 7(2):156. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020156
Chicago/Turabian StyleGarner, Will, Palash Samanta, and Ghady Haidar. 2021. "Invasive Fungal Infections after Anti-CD19 Chimeric Antigen Receptor-Modified T-Cell Therapy: State of the Evidence and Future Directions" Journal of Fungi 7, no. 2: 156. https://0-doi-org.brum.beds.ac.uk/10.3390/jof7020156