
Spontaneous Suppressors against Debilitating Transmembrane Mutants of CaMdr1 Disclose Novel Interdomain Communication via Signature Motifs of the Major Facilitator Superfamily

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Reagents
2.1.2. Strains and Culture Conditions
2.2. Methods
2.2.1. Generation and Sequence Analysis of Suppressor Mutants
2.2.2. Site-Directed Mutagenesis and Yeast Transformation
2.2.3. Confocal Microscopy
2.2.4. Spot Dilution Growth Assay
2.2.5. MIC Assay
2.2.6. Nile Red Accumulation Assay
2.2.7. WebLogo Generation
2.2.8. Generation of 3D Homology Model of CaMdr1
2.2.9. Statistical Analyses
3. Results and Discussion
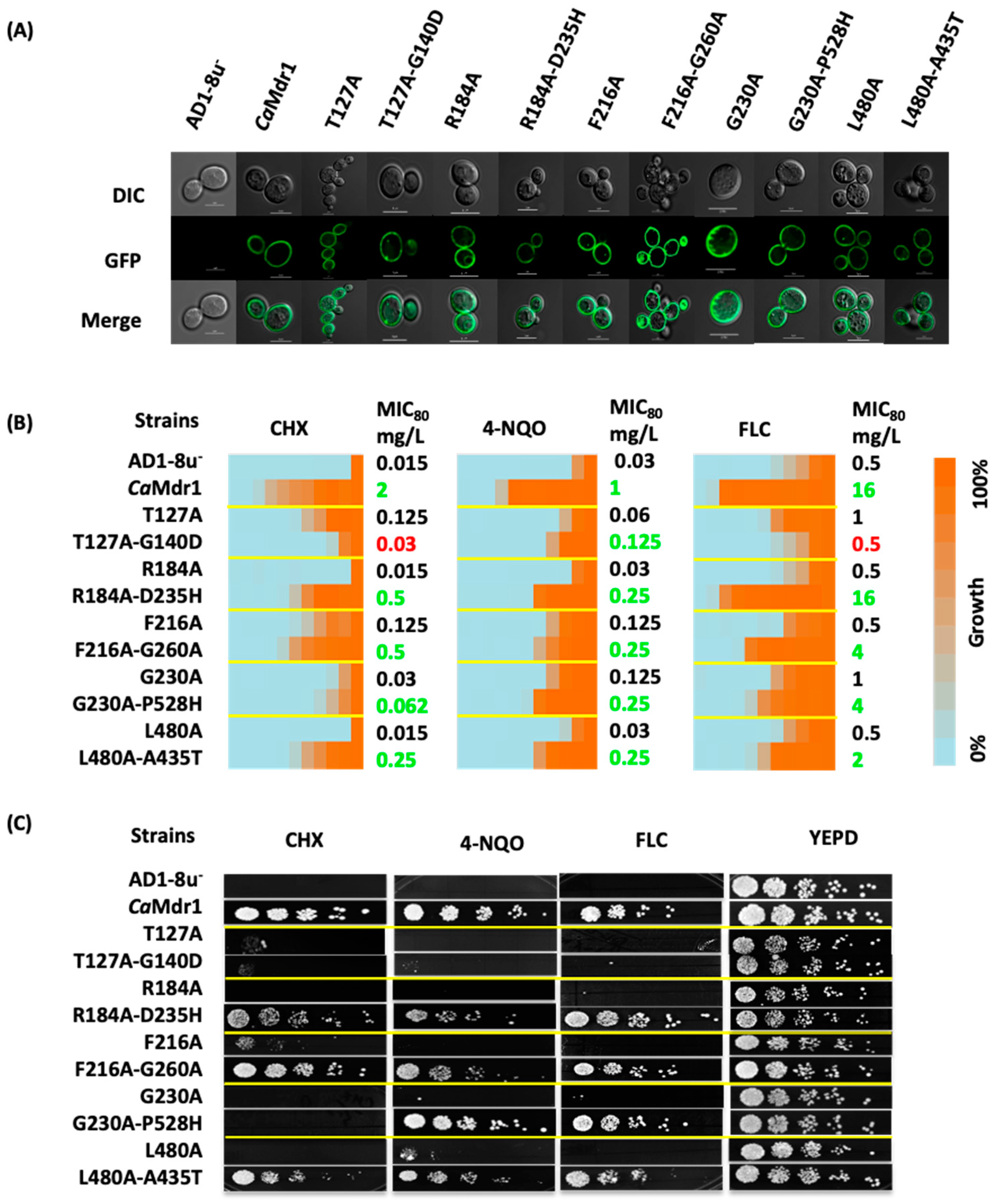
3.1. Generation of CaMdr1 Drug-Sensitive Suppressors
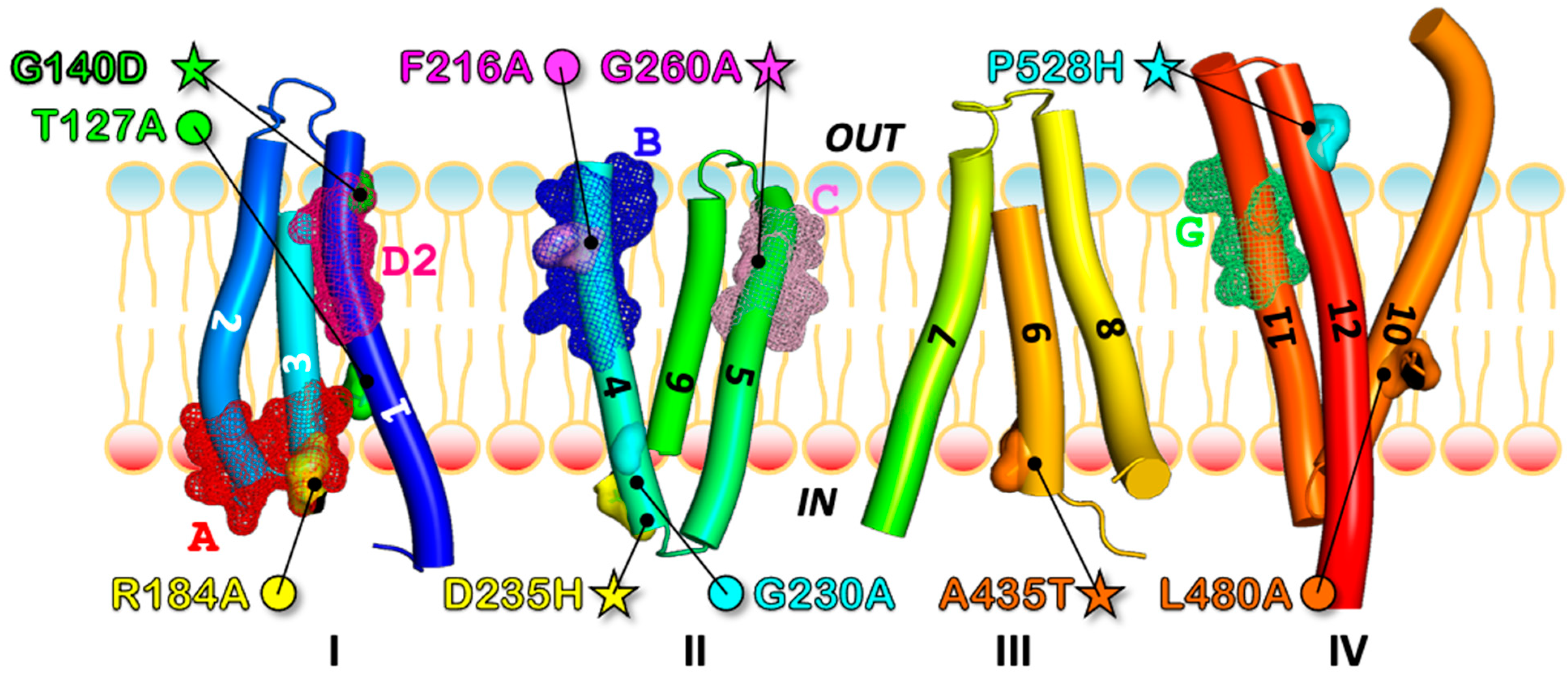
3.2. Most of the Drug-Sensitive Mutants and Suppressors Are Located within the Conserved Signature Motifs of DHA1 MFS
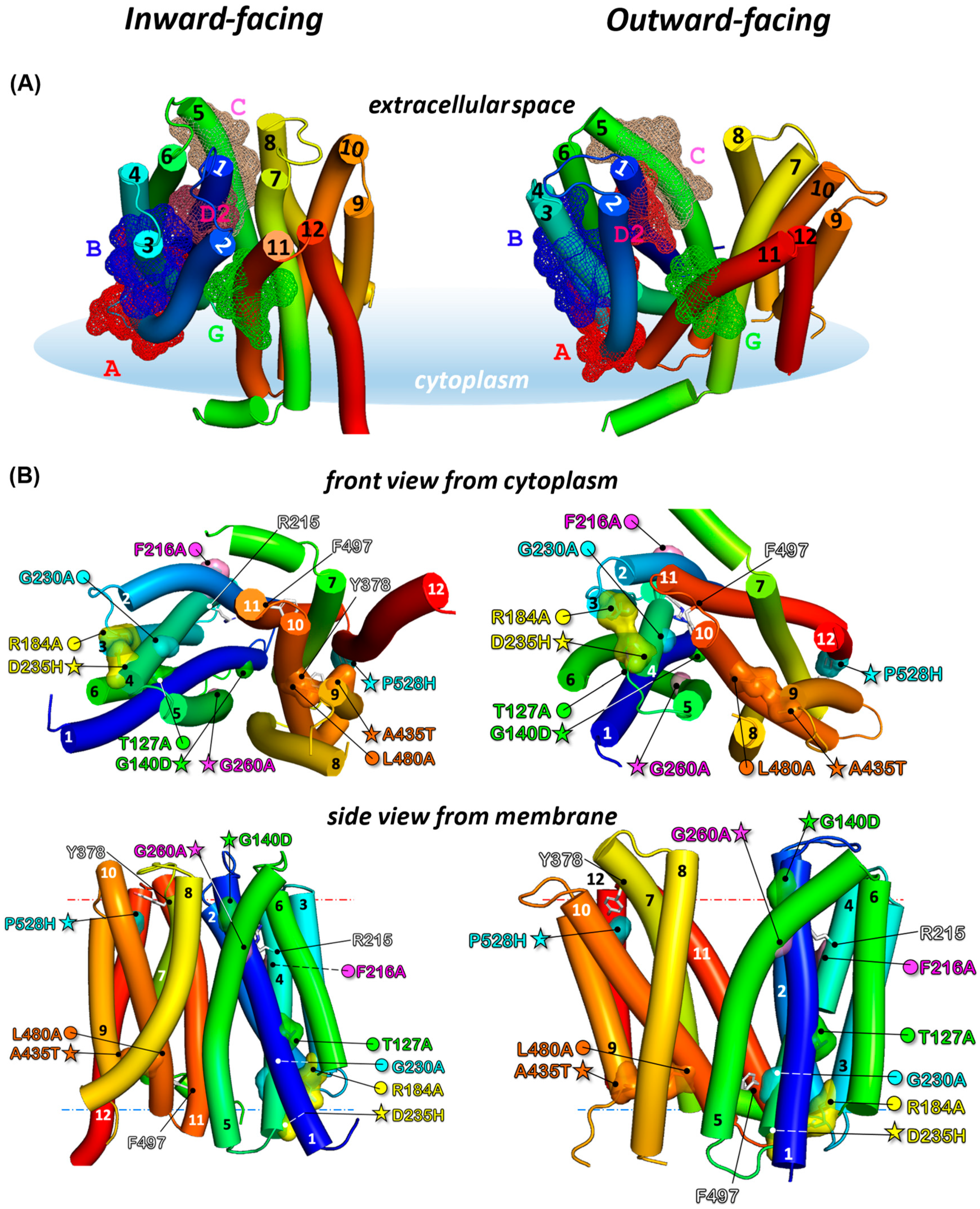
3.3. Localization of Sensitive and Suppressor Mutants in the Outward-Facing Conformation of CaMdr1
3.4. Local Compensations Restore 3-TMHs Repeat Tandem Interactions in N- and C-Domains and Highlight the Role of Motif A
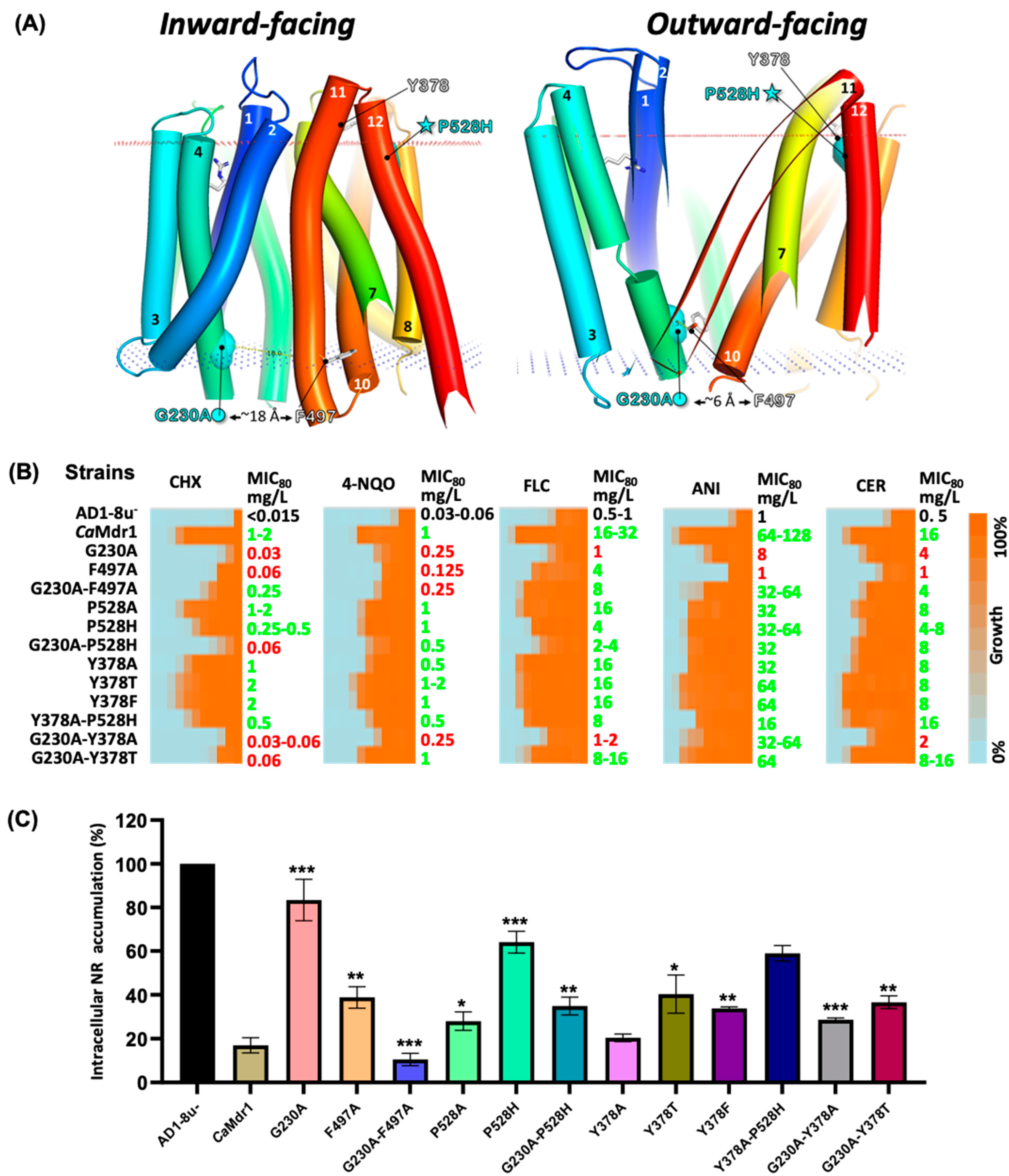
3.5. Distant Compensations in the N-Domain Highlight Interplay between Motifs A, B, and D2
3.6. Long-Range Compensation between the N- and C-Domains
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- White, T.C.; Holleman, S.; Dy, F.; Mirels, L.F.; Stevens, D.A. Resistance Mechanisms in Clinical Isolates of Candida albicans. Antimicrob. Agents Chemother. 2002, 46, 1704–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, R.; De Wergifosse, P.; Goffeau, A.; Balzi, E. Molecular Cloning and Characterization of a Novel Gene of Candida albicans, CDR1, Conferring Multiple Resistance to Drugs and Antifungals. Curr. Genet. 1995, 27, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Sanglard, D.; Kuchler, K.; Ischer, F.; Pagani, J.L.; Monod, M.; Bille, J. Mechanisms of Resistance to Azole Antifungal Agents in Candida albicans Isolates from AIDS Patients Involve Specific Multidrug Transporters. Antimicrob. Agents Chemother. 1995, 39, 2378–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, T.C. Increased MRNA Levels of ERG16, CDR, and MDR1 Correlate with Increases in Azole Resistance in Candida albicans Isolates from a Patient Infected with Human Immunodeficiency Virus. Antimicrob. Agents Chemother. 1997, 41, 1482–1487. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ribot, J.L.; McAtee, R.K.; Lee, L.N.; Kirkpatrick, W.R.; White, T.C.; Sanglard, D.; Patterson, T.F. Distinct Patterns of Gene Expression Associated with Development of Fluconazole Resistance in Serial Candida albicans Isolates from Human Immunodeficiency Virus-Infected Patients with Oropharyngeal Candidiasis. Antimicrob. Agents Chemother. 1998, 42, 2932–2937. [Google Scholar] [CrossRef] [Green Version]
- Harry, J.B.; Song, J.L.; Lyons, C.N.; White, T.C. Transcription Initiation of Genes Associated with Azole Resistance in Candida albicans. Med. Mycol. 2002, 40, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Saier, M.H., Jr.; Reddy, V.S.; Tamang, D.G.; Västermark, Å. The Transporter Classification Database. Nucleic Acids Res. 2014, 42, D251–D258. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Wang, S.C.; Davejan, P.; Hendargo, K.J.; Javadi-Razaz, I.; Chou, A.; Yee, D.C.; Ghazi, F.; Lam, K.J.K.; Conn, A.M.; Madrigal, A.; et al. Expansion of the Major Facilitator Superfamily (MFS) to Include Novel Transporters as Well as Transmembrane-Acting Enzymes. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183277. [Google Scholar] [CrossRef]
- Lee, J.; Sands, Z.A.; Biggin, P.C. A Numbering System for MFS Transporter Proteins. Front. Mol. Biosci. 2016, 3, 21. [Google Scholar] [CrossRef]
- Shi, Y. Common Folds and Transport Mechanisms of Secondary Active Transporters. Annu. Rev. Biophys. 2013, 42, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Law, C.J.; Maloney, P.C.; Wang, D.-N. Ins and Outs of Major Facilitator Superfamily Antiporters. Annu. Rev. Microbiol. 2008, 62, 289–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaur, M.; Puri, N.; Manoharlal, R.; Rai, V.; Mukhopadhayay, G.; Choudhury, D.; Prasad, R. MFS Transportome of the Human Pathogenic Yeast Candida albicans. BMC Genom. 2008, 9, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Yaacov, R.; Knoller, S.; Caldwell, G.A.; Becker, J.M.; Koltin, Y. Candida albicans Gene Encoding Resistance to Benomyl and Methotrexate Is a Multidrug Resistance Gene. Antimicrob. Agents Chemother. 1994, 38, 648–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, A.; Gupta, V.; Krishnamurthy, S.; Hasnain, S.E.; Prasad, R. Specificity of Drug Transport Mediated ByCaMDR1: A Major Facilitator of Candida albicans. J. Biosci. 2001, 26, 333–339. [Google Scholar] [CrossRef]
- Gupta, V.; Kohli, A.; Krishnamurthy, S.; Puri, N.; Aalamgeer, S.A.; Panwar, S.; Prasad, R. Identification of Polymorphic Mutant Alleles of CaMDR1, a Major Facilitator of Candida albicans Which Confers Multidrug Resistance, and Its in Vitro Transcriptional Activation. Curr. Genet. 1998, 34, 192–199. [Google Scholar] [CrossRef]
- Prasad, R.; Kapoor, K. Multidrug Resistance in Yeast Candida. Int. Rev. Cytol. 2005, 242, 215–248. [Google Scholar] [CrossRef]
- Huang, Y.; Lemieux, M.J.; Song, J.; Auer, M.; Wang, D.-N. Structure and Mechanism of the Glycerol-3-Phosphate Transporter from Escherichia coli. Science 2003, 301, 616–620. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; He, X.; Szewczyk, P.; Nguyen, T.; Chang, G. Structure of the Multidrug Transporter EmrD from Escherichia coli. Science 2006, 312, 741–744. [Google Scholar] [CrossRef]
- Sun, L.; Zeng, X.; Yan, C.; Sun, X.; Gong, X.; Rao, Y.; Yan, N. Crystal Structure of a Bacterial Homologue of Glucose Transporters GLUT1–4. Nature 2012, 490, 361–366. [Google Scholar] [CrossRef]
- Jiang, D.; Zhao, Y.; Wang, X.; Fan, J.; Heng, J.; Liu, X.; Feng, W.; Kang, X.; Huang, B.; Liu, J.; et al. Structure of the YajR Transporter Suggests a Transport Mechanism Based on the Conserved Motif A. Proc. Natl. Acad. Sci. USA 2013, 110, 14664–14669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorens, B.; Mueckler, M. Glucose Transporters in the 21st Century. Am. J. Physiol. Metab. 2009, 298, E141–E145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, D.; Xu, C.; Sun, P.; Wu, J.; Yan, C.; Hu, M.; Yan, N. Crystal Structure of the Human Glucose Transporter GLUT1. Nature 2014, 510, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Wisedchaisri, G.; Gonen, T. Crystal Structure of a Nitrate/Nitrite Exchanger. Nature 2013, 497, 647–651. [Google Scholar] [CrossRef] [PubMed]
- Yan, N. Structural Biology of the Major Facilitator Superfamily Transporters. Annu. Rev. Biophys. 2015, 44, 257–283. [Google Scholar] [CrossRef] [PubMed]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and General Transport Mechanisms by the Major Facilitator Superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Huang, W.; Yan, C.; Gong, X.; Jiang, S.; Zhao, Y.; Wang, J.; Shi, Y. Structure and Mechanism of a Nitrate Transporter. Cell Rep. 2013, 3, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Radestock, S.; Forrest, L.R. The Alternating-Access Mechanism of MFS Transporters Arises from Inverted-Topology Repeats. J. Mol. Biol. 2011, 407, 698–715. [Google Scholar] [CrossRef]
- Madej, M.G.; Kaback, H.R. Evolutionary Mix-and-Match with MFS Transporters II. Proc. Natl. Acad. Sci. USA 2013, 110, E4831–E4838. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-Dependent Multidrug Efflux Systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef]
- Putman, M.; van Veen, H.W.; Konings, W. Molecular Properties of Bacterial Multidrug Transporters. Microbiol. Mol. Biol. Rev. 2000, 64, 672–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakarla, P.; KC, R.; Shrestha, U.; Ranaweera, I.; Mukherjee, M.M.; Willmon, T.M.; Hernandez, A.J.; Barr, S.R.; Varela, M.F. Functional Roles of Highly Conserved Amino Acid Sequence Motifs A and C in Solute Transporters of the Major Facilitator Superfamily. In Drug Resistance in Bacteria, Fungi, Malaria, and Cancer; Arora, G., Sajid, A., Kalia, V.C., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 111–140. ISBN 978-3-319-48683-3. [Google Scholar]
- Kumar, S.; Lekshmi, M.; Parvathi, A.; Ojha, M.; Wenzel, N.; Varela, M.F. Functional and Structural Roles of the Major Facilitator Superfamily Bacterial Multidrug Efflux Pumps. Microorganisms 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redhu, A.K.; Khandelwal, N.K.; Banerjee, A.; Moreno, A.; Falson, P.; Prasad, R. PHluorin Enables Insights into the Transport Mechanism of Antiporter Mdr1: R215 Is Critical for Drug/H+ Antiport. Biochem. J. 2016, 473, 3127–3145. [Google Scholar] [CrossRef]
- Russ, W.P.; Engelman, D.M. The GxxxG Motif: A Framework for Transmembrane Helix-Helix Association Edited by G. von Heijne. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Brown, M.H.; Skurray, R.A. The TetA(K) Tetracycline/H+ Antiporter from Staphylococcus aureus: Mutagenesis and Functional Analysis of Motif C. J. Bacteriol. 2000, 182, 1492–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritu, P.; Dibyendu, B.; Rajendra, P. Structure and Function Analysis of CaMdr1p, a Major Facilitator Superfamily Antifungal Efflux Transporter Protein of Candida albicans: Identification of Amino Acid Residues Critical for Drug/H+ Transport. Eukaryot. Cell 2007, 6, 443–453. [Google Scholar] [CrossRef] [Green Version]
- Redhu, A.K.; Banerjee, A.; Shah, A.H.; Moreno, A.; Rawal, M.K.; Nair, R.; Falson, P.; Prasad, R. Molecular Basis of Substrate Polyspecificity of the Candida albicans Mdr1p Multidrug/H+ Antiporter. J. Mol. Biol. 2018, 430, 682–694. [Google Scholar] [CrossRef]
- Shuman, H.A. The Use of Gene Fusions to Study Bacterial Transport Proteins. J. Membr. Biol. 1981, 61, 1–11. [Google Scholar] [CrossRef]
- Abramson, J.; Smirnova, I.; Kasho, V.; Verner, G.; Kaback, H.R.; Iwata, S. Structure and Mechanism of the Lactose Permease of Escherichia coli. Science 2003, 301, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Guan, L.; Mirza, O.; Verner, G.; Iwata, S.; Kaback, H.R. Structural Determination of Wild-Type Lactose Permease. Proc. Natl. Acad. Sci. USA 2007, 104, 15294–15298. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Finer-Moore, J.S.; Jiang, X.; Smirnova, I.; Kasho, V.; Pardon, E.; Steyaert, J.; Kaback, H.R.; Stroud, R.M. Crystal Structure of a Ligand-Bound LacY–Nanobody Complex. Proc. Natl. Acad. Sci. USA 2018, 115, 8769–8774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.C.; Zhao, Y.; Heng, J.; Jiang, D. Energy Coupling Mechanisms of MFS Transporters. Protein Sci. 2015, 24, 1560–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisedchaisri, G.; Park, M.-S.; Iadanza, M.G.; Zheng, H.; Gonen, T. Proton-Coupled Sugar Transport in the Prototypical Major Facilitator Superfamily Protein XylE. Nat. Commun. 2014, 5, 4521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Sun, B.; Zhou, Y.; Wang, C.; Guo, L.; He, J.; Deng, D. Visualizing the Nonlinear Changes of a Drug-Proton Antiporter from Inward-Open to Occluded State. Biochem. Biophys. Res. Commun. 2021, 534, 272–278. [Google Scholar] [CrossRef]
- Tan, X.; Wang, B. Inward Open Characterization of EmrD Transporter with Molecular Dynamics Simulation. Biochem. Biophys. Res. Commun. 2016, 474, 640–645. [Google Scholar] [CrossRef]
- Debruycker, V.; Hutchin, A.; Masureel, M.; Ficici, E.; Martens, C.; Legrand, P.; Stein, R.A.; Mchaourab, H.S.; Faraldo-Gómez, J.D.; Remaut, H.; et al. An Embedded Lipid in the Multidrug Transporter LmrP Suggests a Mechanism for Polyspecificity. Nat. Struct. Mol. Biol. 2020, 27, 829–835. [Google Scholar] [CrossRef]
- Fluman, N.; Ryan, C.M.; Whitelegge, J.P.; Bibi, E. Dissection of Mechanistic Principles of a Secondary Multidrug Efflux Protein. Mol. Cell 2012, 47, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Heng, J.; Zhao, Y.; Liu, M.; Liu, Y.; Fan, J.; Wang, X.; Zhao, Y.; Zhang, X.C. Substrate-Bound Structure of the E. Coli Multidrug Resistance Transporter MdfA. Cell Res. 2015, 25, 1060–1073. [Google Scholar] [CrossRef]
- Schaedler, T.A.; Van Veen, H.W. A Flexible Cation Binding Site in the Multidrug Major Facilitator Superfamily Transporter LmrP Is Associated with Variable Proton Coupling. FASEB J. 2010, 24, 3653–3661. [Google Scholar] [CrossRef]
- Wu, H.-H.; Symersky, J.; Lu, M. Structure and Mechanism of a Redesigned Multidrug Transporter from the Major Facilitator Superfamily. Sci. Rep. 2020, 10, 3949. [Google Scholar] [CrossRef] [Green Version]
- Mandal, A.; Kumar, A.; Singh, A.; Lynn, A.M.; Kapoor, K.; Prasad, R. A Key Structural Domain of the Candida albicans Mdr1 Protein. Biochem. J. 2012, 445, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, K.; Rehan, M.; Kaushiki, A.; Pasrija, R.; Lynn, A.M.; Prasad, R. Rational Mutational Analysis of a Multidrug MFS Transporter CaMdr1p of Candida albicans by Employing a Membrane Environment Based Computational Approach. PLoS Comput. Biol. 2009, 5, e1000624. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, K.; Rehan, M.; Lynn, A.M.; Prasad, R. Employing Information Theoretic Measures and Mutagenesis to Identify Residues Critical for Drug-Proton Antiport Function in Mdr1p of Candida albicans. PLoS ONE 2010, 5, e11041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stirling, D. DNA Extraction from Fungi, Yeast, and Bacteria. In PCR Protocols; Bartlett, J.M.S., Stirling, D., Eds.; Humana Press: Totowa, NJ, USA, 2003; pp. 53–54. ISBN 978-1-59259-384-2. [Google Scholar]
- Lamping, E.; Monk, B.C.; Niimi, K.; Holmes, A.R.; Tsao, S.; Tanabe, K.; Niimi, M.; Uehara, Y.; Cannon, R.D. Characterization of Three Classes of Membrane Proteins Involved in Fungal Azole Resistance by Functional Hyperexpression in Saccharomyces cerevisiae. Eukaryot. Cell 2007, 6, 1150–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, K.; Kohli, A.; Prasad, R. Drug Susceptibilities of Yeast Cells Are Affected by Membrane Lipid Composition. Antimicrob. Agents Chemother. 2002, 46, 3695–3705. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Moreno, A.; Khan, M.F.; Nair, R.; Sharma, S.; Sen, S.; Mondal, A.K.; Pata, J.; Orelle, C.; Falson, P.; et al. Cdr1p Highlights the Role of the Non-Hydrolytic ATP-Binding Site in Driving Drug Translocation in Asymmetric ABC Pumps. Biochim. Biophys. Acta-Biomembr. 2020, 1862, 183131. [Google Scholar] [CrossRef]
- Ivnitski-Steele, I.; Holmes, A.R.; Lamping, E.; Monk, B.C.; Cannon, R.D.; Sklar, L.A. Identification of Nile Red as a Fluorescent Substrate of the Candida albicans ATP-Binding Cassette Transporters Cdr1p and Cdr2p and the Major Facilitator Superfamily Transporter Mdr1p. Anal. Biochem. 2009, 394, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Stamm, M.; Staritzbichler, R.; Khafizov, K.; Forrest, L.R. AlignMe—A Membrane Protein Sequence Alignment Web Server. Nucleic Acids Res. 2014, 42, W246–W251. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Structural Genomics: General Applications; Chen, Y.W., Yiu, C.-P.B., Eds.; Springer: New York, NY, USA, 2021; pp. 239–255. ISBN 978-1-0716-0892-0. [Google Scholar]
- Decottignies, A.; Grant, A.M.; Nichols, J.W.; de Wet, H.; McIntosh, D.B.; Goffeau, A. ATPase and Multidrug Transport Activities of the Overexpressed Yeast ABC Protein Yor1p. J. Biol. Chem. 1998, 273, 12612–12622. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Niimi, M.; Niimi, K.; Holmes, A.R.; Yates, J.E.; Decottignies, A.; Monk, B.C.; Goffeau, A.; Cannon, R.D. Functional Expression of Candida albicans Drug Efflux Pump Cdr1p in a Saccharomyces cerevisiae Strain Deficient in Membrane Transporters. Antimicrob. Agents Chemother. 2001, 45, 3366–3374. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Pata, J.; Sharma, S.; Monk, B.C.; Falson, P.; Prasad, R. Directed Mutational Strategies Reveal Drug Binding and Transport by the MDR Transporters of Candida albicans. J. Fungi 2021, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Someya, Y.; Kimura-Someya, T.; Yamaguchi, A. Role of the Charge Interaction between Arg70 and Asp120 in the Tn10-Encoded Metal-Tetracycline/H+ Antiporter of Escherichia coli. J. Biol. Chem. 2000, 275, 210–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Mutant | Suppressor | |||||
|---|---|---|---|---|---|---|
| Categories | Mutation | Location | Phenotype | Function (Redhu et al., 2018) | Drug Leading to Resistance | Phenotype |
| Transiently resistant strains | I123A | TMH-1 | SS | drug/H+ antiport mechanism | CHX, 4-NQO | TS |
| W249A | TMH-5 | TS | drug binding and transport | CHX | TS | |
| Y365A | TMH-7 | TS | drug binding and transport | CHX, 4-NQO | TS | |
| Y369A | TMH-7 | TS | drug binding and transport | CHX | PR | |
| F371A | TMH-7 | TS | drug binding and transport | CHX, 4-NQO | PR | |
| Y408A | TMH-8 | TS | exposed to lipid interface | 4-NQO | PR | |
| Resistance phenotype (intergenic mutation) | P257A | TMH-5 | TS | structural integrity | CHX | TR |
| I448A | TMH-9 | TS | Crucial for plasma membrane localization | CHX, 4-NQO | TR | |
| F474A | TMH-10 | TS | drug binding and transport | CHX | SR | |
| Q478A | TMH-10 | TS | drug binding and transport | CHX | SR | |
| V506A | TMH-11 | TS | Crucial role in polyspecific substrate binding and transport | CHX | TR | |
| Resistance phenotype (intragenic mutation) | T127A | TMH-1 | TS | drug/H+ antiport mechanism | CHX | SR |
| R184A | TMH-3 | TS | drug/H+ antiport mechanism | CHX | TR | |
| F216A | TMH-4 | TS | exposed to lipid interface | CHX | TR | |
| G230A | TMH-4 | TS | structural integrity | CHX | SR | |
| L480A | TMH-10 | TS | structural integrity | 4-NQO | TR | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, S.; Banerjee, A.; Moreno, A.; Redhu, A.K.; Falson, P.; Prasad, R. Spontaneous Suppressors against Debilitating Transmembrane Mutants of CaMdr1 Disclose Novel Interdomain Communication via Signature Motifs of the Major Facilitator Superfamily. J. Fungi 2022, 8, 538. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8050538
Sharma S, Banerjee A, Moreno A, Redhu AK, Falson P, Prasad R. Spontaneous Suppressors against Debilitating Transmembrane Mutants of CaMdr1 Disclose Novel Interdomain Communication via Signature Motifs of the Major Facilitator Superfamily. Journal of Fungi. 2022; 8(5):538. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8050538
Chicago/Turabian StyleSharma, Suman, Atanu Banerjee, Alexis Moreno, Archana Kumari Redhu, Pierre Falson, and Rajendra Prasad. 2022. "Spontaneous Suppressors against Debilitating Transmembrane Mutants of CaMdr1 Disclose Novel Interdomain Communication via Signature Motifs of the Major Facilitator Superfamily" Journal of Fungi 8, no. 5: 538. https://0-doi-org.brum.beds.ac.uk/10.3390/jof8050538