Human Brain Shows Recurrent Non-Canonical MicroRNA Editing Events Enriched for Seed Sequence with Possible Functional Consequence


Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Samples
3.2. RNA Isolation, Library Preparation and Sequencing
3.3. Data Analysis
3.4. Target Prediction and Functional Enrichment Analysis
3.5. Sequence Context for the Edited miRNAs
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Li, J.B.; Levanon, E.Y.; Yoon, J.K.; Aach, J.; Xie, B.; Leproust, E.; Zhang, K.; Gao, Y.; Church, G.M. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science 2009, 324, 1210–1213. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.J.; Platas, A.A.; Hawley, D.K. Transcriptional fidelity and proofreading by RNA polymerase II. Cell 1998, 93, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Zenkin, N.; Yuzenkova, Y.; Severinov, K. Transcript-assisted transcriptional proofreading. Science 2006, 313, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Paul, M.S.; Bass, B.L. Inosine exists in mRNA at tissue-specific levels and is most abundant in brain mRNA. EMBO J. 1998, 17, 1120–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, T.; Park, C.K.; Leung, A.K.; Gao, Y.; Hyde, T.M.; Kleinman, J.E.; Rajpurohit, A.; Tao, R.; Shin, J.H.; Weinberger, D.R. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Behm, M.; Ohman, M. RNA Editing: A Contributor to Neuronal Dynamics in the Mammalian Brain. Trends Genet. 2016, 32, 165–175. [Google Scholar] [CrossRef]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [Green Version]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [Green Version]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, C.R.; Li, J.B. Rewriting the transcriptome: Adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, V.; Davidson, N.O. C-to-U RNA editing: Mechanisms leading to genetic diversity. J. Biol. Chem. 2003, 278, 1395–1398. [Google Scholar] [CrossRef] [Green Version]
- Keegan, L.P.; Gallo, A.; O’Connell, M.A. The many roles of an RNA editor. Nat. Rev. Genet. 2001, 2, 869–878. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Torres, A.G.; Pineyro, D.; Filonava, L.; Stracker, T.H.; Batlle, E.; de Pouplana, L.R. A-to-I editing on tRNAs: Biochemical, biological and evolutionary implications. FEBS Lett. 2014, 588, 4279–4286. [Google Scholar] [CrossRef]
- Luciano, D.J.; Mirsky, H.; Vendetti, N.J.; Maas, S. RNA editing of a miRNA precursor. RNA 2004, 10, 1174–1177. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; Nishikura, K. Extensive adenosine-to-inosine editing detected in Alu repeats of antisense RNAs reveals scarcity of sense-antisense duplex formation. FEBS Lett. 2006, 580, 2301–2305. [Google Scholar] [CrossRef] [Green Version]
- Carmi, S.; Borukhov, I.; Levanon, E.Y. Identification of widespread ultra-edited human RNAs. PLoS Genet. 2011, 7, e1002317. [Google Scholar] [CrossRef]
- Paz-Yaacov, N.; Bazak, L.; Buchumenski, I.; Porath, H.T.; Danan-Gotthold, M.; Knisbacher, B.A.; Eisenberg, E.; Levanon, E.Y. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell Rep. 2015, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, I.X.; Li, Y.; Bruzel, A.; Richards, A.L.; Toung, J.M.; Cheung, V.G. Widespread RNA and DNA sequence differences in the human transcriptome. Science 2011, 333, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, C.L.; Majewski, J. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302, author reply 1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, J.K.; Gilad, Y.; Pritchard, J.K. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302, author reply 1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.; Piskol, R.; Tan, M.H.; Li, J.B. Comment on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302, author reply 1302. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, I.X.; Cheung, V.G. Response to comments on “Widespread RNA and DNA sequence differences in the human transcriptome”. Science 2012, 335, 1302. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.J.; Aggarwal, P.; Miller, H.E.; Waukau, J.; Routes, J.M.; Broeckel, U.; Robinson, R.T. The introduction of RNA-DNA differences underlies interindividual variation in the human IL12RB1 mRNA repertoire. Proc. Natl. Acad. Sci. USA 2015, 112, 15414–15419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustamante, J.; Boisson-Dupuis, S.; Abel, L.; Casanova, J.L. Mendelian susceptibility to mycobacterial disease: Genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin. Immunol. 2014, 26, 454–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, R.T. IL12Rbeta1: The cytokine receptor that we used to know. Cytokine 2015, 71, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.; Lin, Y.; Zhang, R.R. RNA-DNA differences are rarer in proto-oncogenes than in tumor suppressor genes. Sci. Rep. 2012, 2, 245. [Google Scholar] [CrossRef]
- Ju, Y.S.; Kim, J.I.; Kim, S.; Hong, D.; Park, H.; Shin, J.Y.; Lee, S.; Lee, W.C.; Yu, S.B.; Park, S.S.; et al. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat. Genet. 2011, 43, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Lee, J.H.; Li, G.; Greer, C.; Peng, G.; Xiao, X. Accurate identification of A-to-I RNA editing in human by transcriptome sequencing. Genome Res. 2012, 22, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silberberg, G.; Lundin, D.; Navon, R.; Ohman, M. Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum. Mol. Genet. 2012, 21, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Zamore, P.D. A microRNA in a multiple-turnover RNAi enzyme complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawahara, Y.; Zinshteyn, B.; Chendrimada, T.P.; Shiekhattar, R.; Nishikura, K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007, 8, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, Y.; Tay, F.C.; Lam, D.H.; Sandanaraj, E.; Tang, C.; Ang, B.T.; Wang, S. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Investig. 2012, 122, 4059–4076. [Google Scholar] [CrossRef] [Green Version]
- Negi, V.; Paul, D.; Das, S.; Bajpai, P.; Singh, S.; Mukhopadhyay, A.; Agrawal, A.; Ghosh, B. Altered expression and editing of miRNA-100 regulates iTreg differentiation. Nucleic Acids Res. 2015, 43, 8057–8065. [Google Scholar] [CrossRef]
- Tomaselli, S.; Galeano, F.; Alon, S.; Raho, S.; Galardi, S.; Polito, V.A.; Presutti, C.; Vincenti, S.; Eisenberg, E.; Locatelli, F.; et al. Modulation of microRNA editing, expression and processing by ADAR2 deaminase in glioblastoma. Genome Biol. 2015, 16, 5. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Sinha, A.N.; Ray, A.; Lal, M.; Nayak, S.; Sharma, A.; Mehani, B.; Mukherjee, D.; Laddha, S.V.; Suri, A.; et al. A-to-I editing in human miRNAs is enriched in seed sequence, influenced by sequence contexts and significantly hypoedited in glioblastoma multiforme. Sci. Rep. 2017, 7, 2466. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, X.; Yu, S.; Jeong, K.J.; Zhou, Z.; Han, L.; Tsang, Y.H.; Li, J.; Chen, H.; Mangala, L.S.; et al. Systematic characterization of A-to-I RNA editing hotspots in microRNAs across human cancers. Genome Res. 2017, 27, 1112–1125. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Song, Y.; Shi, X.; Liu, J.; Xiong, S.; Chen, W.; Fu, Q.; Huang, Z.; Gu, N.; Zhang, R. The landscape of miRNA editing in animals and its impact on miRNA biogenesis and targeting. Genome Res. 2018, 28, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarini, V.; Silvestris, D.A.; Tassinari, V.; Tomaselli, S.; Alon, S.; Eisenberg, E.; Locatelli, F.; Gallo, A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids Res. 2018, 46, 2045–2059. [Google Scholar] [CrossRef]
- Joyce, C.E.; Zhou, X.; Xia, J.; Ryan, C.; Thrash, B.; Menter, A.; Zhang, W.; Bowcock, A.M. Deep sequencing of small RNAs from human skin reveals major alterations in the psoriasis miRNAome. Hum. Mol. Genet. 2011, 20, 4025–4040. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wu, Y.; Zhang, X.; Liao, Y.; Sibanda, V.L.; Liu, W.; Guo, A.Y. Comprehensive analysis of human small RNA sequencing data provides insights into expression profiles and miRNA editing. RNA Biol. 2014, 11, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ji, B.; Song, R.; Wang, S.; Li, T.; Zhang, X.; Chen, K.; Li, J. Accurate detection for a wide range of mutation and editing sites of microRNAs from small RNA high-throughput sequencing profiles. Nucleic Acids Res. 2016, 44, e123. [Google Scholar] [CrossRef] [PubMed]
- Oak, N.; Ghosh, R.; Huang, K.L.; Wheeler, D.A.; Ding, L.; Plon, S.E. Framework for microRNA variant annotation and prioritization using human population and disease datasets. Hum. Mutat. 2019, 40, 73–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Carvalho, J.B.; de Morais, G.L.; Vieira, T.; Rabelo, N.C.; Llerena, J.C., Jr.; Gonzalez, S.M.C.; de Vasconcelos, A.T.R. miRNA Genetic Variants Alter Their Secondary Structure and Expression in Patients With RASopathies Syndromes. Front. Genet. 2019, 10, 1144. [Google Scholar] [CrossRef] [PubMed]
- Dohm, J.C.; Lottaz, C.; Borodina, T.; Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008, 36, e105. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011, 39, e90. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; O’Connor, M.D.; Griffith, M.; Kuchenbauer, F.; Delaney, A.; Prabhu, A.L.; Zhao, Y.; McDonald, H.; Zeng, T.; Hirst, M.; et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008, 18, 610–621. [Google Scholar] [CrossRef] [Green Version]
- Marti, E.; Pantano, L.; Banez-Coronel, M.; Llorens, F.; Minones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef]
- Alon, S.; Mor, E.; Vigneault, F.; Church, G.M.; Locatelli, F.; Galeano, F.; Gallo, A.; Shomron, N.; Eisenberg, E. Systematic identification of edited microRNAs in the human brain. Genome Res. 2012, 22, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Distefano, R.; Nigita, G.; Veneziano, D.; Romano, G.; Croce, C.M.; Acunzo, M. isoTar: Consensus Target Prediction with Enrichment Analysis for MicroRNAs Harboring Editing Sites and Other Variations. Methods Mol. Biol. 2019, 1970, 211–235. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]




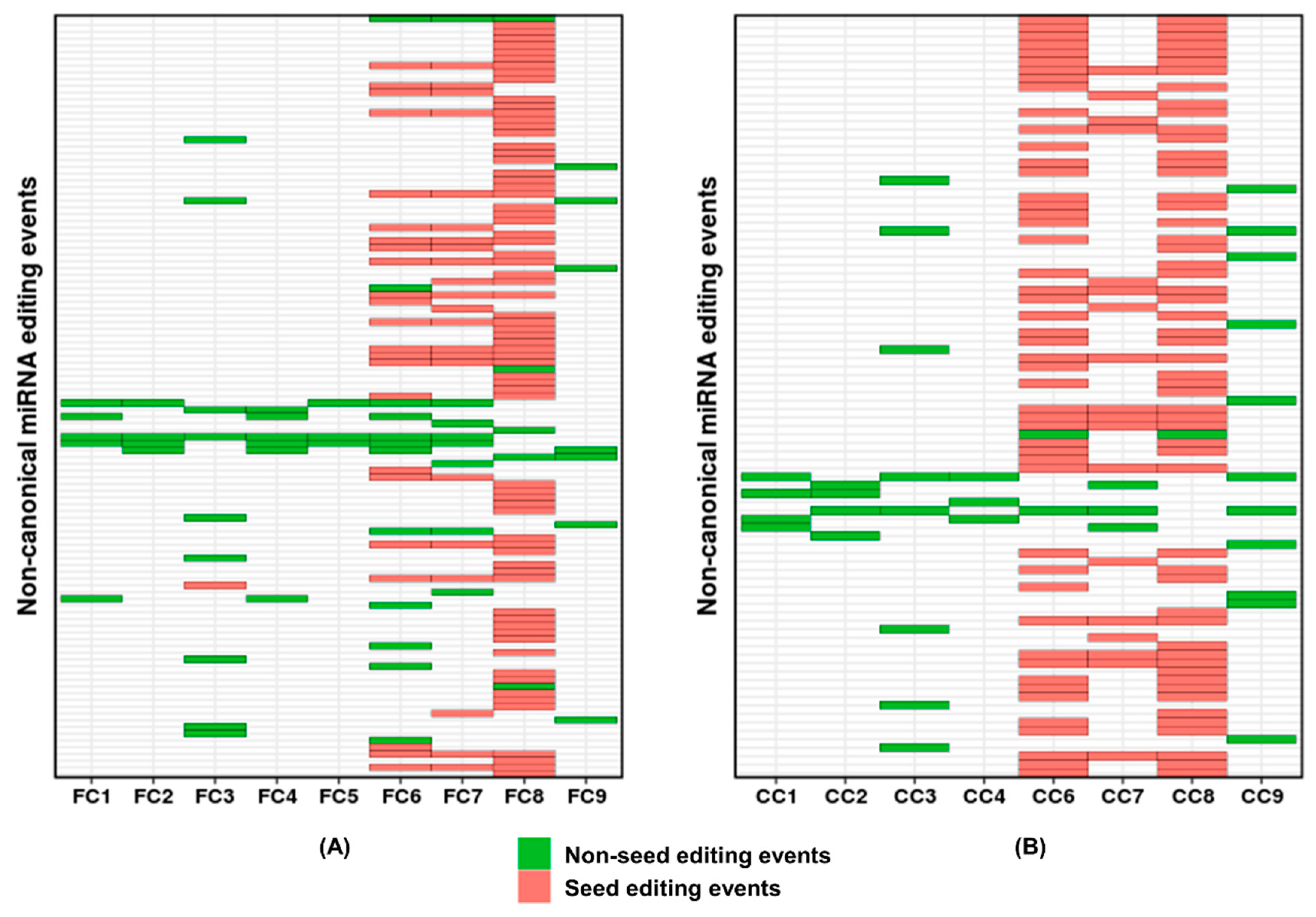{kind=link}
{kind=link}
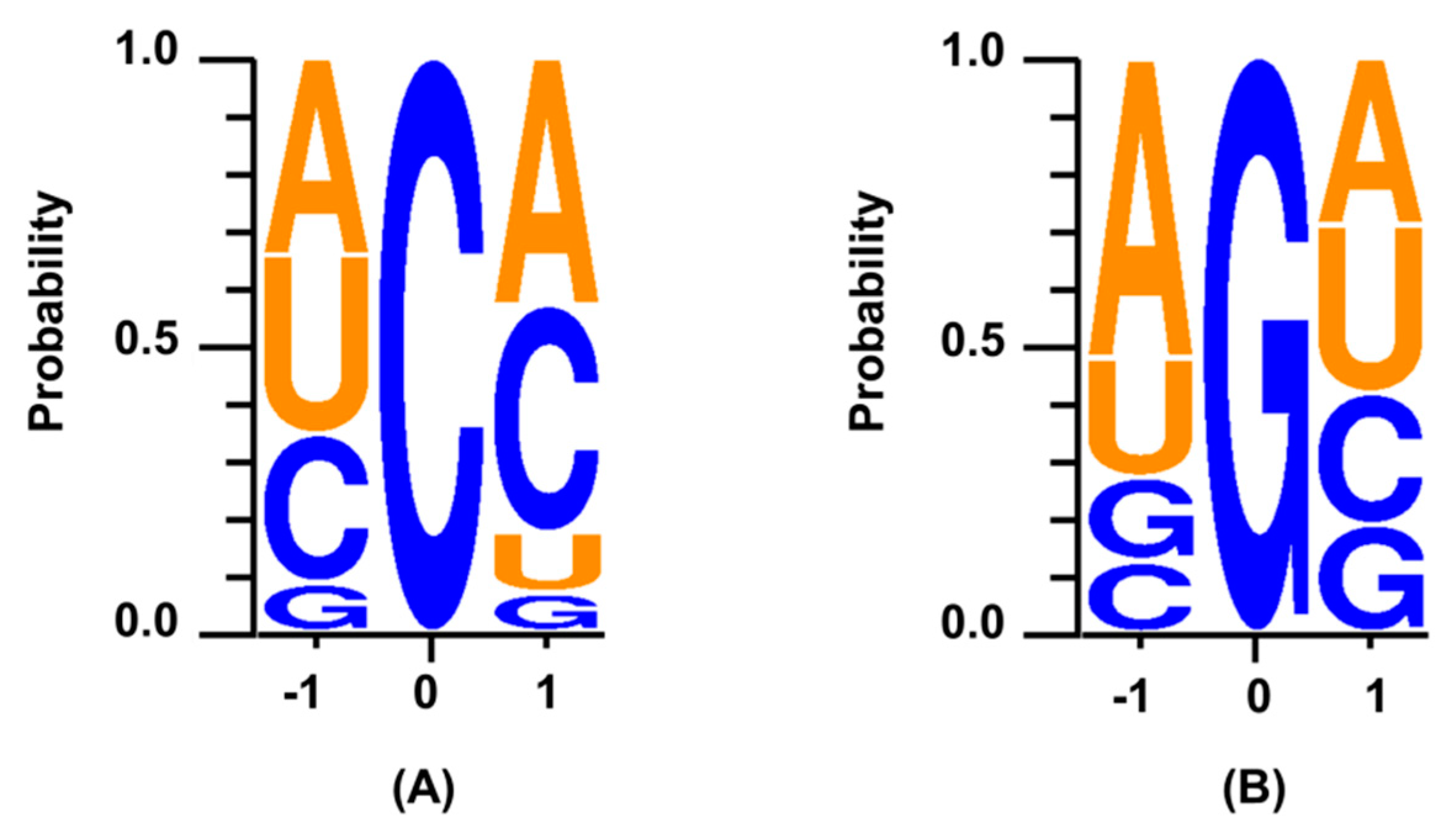{kind=link}
| MiRNA | Strand/Position | Seed | Editing Types | Presence in Samples (FC/CC) | a Target Prediction (Before/After Editing) | Overlap (%) |
|---|---|---|---|---|---|---|
| hsa-miR-100 | 5p/3 | Yes | C-to-A | 3/3 | 30/16 | 0 (0) |
| hsa-miR-127 | 3p/4 | Yes | G-to-U | 3/3 | 128/60 | 0 (0) |
| hsa-miR-146b | 5p/4 | Yes | G-to-U | 3/2 | 502/111 | 9 (1.79) |
| hsa-miR-181c | 5p/3 | Yes | C-to-A | 3/3 | 207/72 | 1 (0.48) |
| hsa-miR-181d | 5p/3 | Yes | C-to-A | 3/2 | 580/269 | 21 (3.62) |
| hsa-miR-204 | 5p/3 | Yes | C-to-A | 3/3 | 498/90 | 6 (1.20) |
| hsa-miR-221 | 3p/4 | Yes | U-to-G | 3/0 | 378/103 | 5 (1.32) |
| hsa-miR-23a | 3p/3 | Yes | C-to-A | 3/3 | 258/73 | 2 (0.78) |
| hsa-miR-23b | 3p/3 | Yes | C-to-A | 3/3 | 372/106 | 5 (1.34) |
| hsa-miR-26b | 5p/3 | Yes | C-to-A | 3/3 | 93/41 | 0 (0) |
| hsa-miR-301a | 3p/4 | Yes | U-to-G | 2/3 | 184/508 | 7 (3.80) |
| hsa-miR-421 | Mature/3 | Yes | C-to-A | 3/3 | 248/122 | 5 (2.02) |
| hsa-miR-433 | 3p/3 | Yes | C-to-A | 3/3 | 403/166 | 10 (2.48) |
| hsa-miR-99a | 5p/3 | Yes | C-to-A | 3/3 | 35/23 | 0 (0) |
| hsa-miR-99b | 5p/3 | Yes | C-to-A | 3/2 | 49/22 | 0 (0) |
| hsa-miR-454 | 3p/4 | Yes | U-to-G | 0/3 | 232/622 | 21 (9.05) |
| hsa-let-7a-1 | 5p/9 | No | U-to-G | 3/0 | NA | NA |
| hsa-miR-30a | 5p/19 | No | G-to-C | 5/2 | NA | NA |
| hsa-miR-30a | 5p/19 | No | G-to-U | 2/4 | NA | NA |
| hsa-miR-30d | 5p/19 | No | G-to-C | 3/2 | NA | NA |
| hsa-miR-30e | 5p/19 | No | G-to-C | 7/5 | NA | NA |
| hsa-miR-30e | 5p/17 | No | U-to-C | 6/2 | NA | NA |
| hsa-miR-30e | 5p/18 | No | G-to-A | 4/1 | NA | NA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, D.; Ansari, A.H.; Lal, M.; Mukhopadhyay, A. Human Brain Shows Recurrent Non-Canonical MicroRNA Editing Events Enriched for Seed Sequence with Possible Functional Consequence. Non-Coding RNA 2020, 6, 21. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020021
Paul D, Ansari AH, Lal M, Mukhopadhyay A. Human Brain Shows Recurrent Non-Canonical MicroRNA Editing Events Enriched for Seed Sequence with Possible Functional Consequence. Non-Coding RNA. 2020; 6(2):21. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020021
Chicago/Turabian StylePaul, Deepanjan, Asgar Hussain Ansari, Megha Lal, and Arijit Mukhopadhyay. 2020. "Human Brain Shows Recurrent Non-Canonical MicroRNA Editing Events Enriched for Seed Sequence with Possible Functional Consequence" Non-Coding RNA 6, no. 2: 21. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020021