Polymer Networks Synthesized from Poly(Sorbitol Adipate) and Functionalized Poly(Ethylene Glycol)

 , ,
, ,  ,
,
Abstract
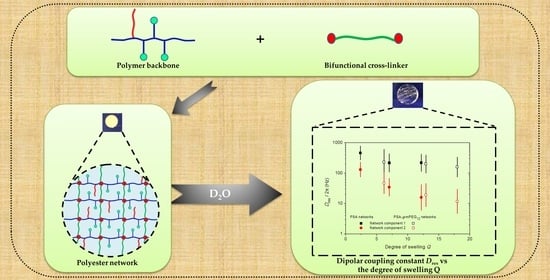
:
1. Introduction
2. Results and Discussion
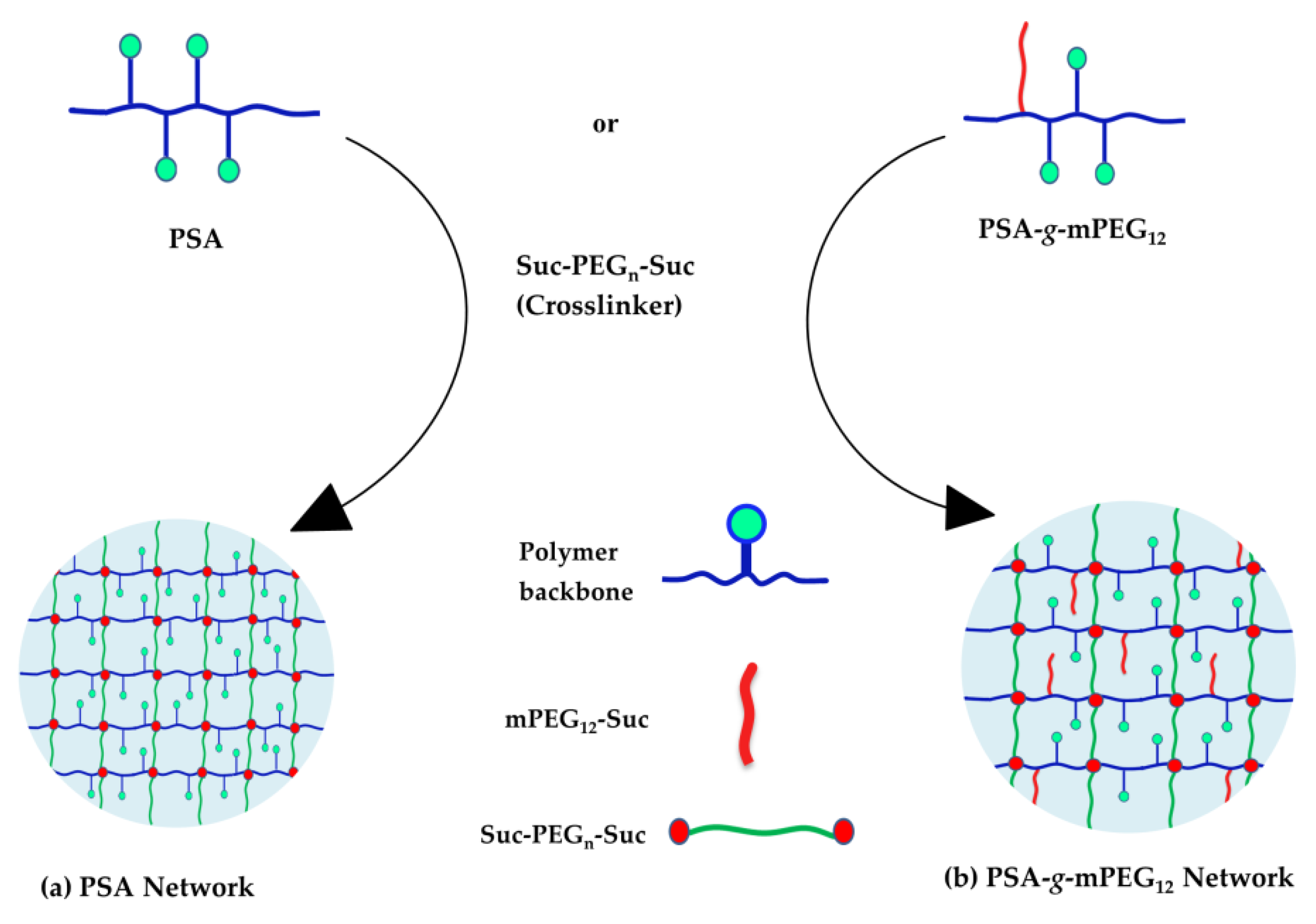
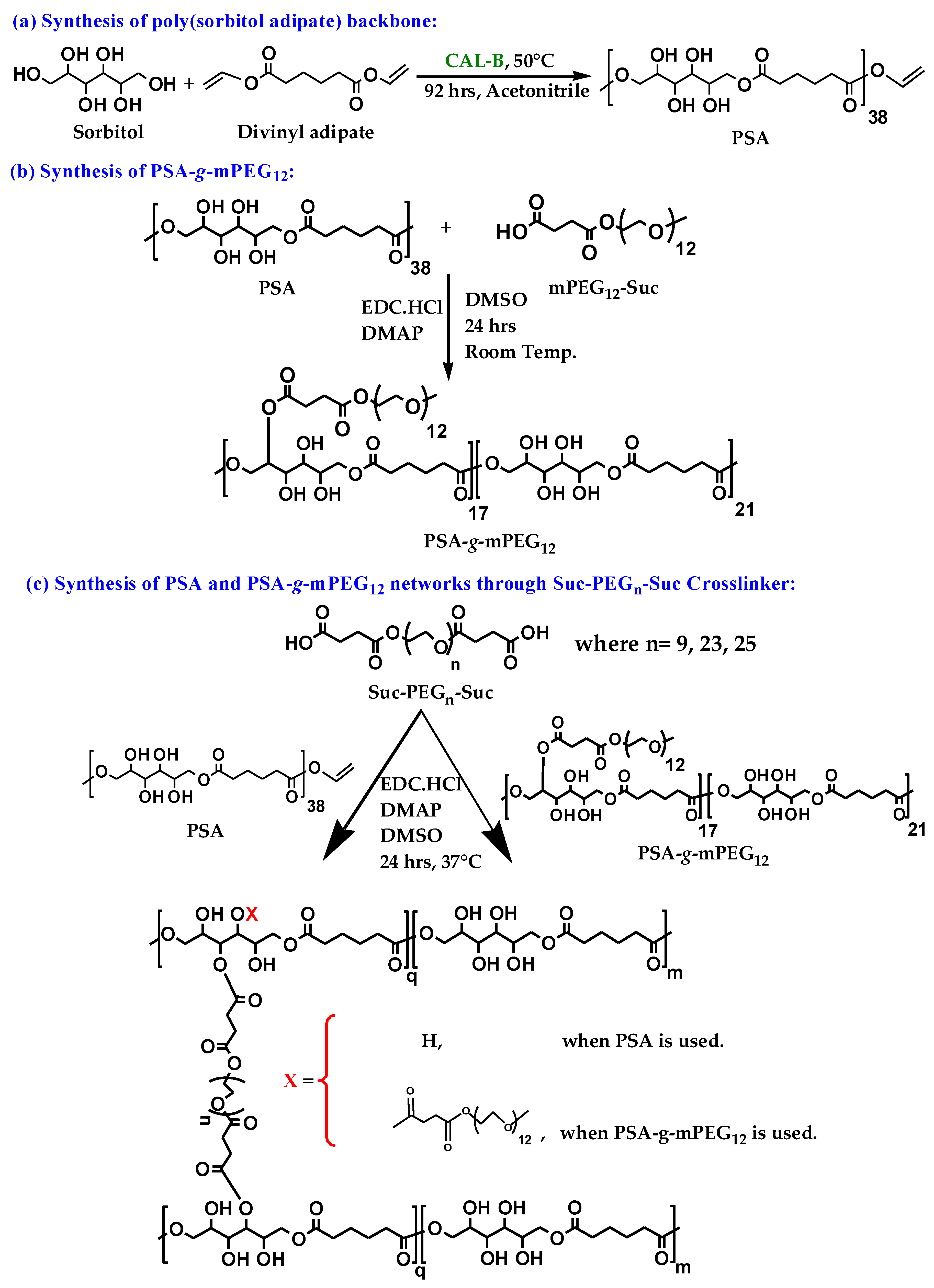
2.1. Polymer Syntheses
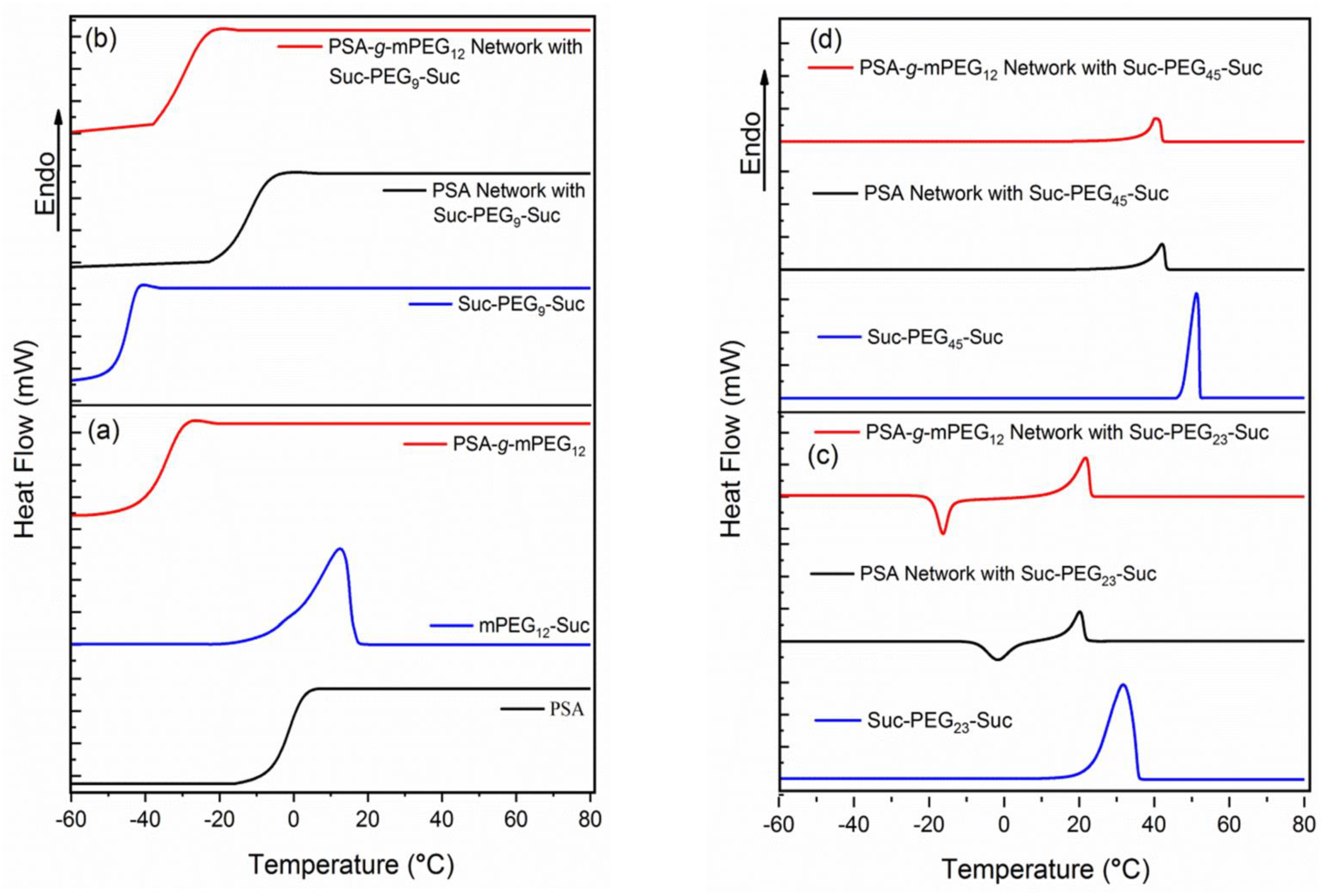
2.2. Differential Scanning Calorimetry
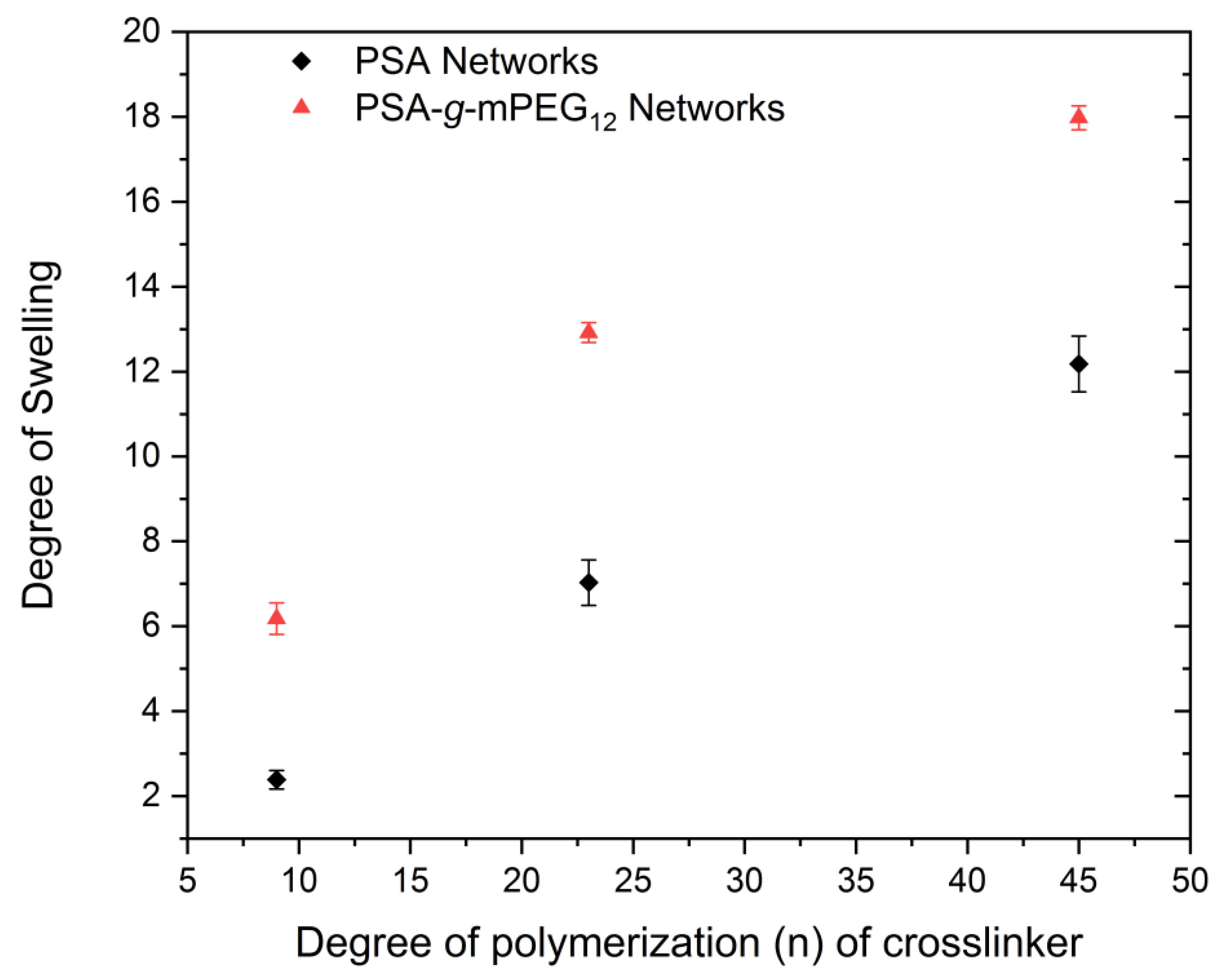
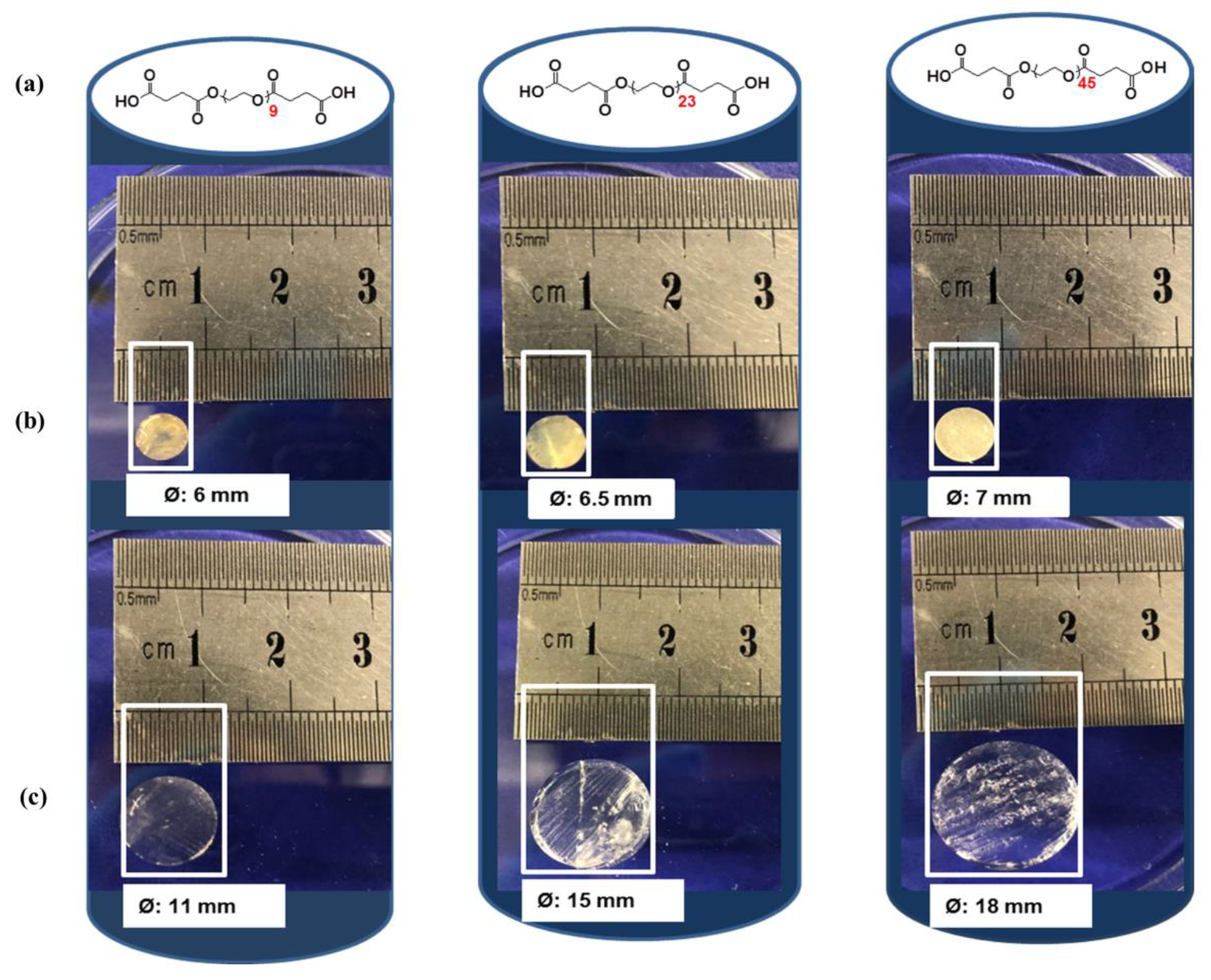
2.3. Swelling Studies
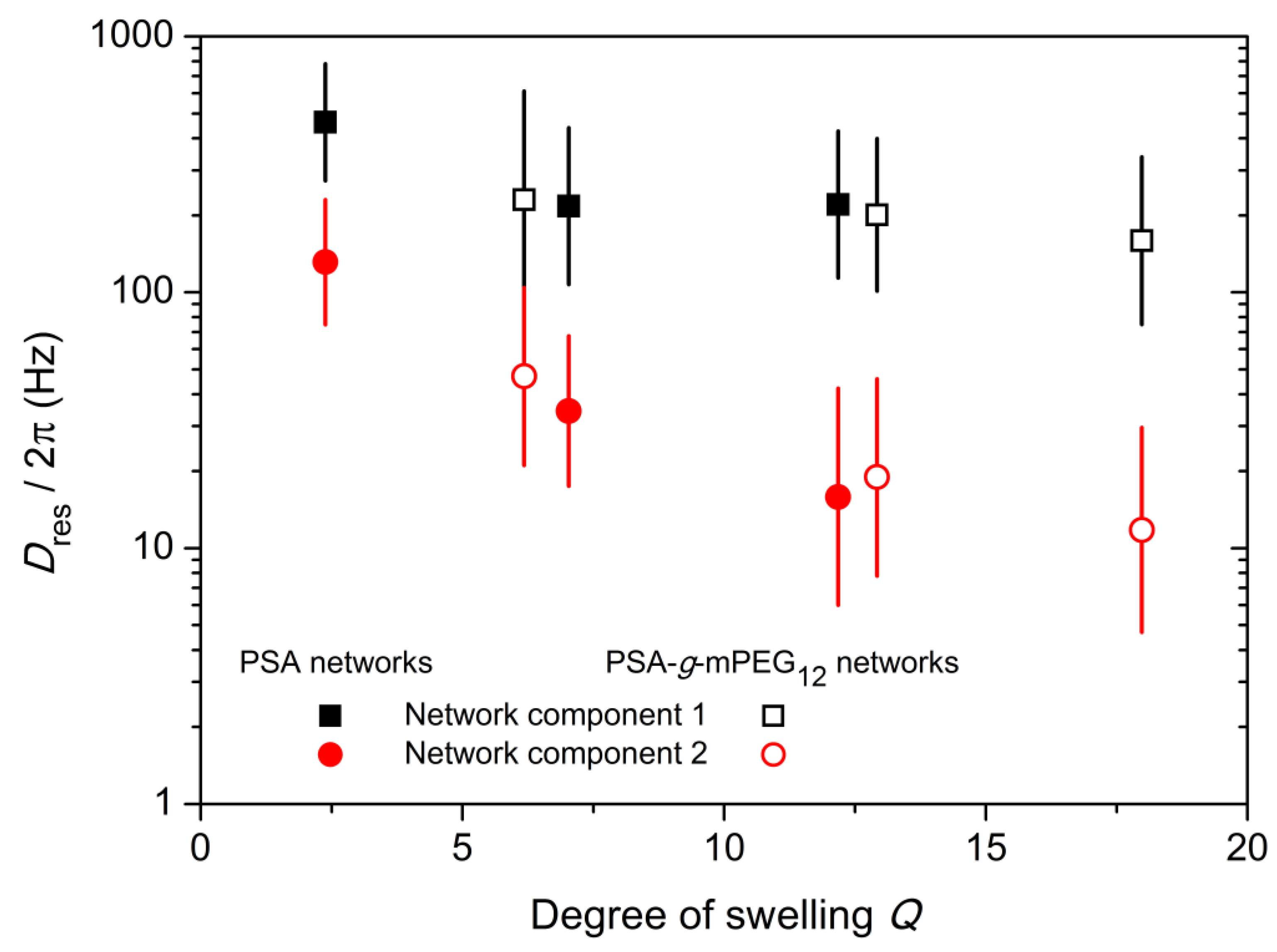
2.4. 1H Double-Quantum (DQ) NMR Spectroscopy
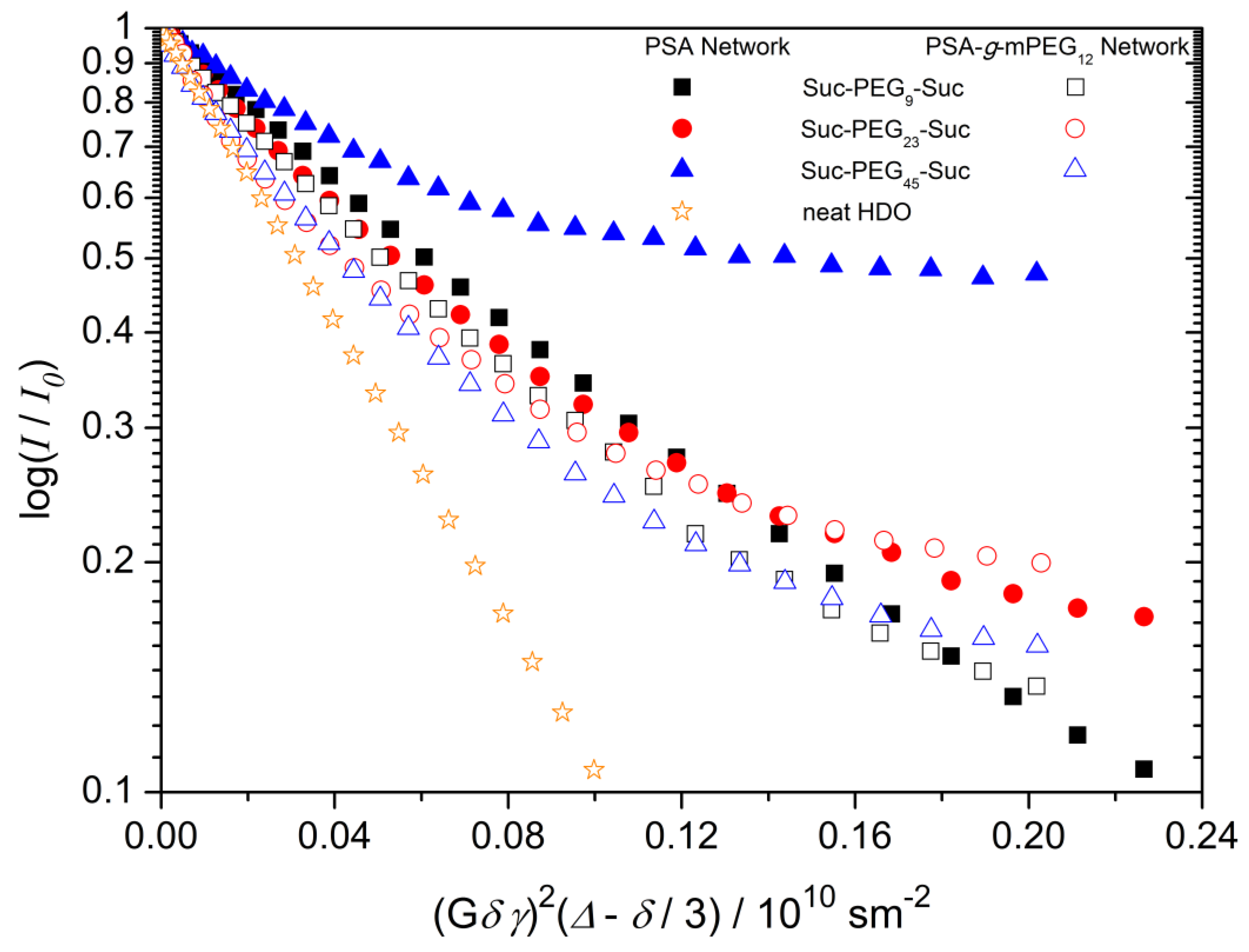
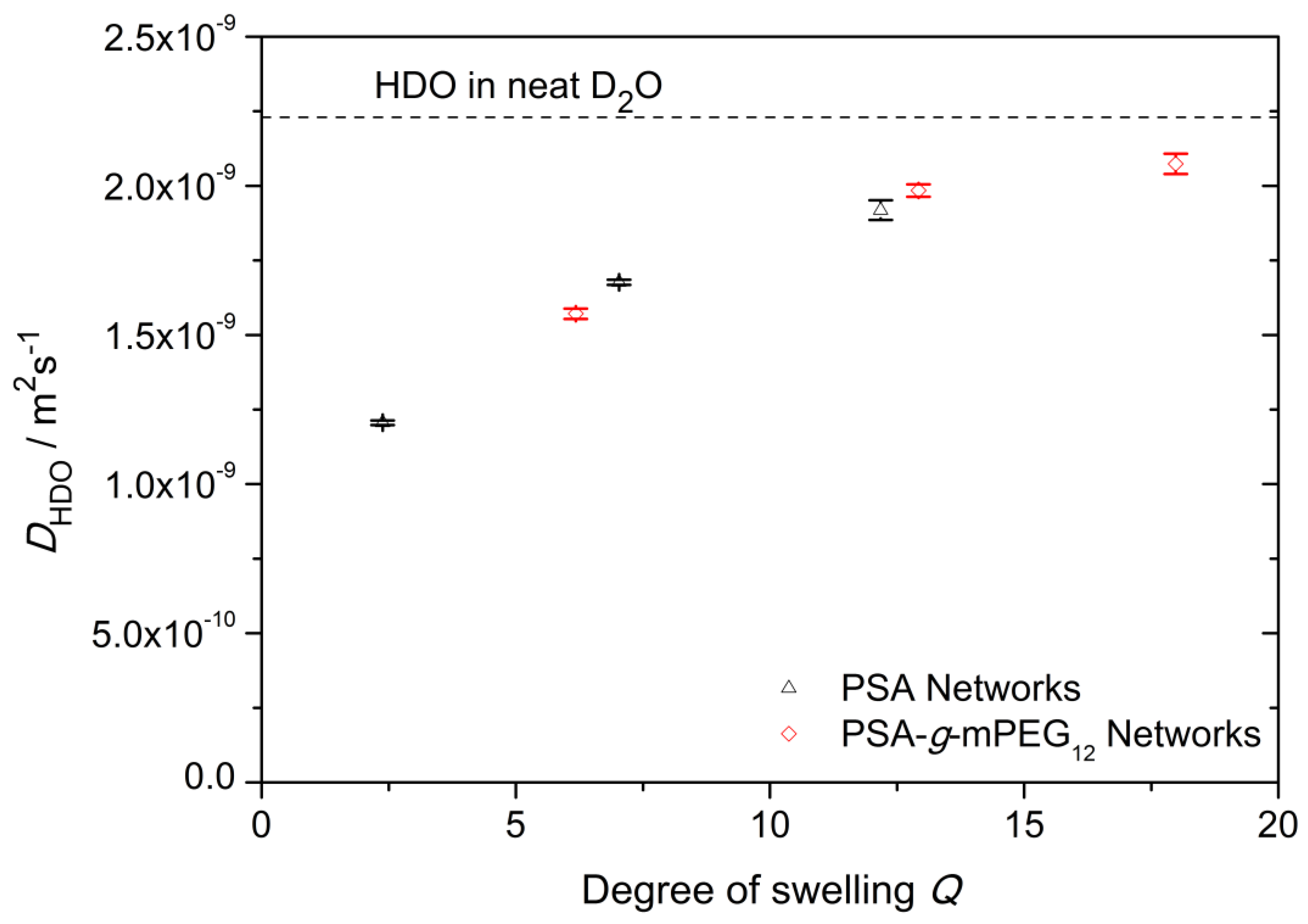
2.5. 1H Pulsed Field Gradient (PFG) NMR Spectroscopy
3. Conclusions
4. Materials and Methods
4.1. Materials
4.2. Synthesis of Poly(Sorbitol Adipate) (PSA)
4.3. Synthesis of Mono- and Bifunctional PEG
4.4. Synthesis of PSA-g-mPEG12
4.5. Network Syntheses of PSA or PSA-g-mPEG12 with Suc-PEGn-Suc
4.6. Nuclear Magnetic Resonance (NMR) Spectroscopy
4.6.1. Solution NMR Spectroscopy
4.6.2. Solid State NMR Spectroscopy
13C Magic Angle Spinning (MAS) NMR Spectroscopy
1H Double Quantum (DQ) NMR Spectroscopy
1H Pulsed Field Gradient (PFG) NMR Spectroscopy
4.7. Gel Permeation Chromatography (GPC)
4.8. Differential Scanning Calorimetry (DSC)
4.9. Swelling Studies
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PSA | Poly(sorbitol adipate) |
| DVA | Divinyl adipate |
| PEG | Poly(ethylene glycol) |
| mPEG12 | PEG monomethyl ether with molar mass 550 g·mol−1 |
| mPEG12-Suc | mPEG12-Succinyl |
| PSA-g-mPEG12 | Poly(sorbitol adipate)-g-α-methoxy,ω-succinyl poly(ethylene glycol)12 |
| Suc-PEGn-Suc | Succinyl-PEGn-Succinyl |
| NMR | Nuclear magnetic resonance spectroscopy |
| GPC | Gel permeation chromatography |
| DSC | Differential scanning calorimetry |
| 13C MAS NMR | 13C magic-angle spinning NMR spectroscopy |
| CP | 13C cross-polarization (CP) MAS NMR |
| SP | 13C single-pulse (SP) MAS NMR |
| 1H DQ NMR | 1H double-quantum (DQ) NMR spectroscopy |
| 1H PFG NMR | 1H pulsed field gradient (PFG) NMR spectroscopy |
References
- Xiang, H.P.; Qian, H.J.; Lu, Z.Y.; Rong, M.Z.; Zhang, M.Q. Crack healing and reclaiming of vulcanized rubber by triggering the rearrangement of inherent sulfur crosslinked networks. Green Chem. 2015, 17, 4315–4325. [Google Scholar] [CrossRef]
- Xu, C.; Cui, R.; Fu, L.; Lin, B. Recyclable and heat-healable epoxidized natural rubber/bentonite composites. Compos. Sci. Technol. 2018, 167, 421–430. [Google Scholar] [CrossRef]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Buwalda, S.J.; Boere, K.W.M.; Dijkstra, P.J.; Feijen, J.; Vermonden, T.; Hennink, W.E. Hydrogels in a historical perspective: From simple networks to smart materials. J. Control. Release 2014, 190, 254–273. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, J.; Johnson, J.A. Polymer Networks: From Plastics and Gels to Porous Frameworks. Angew. Chem.-Int. Ed. 2020, 59, 5022–5049. [Google Scholar] [CrossRef]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 223–236. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009, 103, 655–663. [Google Scholar] [CrossRef] [Green Version]
- Beria, L.; Gevrek, T.N.; Erdog, A.; Sanyal, R.; Pasini, D.; Sanyal, A. “Clickable” hydrogels for all: Facile fabrication and functionalization. Biomater. Sci. 2014, 2, 67–75. [Google Scholar] [CrossRef]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Holback, H.; Yeo, Y.; Park, K. Hydrogel swelling behavior and its biomedical applications. In Biomedical Hydrogels: Biochemistry, Manufacture and Medical Applications; Elsevier: Amsterdam, The Netherlands, 2011; pp. 3–24. ISBN 9781845695903. [Google Scholar]
- Munim, S.A.; Raza, Z.A. Poly(lactic acid) based hydrogels: Formation, characteristics and biomedical applications. J. Porous Mater. 2019, 26, 881–901. [Google Scholar] [CrossRef]
- Vert, M. Aliphatic polyesters: Great degradable polymers that cannot do everything. Biomacromolecules 2005, 6, 538–546. [Google Scholar] [CrossRef]
- Seyednejad, H.; Ghassemi, A.H.; Van Nostrum, C.F.; Vermonden, T.; Hennink, W.E. Functional aliphatic polyesters for biomedical and pharmaceutical applications. J. Control. Release 2011, 152, 168–176. [Google Scholar] [CrossRef]
- Bilal, M.H.; Hussain, H.; Prehm, M.; Baumeister, U.; Meister, A.; Hause, G.; Busse, K.; Mäder, K.; Kressler, J. Synthesis of poly(glycerol adipate)-g-oleate and its ternary phase diagram with glycerol monooleate and water. Eur. Polym. J. 2017, 91, 162–175. [Google Scholar] [CrossRef]
- Albertsson, A.C.; Varma, I.K. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules 2003, 4, 1466–1486. [Google Scholar] [CrossRef]
- Jiang, Y.; Loos, K. Enzymatic synthesis of biobased polyesters and polyamides. Polymers. 2016, 8, 243. [Google Scholar] [CrossRef] [Green Version]
- Naolou, T.; Busse, K.; Kressler, J. Synthesis of well-defined graft copolymers by combination of enzymatic polycondensation and “Click” chemistry. Biomacromolecules 2010, 11, 3660–3667. [Google Scholar] [CrossRef]
- Métrai, G.; Wentland, J.; Thomann, Y.; Tiller, J.C. Biodegradable poly(ester hydrazide)s via enzymatic polymerization. Macromol. Rapid Commun. 2005, 26, 1330–1335. [Google Scholar] [CrossRef]
- Gross, R.A.; Ganesh, M.; Lu, W. Enzyme-catalysis breathes new life into polyester condensation polymerizations. Trends Biotechnol. 2010, 28, 435–443. [Google Scholar] [CrossRef]
- Naolou, T. Green Route to Prepare Renewable Polyesters from Monomers: Enzymatic Polymerization. In Introduction to Renewable Biomaterials: First Principles and Concepts; Wiley: Hoboken, NJ, USA, 2017; pp. 219–237. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Kimura, S. Enzymatic polymerization. Chem. Rev. 2001, 101, 3793–3818. [Google Scholar] [CrossRef]
- Douka, A.; Vouyiouka, S.; Papaspyridi, L.M.; Papaspyrides, C.D. A review on enzymatic polymerization to produce polycondensation polymers: The case of aliphatic polyesters, polyamides and polyesteramides. Prog. Polym. Sci. 2018, 79, 1–25. [Google Scholar] [CrossRef]
- Kobayashi, S. Enzymatic ring-opening polymerization and polycondensation for the green synthesis of polyesters. Polym. Adv. Technol. 2015, 26, 677–686. [Google Scholar] [CrossRef]
- Kobayashi, S. Lipase-catalyzed polyester synthesis—A green polymer chemistry. Proc. Jpn. Acad. Ser. B 2010, 86, 338–365. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Makino, A. Enzymatic polymer synthesis: An opportunity for green polymer chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef]
- Taresco, V.; Suksiriworapong, J.; Creasey, R.; Burley, J.C.; Mantovani, G.; Alexander, C.; Treacher, K.; Booth, J.; Garnett, M.C. Properties of acyl modified poly(glycerol-adipate) comb-like polymers and their self-assembly into nanoparticles. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 3267–3278. [Google Scholar] [CrossRef] [Green Version]
- Orafai, H.; Kallinteri, P.; Garnett, M.; Huggins, S.; Hutcheon, G.; Pourcain, C. Novel poly(glycerol-adipate) polymers used for nanoparticle making: A study of surface free energy. Iran. J. Pharm. Res. 2008, 7, 11–19. [Google Scholar] [CrossRef]
- Wersig, T.; Krombholz, R.; Janich, C.; Meister, A.; Kressler, J.; Mäder, K. Indomethacin functionalised poly(glycerol adipate) nanospheres as promising candidates for modified drug release. Eur. J. Pharm. Sci. 2018, 123, 350–361. [Google Scholar] [CrossRef]
- Weiss, V.M.; Naolou, T.; Hause, G.; Kuntsche, J.; Kressler, J.; Mäder, K. Poly(glycerol adipate)-fatty acid esters as versatile nanocarriers: From nanocubes over ellipsoids to nanospheres. J. Control. Release 2012, 158, 156–164. [Google Scholar] [CrossRef]
- Naolou, T.; Meister, A.; Schöps, R.; Pietzsch, M.; Kressler, J. Synthesis and characterization of graft copolymers able to form polymersomes and worm-like aggregates. Soft Matter 2013, 9, 10364–10372. [Google Scholar] [CrossRef]
- Gaskell, E.E.; Hobbs, G.; Rostron, C.; Hutcheon, G.A. Encapsulation and release of α-chymotrypsin from poly(glycerol adipate-co-ω-pentadecalactone) microparticles. J. Microencapsul. 2008, 25, 187–195. [Google Scholar] [CrossRef]
- Abo-zeid, Y.; Mantovani, G.; Irving, W.L.; Garnett, M.C. Synthesis of nucleoside-boronic esters hydrophobic pro-drugs: A possible route to improve hydrophilic nucleoside drug loading into polymer nanoparticles. J. Drug Deliv. Sci. Technol. 2018, 46, 354–364. [Google Scholar] [CrossRef]
- Suksiriworapong, J.; Taresco, V.; Ivanov, D.P.; Styliari, I.D.; Sakchaisri, K.; Junyaprasert, V.B.; Garnett, M.C. Synthesis and properties of a biodegradable polymer-drug conjugate: Methotrexate-poly(glycerol adipate). Colloids Surfaces B Biointerfaces 2018, 167, 115–125. [Google Scholar] [CrossRef]
- Hu, J.; Gao, W.; Kulshrestha, A.; Gross, R.A. “Sweet polyesters”: Lipase-catalyzed condensation—Polymerizations of alditols. Macromolecules 2006, 39, 6789–6792. [Google Scholar] [CrossRef]
- Uyama, H.; Kobayashi, S. Enzymatic synthesis of polyesters via polycondensation. In Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2006; Volume 194, pp. 133–158. [Google Scholar]
- Nitta, S.; Iwamoto, H. Lipase-catalyzed synthesis of epigallocatechin gallate-based polymer for long-term release of epigallocatechin gallate with antioxidant property. J. Appl. Polym. Sci. 2019, 136, 47693. [Google Scholar] [CrossRef]
- Adharis, A.; Loos, K. Synthesis of glycomonomers via biocatalytic methods. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 627, pp. 215–247. ISBN 9780128170953. [Google Scholar]
- Uyama, H.; Wada, S.; Fukui, T.; Kobayashi, S. Lipase-catalyzed synthesis of polyesters from anhydride derivatives involving dehydration. Biochem. Eng. J. 2003, 16, 145–152. [Google Scholar] [CrossRef]
- Bilal, M.H.; Prehm, M.; Njau, A.E.; Samiullah, M.H.; Meister, A.; Kressler, J. Enzymatic synthesis and characterization of hydrophilic sugar based polyesters and their modification with stearic acid. Polymers 2016, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Uyama, H.; Inada, K.; Kobayashi, S. Regioselective polymerization of divinyl sebacate and triols using lipase catalyst. Macromol. Rapid Commun. 1999, 20, 171–174. [Google Scholar] [CrossRef]
- D’souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, B. Theory of cold crystallization of high polymers. J. Chem. Phys. 1958, 29, 1395–1404. [Google Scholar] [CrossRef]
- Wang, J.; Andriamitantsoa, R.S.; Atinafu, D.G.; Gao, H.; Dong, W.; Wang, G. A one-step in-situ assembly strategy to construct PEG@MOG-100-Fe shape-stabilized composite phase change material with enhanced storage capacity for thermal energy storage. Chem. Phys. Lett. 2018, 695, 99–106. [Google Scholar] [CrossRef]
- Pielichowski, K.; Flejtuch, K. Differential scanning calorimetry studies on poly(ethylene glycol) with different molecular weights for thermal energy storage materials. Polym. Adv. Technol. 2002, 13, 690–696. [Google Scholar] [CrossRef]
- Kaşgöz, H.; Aydin, I.; Kaşgöz, A. The effect of PEG(400)DA crosslinking agent on swelling behaviour of acrylamide-maleic acid hydrogels. Polym. Bull. 2005, 54, 387–397. [Google Scholar] [CrossRef]
- Lange, F.; Schwenke, K.; Kurakazu, M.; Akagi, Y.; Chung, U.I.; Lang, M.; Sommer, J.U.; Sakai, T.; Saalwächter, K. Connectivity and structural defects in model hydrogels: A combined proton NMR and Monte Carlo simulation study. Macromolecules 2011, 44, 9666–9674. [Google Scholar] [CrossRef]
- Baum, J.; Pines, A. NMR Studies of Clustering in Solids. J. Am. Chem. Soc. 1986, 108, 7447–7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saalwächter, K. Proton multiple-quantum NMR for the study of chain dynamics and structural constraints in polymeric soft materials. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 1–35. [Google Scholar] [CrossRef]
- Golitsyn, Y.; Pulst, M.; Samiullah, M.H.; Busse, K.; Kressler, J.; Reichert, D. Crystallization in PEG networks: The importance of network topology and chain tilt in crystals. Polymer 2019, 165, 72–82. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin diffusion measurements: Spin echoes in the presence of a time-dependent field gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Matsukawa, S.; Ando, I. Study of self-diffusion of molecules in polymer gel by pulsed-gradient spin-echo 1H NMR. Macromolecules 1996, 29, 7136–7140. [Google Scholar] [CrossRef]
- Bilal, M.H.; Alaneed, R.; Steiner, J.; Mäder, K.; Pietzsch, M.; Kressler, J. Multiple grafting to enzymatically synthesized polyesters. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2019; Volume 627, pp. 57–97. ISBN 9780128170953. [Google Scholar]
- Lu, C.; Zhong, W. Synthesis of Propargyl-Terminated Heterobifunctional Poly(ethylene glycol). Polymers 2010, 2, 407–417. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymers and Networks | Mn (g·mol−1) | Ð | Tg (°C) | Tm (°C) | ΔHm (J·g−1) |
|---|---|---|---|---|---|
| PSA | 11,000 1 | 1.8 3 | −1 | --- | --- |
| mPEG12-Suc | 650 4 | --- | --- | 12 | 45 |
| PSA-g-mPEG12 | 22,000 2 | 1.5 3 | −34 | --- | --- |
| Suc-PEG9-Suc | 600 4 | --- | −45 | --- | --- |
| PSA cross-linked with Suc-PEG9-Suc | --- | --- | −11 | --- | --- |
| PSA-g-mPEG12 cross-linked with Suc-PEG9-Suc | --- | --- | −29 | --- | --- |
| Suc-PEG23-Suc | 1200 4 | --- | --- | 31 | 86 |
| PSA cross-linked with Suc-PEG23-Suc | --- | --- | --- | 20 | 23 |
| PSA-g-mPEG12 cross-linked with Suc-PEG23-Suc | --- | --- | --- | 22 | 40 |
| Suc-PEG45-Suc | 2200 4 | --- | --- | 51 | 123 |
| PSA cross-linked with Suc-PEG45-Suc | --- | --- | --- | 42 | 32 |
| PSA-g-mPEG12 cross-linked with Suc-PEG45-Suc | --- | --- | --- | 41 | 34 |
| Polymer Networks | Crosslinking Precursors | f1 (%) | T2* (ms) | β | Dres/2π (Hz) | σ | f2 (%) | T2* (ms) | β | Dres /2π (Hz) | σ |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PSA Networks | Suc-PEG9-Suc | 37.0 | 5.2 | 2.0 | 462.5 | 0.46 | 63.0 | 7.8 | 1.5 | 131.3 | 0.49 |
| Suc-PEG23-Suc | 31.7 | 10.3 | 1.9 | 217.1 | 0.61 | 68.3 | 26.1 | 1.5 | 34.4 | 0.59 | |
| Suc-PEG45-Suc | 21.8 | 11.3 | 2.0 | 220.3 | 0.57 | 78.2 | 45.7 | 1.2 | 15.9 | 0.85 | |
| PSA-g- mPEG12 Networks | Suc-PEG9-Suc | 33.5 | 8.3 | 1.5 | 230.1 | 0.85 | 66.5 | 15.0 | 1.2 | 47.0 | 0.69 |
| Suc-PEG23-Suc | 18.3 | 11.8 | 1.9 | 200.8 | 0.60 | 81.7 | 40.7 | 1.2 | 19.0 | 0.77 | |
| Suc-PEG45-Suc | 19.9 | 12.2 | 1.9 | 159.1 | 0.65 | 80.1 | 60.0 | 1.2 | 11.8 | 0.80 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashid, H.; Golitsyn, Y.; Bilal, M.H.; Mäder, K.; Reichert, D.; Kressler, J. Polymer Networks Synthesized from Poly(Sorbitol Adipate) and Functionalized Poly(Ethylene Glycol). Gels 2021, 7, 22. https://0-doi-org.brum.beds.ac.uk/10.3390/gels7010022
Rashid H, Golitsyn Y, Bilal MH, Mäder K, Reichert D, Kressler J. Polymer Networks Synthesized from Poly(Sorbitol Adipate) and Functionalized Poly(Ethylene Glycol). Gels. 2021; 7(1):22. https://0-doi-org.brum.beds.ac.uk/10.3390/gels7010022
Chicago/Turabian StyleRashid, Haroon, Yury Golitsyn, Muhammad Humayun Bilal, Karsten Mäder, Detlef Reichert, and Jörg Kressler. 2021. "Polymer Networks Synthesized from Poly(Sorbitol Adipate) and Functionalized Poly(Ethylene Glycol)" Gels 7, no. 1: 22. https://0-doi-org.brum.beds.ac.uk/10.3390/gels7010022