LncRNA NEAT1 in Paraspeckles: A Structural Scaffold for Cellular DNA Damage Response Systems?

 , , ,
, , ,
Abstract
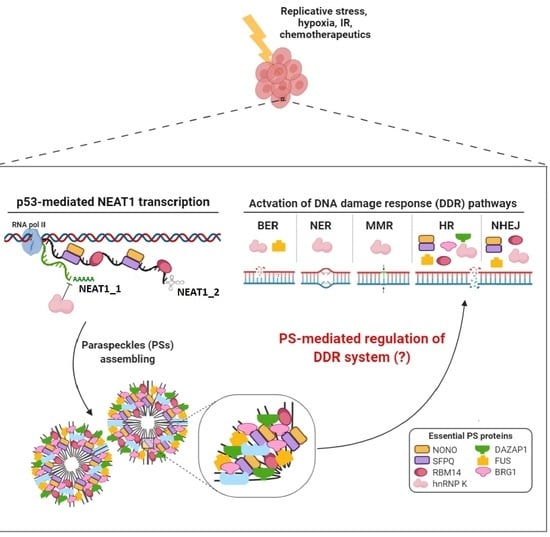
:
1. Introduction
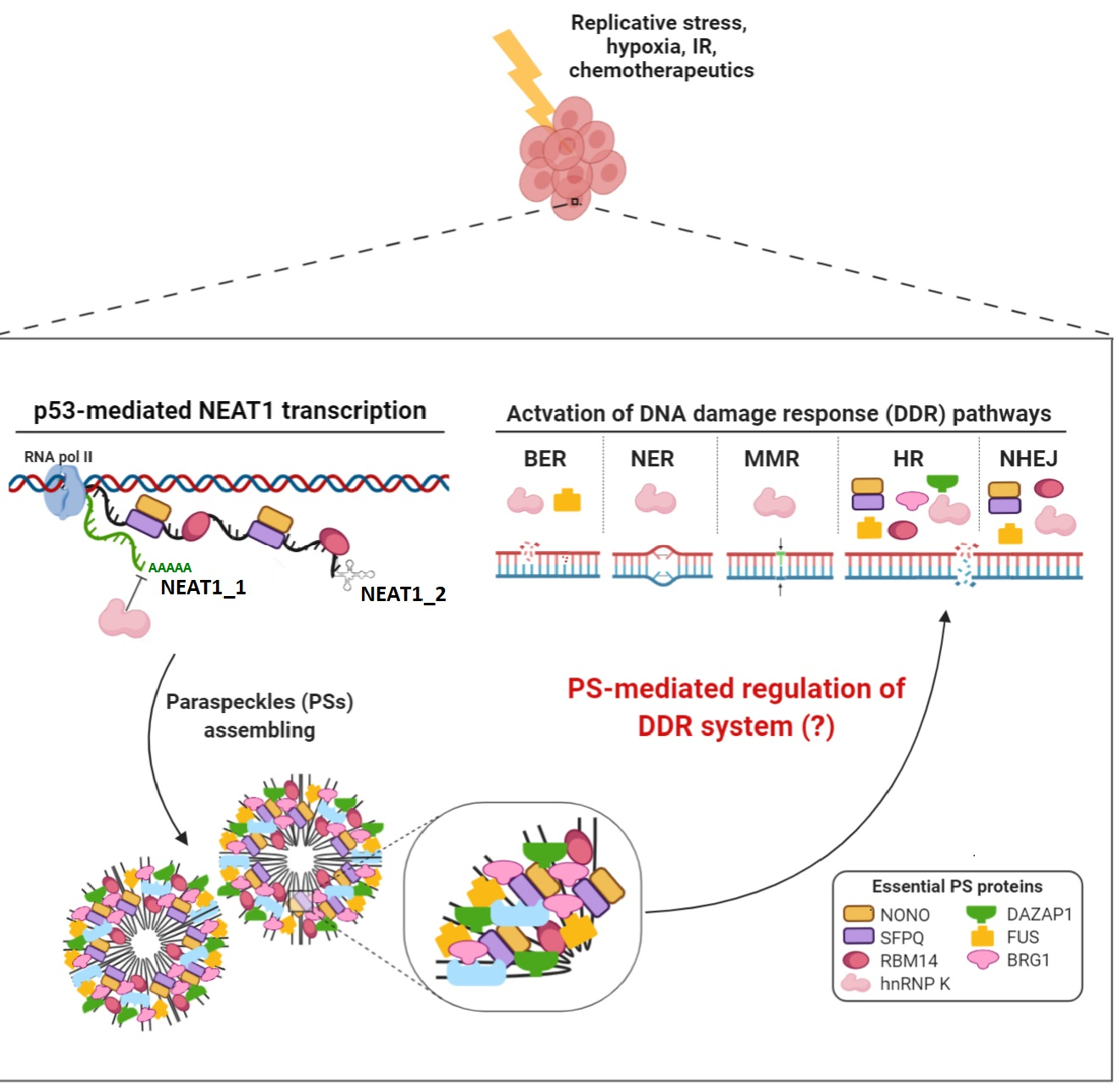
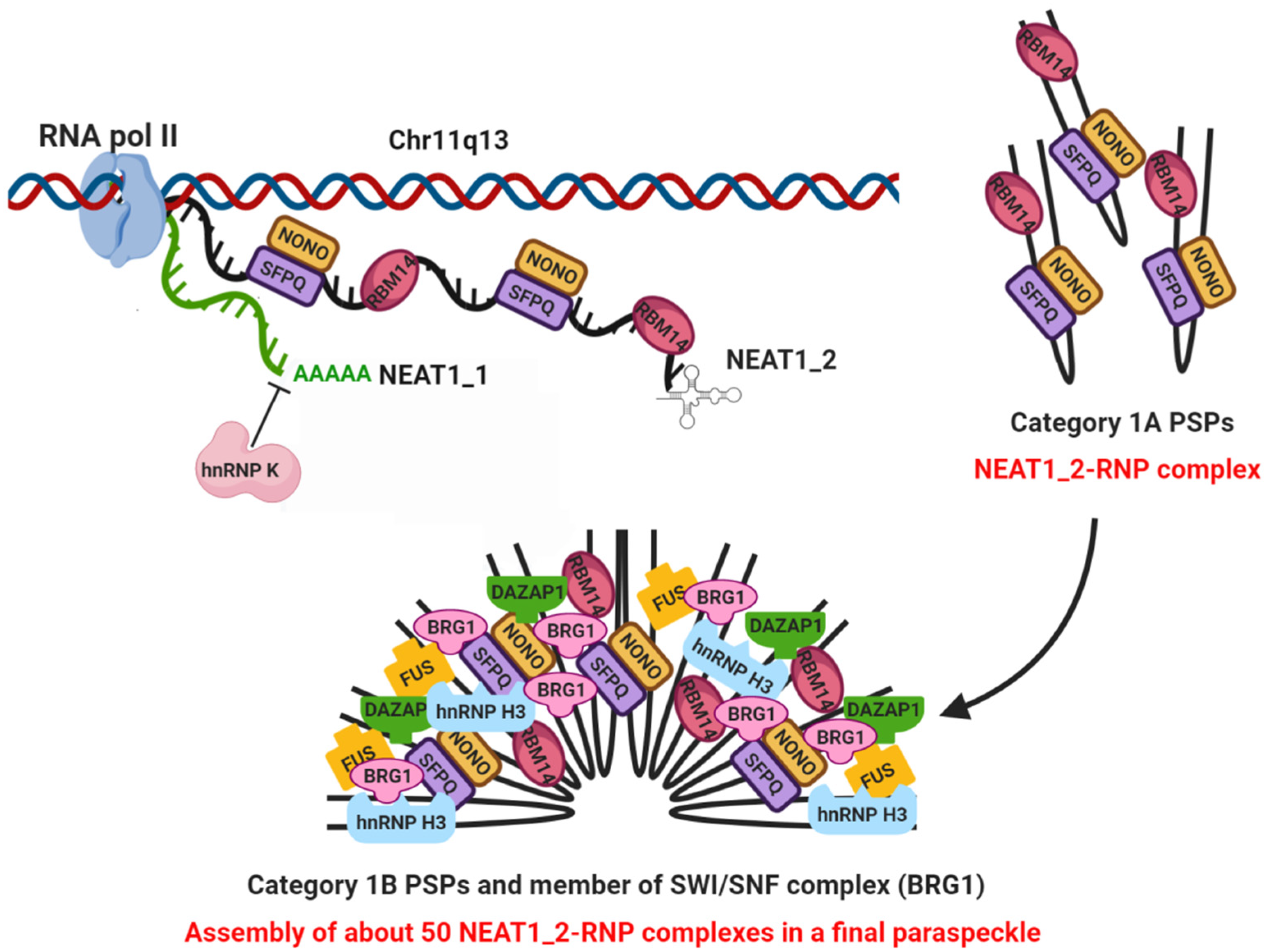
2. NEAT1 as Essential Structural Scaffold for Paraspeckle Assembling
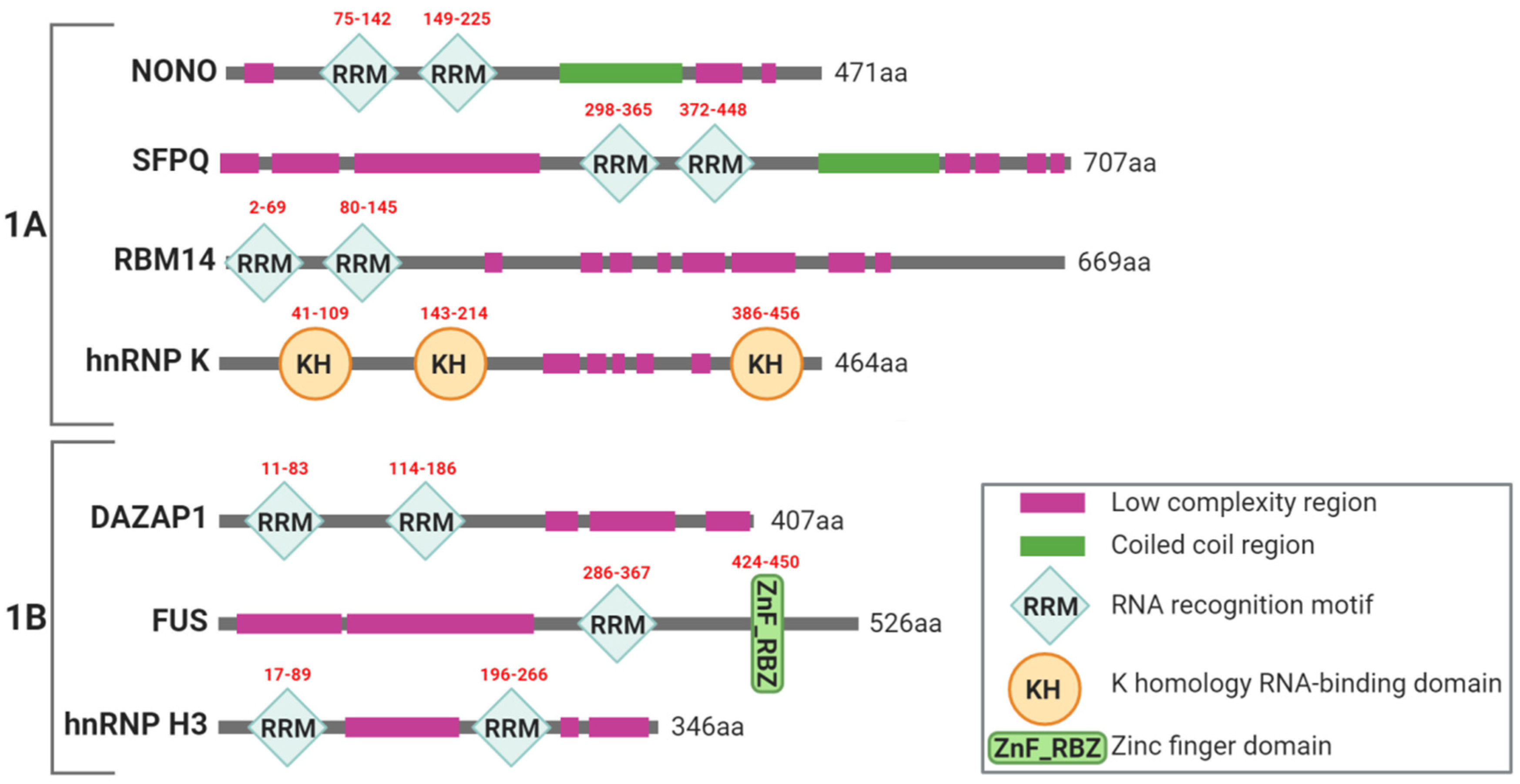
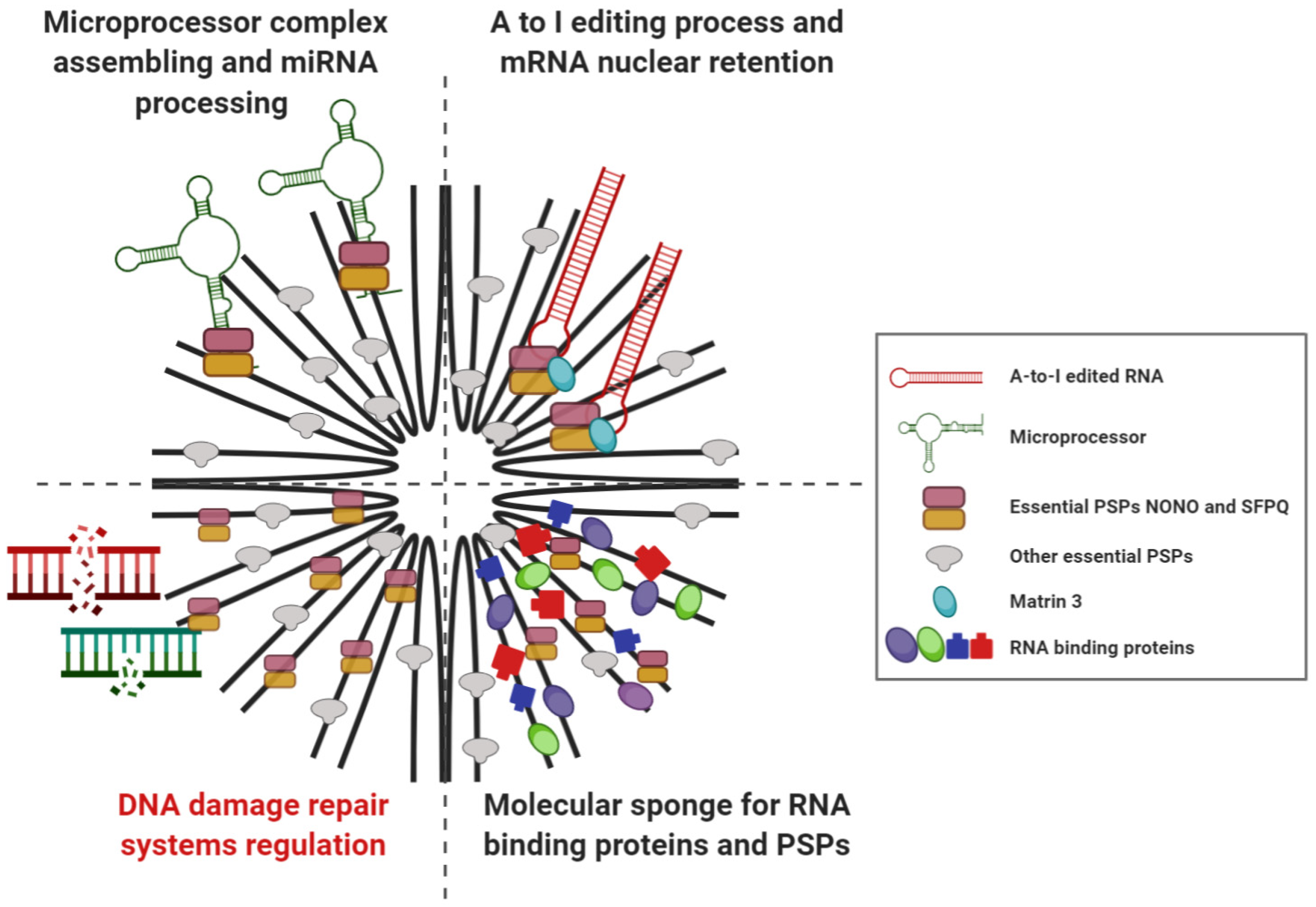
2.1. Paraspeckles Biogenesis and Essential Factors for Their Assembling
2.2. Cellular Functions of Paraspeckle
3. Involvement of NEAT1 in DNA Damage Response
4. Involvement of Paraspeckle Proteins in the DNA Damage Response
4.1. NONO
4.2. SFPQ
4.3. RBM14
4.4. HnRNP K
4.5. FUS/TLS
4.6. BRG1 (SMARCA4)
4.7. DAZAP1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Essential PSP | PSP Category | Role in PS Formation | Molecular Function | DDR Regulated | Reference |
|---|---|---|---|---|---|
| NONO | 1A | N1_2 stability | RNA BP | HR/NHEJ | [55,56,57,58,59,60,61,62] |
| SFPQ | 1A | N1_2 stability | RNA BP | HR/NHEJ | [56,60,62,64,65,66,67,68,69] |
| RBM14 | 1A | N1_2 stability | RNA BP | NHEJ | [70,71,72] |
| HnRNP K | 1A | Inhibition of N1_1 polyadenylation | RNA BP | NER/BER/MMRHR/NHEJ (?) | [74,75,76] |
| FUS | 1B | PS assembly | RNA BP | HR/NHEJ/BER | [81,83,84,85,86] |
| BRG1 | PS assembly | ATPase | HR | [90,92,93,94,95,96,97,98,100] | |
| DAZAP1 | 1B | PS assembly | RNA BP | HR (?) | [102,103,104,105] |
| HnRNP H3 | 1B | PS assembly | RNA BP | N/A | N/A |
4.8. HnRNP H3
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ghafouri-Fard, S.; Taheri, M. Nuclear Enriched Abundant Transcript 1 (NEAT1): A long non-coding RNA with diverse functions in tumorigenesis. Biomed. Pharmacother. 2019, 111, 51–59. [Google Scholar] [CrossRef]
- Klec, C.; Prinz, F.; Pichler, M. Involvement of the long noncoding RNA NEAT1 in carcinogenesis. Mol. Oncol. 2019, 13, 46–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.; Radaelli, E.; Vermi, W.; et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016, 22, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Blume, C.J.; Hotz-Wagenblatt, A.; Hullein, J.; Sellner, L.; Jethwa, A.; Stolz, T.; Slabicki, M.; Lee, K.; Sharathchandra, A.; Benner, A.; et al. p53-dependent non-coding RNA networks in chronic lymphocytic leukemia. Leukemia 2015, 29, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Favasuli, V.; Ronchetti, D.; Todoerti, K.; Pelizzoni, F.; Manzoni, M.; Barbieri, M.; Fabris, S.; Silvestris, I.; Gallo Cantafio, M.E.; et al. Long non-coding RNA NEAT1 targeting impairs the DNA repair machinery and triggers anti-tumor activity in multiple myeloma. Leukemia 2020, 34, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R. Long non-coding RNAs in Huntington’s disease neurodegeneration. Neurobiol. Dis. 2012, 46, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.S.; Lee, S.T.; Im, W.; Lee, M.; Byun, J.I.; Jung, K.H.; Park, K.I.; Jung, K.Y.; Lee, S.K.; Chu, K.; et al. Altered Expression of the Long Noncoding RNA NEAT1 in Huntington’s Disease. Mol. Neurobiol. 2017, 54, 1577–1586. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Nakagawa, S.; Hirose, T.; Okano, H.J.; Takao, M.; Shibata, S.; Suyama, S.; Kuwako, K.; Imai, T.; Murayama, S.; et al. The long non-coding RNA nuclear-enriched abundant transcript 1_2 induces paraspeckle formation in the motor neuron during the early phase of amyotrophic lateral sclerosis. Mol. Brain 2013, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Shelkovnikova, T.A.; Kukharsky, M.S.; An, H.; Dimasi, P.; Alexeeva, S.; Shabir, O.; Heath, P.R.; Buchman, V.L. Protective paraspeckle hyper-assembly downstream of TDP-43 loss of function in amyotrophic lateral sclerosis. Mol. Neurodegener. 2018, 13, 30. [Google Scholar] [CrossRef]
- Simchovitz, A.; Hanan, M.; Niederhoffer, N.; Madrer, N.; Yayon, N.; Bennett, E.R.; Greenberg, D.S.; Kadener, S.; Soreq, H. NEAT1 is overexpressed in Parkinson’s disease substantia nigra and confers drug-inducible neuroprotection from oxidative stress. FASEB J. 2019, 33, 11223–11234. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Williams, N.G.; Shelkovnikova, T.A. NEAT1 and paraspeckles in neurodegenerative diseases: A missing lnc found? Non-Coding Rna Res. 2018, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visa, N.; Puvion-Dutilleul, F.; Bachellerie, J.P.; Puvion, E. Intranuclear distribution of U1 and U2 snRNAs visualized by high resolution in situ hybridization: Revelation of a novel compartment containing U1 but not U2 snRNA in HeLa cells. Eur. J. Cell Biol. 1993, 60, 308–321. [Google Scholar] [PubMed]
- Dettwiler, S.; Aringhieri, C.; Cardinale, S.; Keller, W.; Barabino, S.M. Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. J. Biol. Chem. 2004, 279, 35788–35797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, A.H.; Bond, C.S.; Lamond, A.I. P54nrb forms a heterodimer with PSP1 that localizes to paraspeckles in an RNA-dependent manner. Mol. Biol. Cell 2005, 16, 5304–5315. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.H.; Lam, Y.W.; Leung, A.K.; Lyon, C.E.; Andersen, J.; Mann, M.; Lamond, A.I. Paraspeckles: A novel nuclear domain. Curr. Biol. 2002, 12, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L.; Carmichael, G.G. Altered nuclear retention of mRNAs containing inverted repeats in human embryonic stem cells: Functional role of a nuclear noncoding RNA. Mol. Cell 2009, 35, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.T.; Ideue, T.; Sano, M.; Mituyama, T.; Hirose, T. MENepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 2009, 106, 2525–2530. [Google Scholar] [CrossRef] [Green Version]
- Sunwoo, H.; Dinger, M.E.; Wilusz, J.E.; Amaral, P.P.; Mattick, J.S.; Spector, D.L. MEN epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res. 2009, 19, 347–359. [Google Scholar] [CrossRef] [Green Version]
- Bond, C.S.; Fox, A.H. Paraspeckles: Nuclear bodies built on long noncoding RNA. J. Cell Biol. 2009, 186, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, A.H.; Lamond, A.I. Paraspeckles. Cold Spring Harb. Perspect. Biol. 2010, 2, a000687. [Google Scholar] [CrossRef] [PubMed]
- Guru, S.C.; Agarwal, S.K.; Manickam, P.; Olufemi, S.E.; Crabtree, J.S.; Weisemann, J.M.; Kester, M.B.; Kim, Y.S.; Wang, Y.; Emmert-Buck, M.R.; et al. A transcript map for the 2.8-Mb region containing the multiple endocrine neoplasia type 1 locus. Genome Res. 1997, 7, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Valenstein, M.L.; Yario, T.A.; Tycowski, K.T.; Steitz, J.A. Formation of triple-helical structures by the 3’-end sequences of MALAT1 and MENbeta noncoding RNAs. Proc. Natl. Acad. Sci. USA 2012, 109, 19202–19207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3’ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampetaki, A.; Albrecht, A.; Steinhofel, K. Long Non-coding RNA Structure and Function: Is There a Link? Front. Physiol. 2018, 9, 1201. [Google Scholar] [CrossRef] [PubMed]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adriaens, C.; Rambow, F.; Bervoets, G.; Silla, T.; Mito, M.; Chiba, T.; Asahara, H.; Hirose, T.; Nakagawa, S.; Jensen, T.H.; et al. The long noncoding RNA NEAT1_1 is seemingly dispensable for normal tissue homeostasis and cancer cell growth. RNA 2019, 25, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Yamazaki, T.; Nakagawa, S. Molecular anatomy of the architectural NEAT1 noncoding RNA: The domains, interactors, and biogenesis pathway required to build phase-separated nuclear paraspeckles. Wiley Interdiscip. Rev. RNA 2019, 10, e1545. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakagawa, S.; Tanigawa, A.; Sasaki, Y.F.; Goshima, N.; Hirose, T. Alternative 3’-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 2012, 31, 4020–4034. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Harvey, A.R.; Hodgetts, S.I.; Fox, A.H. Functional dissection of NEAT1 using genome editing reveals substantial localization of the NEAT1_1 isoform outside paraspeckles. RNA 2017, 23, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naganuma, T.; Hirose, T. Paraspeckle formation during the biogenesis of long non-coding RNAs. RNA Biol. 2013, 10, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burd, C.G.; Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 1994, 265, 615–621. [Google Scholar] [CrossRef]
- Brown, R.S. Zinc finger proteins: Getting a grip on RNA. Curr. Opin. Struct. Biol. 2005, 15, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.H.; Nakagawa, S.; Hirose, T.; Bond, C.S. Paraspeckles: Where Long Noncoding RNA Meets Phase Separation. Trends Biochem. Sci. 2018, 43, 124–135. [Google Scholar] [CrossRef] [Green Version]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Souquere, S.; Chujo, T.; Kobelke, S.; Chong, Y.S.; Fox, A.H.; Bond, C.S.; Nakagawa, S.; Pierron, G.; Hirose, T. Functional Domains of NEAT1 Architectural lncRNA Induce Paraspeckle Assembly through Phase Separation. Mol. Cell 2018, 70, 1038–1053 e1037. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Tanigawa, A.; Naganuma, T.; Ohkawa, Y.; Souquere, S.; Pierron, G.; Hirose, T. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4304–4309. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, S.; Naganuma, T.; Shioi, G.; Hirose, T. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 2011, 193, 31–39. [Google Scholar] [CrossRef]
- Nakagawa, S.; Shimada, M.; Yanaka, K.; Mito, M.; Arai, T.; Takahashi, E.; Fujita, Y.; Fujimori, T.; Standaert, L.; Marine, J.C.; et al. The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development 2014, 141, 4618–4627. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Carmichael, G.G. The fate of dsRNA in the nucleus: A p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 2001, 106, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Benard, M.; Fox, A.H.; et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 2014, 25, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Imamachi, N.; Akizuki, G.; Kumakura, M.; Kawaguchi, A.; Nagata, K.; Kato, A.; Kawaguchi, Y.; Sato, H.; Yoneda, M.; et al. Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol. Cell 2014, 53, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhry, H.; Albukhari, A.; Morotti, M.; Haider, S.; Moralli, D.; Smythies, J.; Schodel, J.; Green, C.M.; Camps, C.; Buffa, F.; et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2alpha dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene 2015, 34, 4482–4490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Shao, C.; Wu, Q.J.; Chen, G.; Zhou, J.; Yang, B.; Li, H.; Gou, L.T.; Zhang, Y.; Wang, Y.; et al. NEAT1 scaffolds RNA-binding proteins and the Microprocessor to globally enhance pri-miRNA processing. Nat. Struct. Mol. Biol. 2017, 24, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, S.; Harris, C.C. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Reviews. Mol. Cell Biol. 2005, 6, 44–55. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Huarte, M.; Guttman, M.; Feldser, D.; Garber, M.; Koziol, M.J.; Kenzelmann-Broz, D.; Khalil, A.M.; Zuk, O.; Amit, I.; Rabani, M.; et al. A large intergenic noncoding RNA induced by p53 mediates global gene repression in the p53 response. Cell 2010, 142, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet. 2011, 43, 621–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchetti, D.; Favasuli, V.; Monti, P.; Cutrona, G.; Fabris, S.; Silvestris, I.; Agnelli, L.; Colombo, M.; Menichini, P.; Matis, S.; et al. NEAT1 Long Isoform Is Highly Expressed in Chronic Lymphocytic Leukemia Irrespectively of Cytogenetic Groups or Clinical Outcome. Non-Coding RNA 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shav-Tal, Y.; Zipori, D. PSF and p54(nrb)/NonO--multi-functional nuclear proteins. FEBS Lett. 2002, 531, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Knott, G.J.; Bond, C.S.; Fox, A.H. The DBHS proteins SFPQ, NONO and PSPC1: A multipurpose molecular scaffold. Nucleic Acids Res. 2016, 44, 3989–4004. [Google Scholar] [CrossRef] [PubMed]
- Alfano, L.; Costa, C.; Caporaso, A.; Altieri, A.; Indovina, P.; Macaluso, M.; Giordano, A.; Pentimalli, F. NONO regulates the intra-S-phase checkpoint in response to UV radiation. Oncogene 2016, 35, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Bladen, C.L.; Udayakumar, D.; Takeda, Y.; Dynan, W.S. Identification of the polypyrimidine tract binding protein-associated splicing factor.p54(nrb) complex as a candidate DNA double-strand break rejoining factor. J. Biol. Chem. 2005, 280, 5205–5210. [Google Scholar] [CrossRef] [Green Version]
- Deshar, R.; Yoo, W.; Cho, E.B.; Kim, S.; Yoon, J.B. RNF8 mediates NONO degradation following UV-induced DNA damage to properly terminate ATR-CHK1 checkpoint signaling. Nucleic Acids Res. 2019, 47, 762–778. [Google Scholar] [CrossRef]
- Jaafar, L.; Li, Z.; Li, S.; Dynan, W.S. SFPQ*NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining. Nucleic Acids Res. 2017, 45, 1848–1859. [Google Scholar] [CrossRef]
- Krietsch, J.; Caron, M.C.; Gagne, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [Green Version]
- Kuhnert, A.; Schmidt, U.; Monajembashi, S.; Franke, C.; Schlott, B.; Grosse, F.; Greulich, K.O.; Saluz, H.P.; Hanel, F. Proteomic identification of PSF and p54(nrb) as TopBP1-interacting proteins. J. Cell. Biochem. 2012, 113, 1744–1753. [Google Scholar] [CrossRef]
- Li, S.; Kuhne, W.W.; Kulharya, A.; Hudson, F.Z.; Ha, K.; Cao, Z.; Dynan, W.S. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009, 37, 6746–6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salton, M.; Lerenthal, Y.; Wang, S.Y.; Chen, D.J.; Shiloh, Y. Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle 2010, 9, 1568–1576. [Google Scholar] [CrossRef] [Green Version]
- Morishima, K.; Sakamoto, S.; Kobayashi, J.; Izumi, H.; Suda, T.; Matsumoto, Y.; Tauchi, H.; Ide, H.; Komatsu, K.; Matsuura, S. TopBP1 associates with NBS1 and is involved in homologous recombination repair. Biochem. Biophys. Res. Commun. 2007, 362, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Akhmedov, A.T.; Lopez, B.S. Human 100-kDa homologous DNA-pairing protein is the splicing factor PSF and promotes DNA strand invasion. Nucleic Acids Res. 2000, 28, 3022–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Takeda, Y.; Dynan, W.S. Sequences in PSF/SFPQ mediate radioresistance and recruitment of PSF/SFPQ-containing complexes to DNA damage sites in human cells. DNA Repair 2011, 10, 252–259. [Google Scholar] [CrossRef] [Green Version]
- Morozumi, Y.; Takizawa, Y.; Takaku, M.; Kurumizaka, H. Human PSF binds to RAD51 and modulates its homologous-pairing and strand-exchange activities. Nucleic Acids Res. 2009, 37, 4296–4307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, C.; Baker, D.K.; Pierce, A.J.; Pittman, D.L. The splicing-factor related protein SFPQ/PSF interacts with RAD51D and is necessary for homology-directed repair and sister chromatid cohesion. Nucleic Acids Res. 2011, 39, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Sun, Y.; Garen, A. Roles of PSF protein and VL30 RNA in reversible gene regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 12189–12193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, R.J.; Bodenburg, Y.H.; Wood, T.G. NH2 terminus of PTB-associated splicing factor binds to the porcine P450scc IGF-I response element. Am. J. Physiology. Endocrinol. Metab. 2002, 283, E423–E427. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, T.; Chin, W.W.; Ko, L. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM). J. Biol. Chem. 2001, 276, 33375–33383. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Eberhart, C.G.; Kai, M. RNA binding protein RBM14 promotes radio-resistance in glioblastoma by regulating DNA repair and cell differentiation. Oncotarget 2014, 5, 2820–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, N.E.; Yuan, M.; Kai, M. RNA-binding protein RBM14 regulates dissociation and association of non-homologous end joining proteins. Cell Cycle 2017, 16, 1175–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matunis, M.J.; Michael, W.M.; Dreyfuss, G. Characterization and primary structure of the poly(C)-binding heterogeneous nuclear ribonucleoprotein complex K protein. Mol. Cell. Biol. 1992, 12, 164–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moumen, A.; Masterson, P.; O’Connor, M.J.; Jackson, S.P. hnRNP K: An HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell 2005, 123, 1065–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesmann, N.; Strozynski, J.; Beck, C.; Zimmermann, N.; Mendler, S.; Gieringer, R.; Schmidtmann, I.; Brieger, J. Knockdown of hnRNPK leads to increased DNA damage after irradiation and reduces survival of tumor cells. Carcinogenesis 2017, 38, 321–328. [Google Scholar] [CrossRef]
- Gallardo, M.; Lee, H.J.; Zhang, X.; Bueso-Ramos, C.; Pageon, L.R.; McArthur, M.; Multani, A.; Nazha, A.; Manshouri, T.; Parker-Thornburg, J.; et al. hnRNP K Is a Haploinsufficient Tumor Suppressor that Regulates Proliferation and Differentiation Programs in Hematologic Malignancies. Cancer Cell 2015, 28, 486–499. [Google Scholar] [CrossRef] [Green Version]
- Helton, E.S.; Chen, X. p53 modulation of the DNA damage response. J. Cell. Biochem. 2007, 100, 883–896. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145–146, 78–97. [Google Scholar] [CrossRef] [Green Version]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sama, R.R.; Ward, C.L.; Bosco, D.A. Functions of FUS/TLS from DNA repair to stress response: Implications for ALS. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, M.; Toth, R.; Vandermoere, F.; Morrice, N.A.; Rouse, J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef]
- Tang, L.; Nogales, E.; Ciferri, C. Structure and function of SWI/SNF chromatin remodeling complexes and mechanistic implications for transcription. Prog. Biophys. Mol. Biol. 2010, 102, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef] [Green Version]
- Chai, B.; Huang, J.; Cairns, B.R.; Laurent, B.C. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev. 2005, 19, 1656–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Castro, R.O.; Previato, L.; Goitea, V.; Felberg, A.; Guiraldelli, M.F.; Filiberti, A.; Pezza, R.J. The chromatin-remodeling subunit Baf200 promotes homology-directed DNA repair and regulates distinct chromatin-remodeling complexes. J. Biol. Chem. 2017, 292, 8459–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, D.A.; de la Serna, I.L.; Veal, T.M.; Imbalzano, A.N. BRCA1 interacts with dominant negative SWI/SNF enzymes without affecting homologous recombination or radiation-induced gene activation of p21 or Mdm2. J. Cell. Biochem. 2004, 91, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Kunzel, J.; Lobrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.J.; Park, J.H.; Park, E.J.; Lee, S.A.; Lee, H.S.; Kang, S.W.; Kwon, J. ATM-mediated phosphorylation of the chromatin remodeling enzyme BRG1 modulates DNA double-strand break repair. Oncogene 2015, 34, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Park, E.J.; Lee, H.S.; Kim, S.J.; Hur, S.K.; Imbalzano, A.N.; Kwon, J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006, 25, 3986–3997. [Google Scholar] [CrossRef]
- Qi, W.; Wang, R.; Chen, H.; Wang, X.; Xiao, T.; Boldogh, I.; Ba, X.; Han, L.; Zeng, X. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J. Cell Sci. 2015, 128, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Velez-Cruz, R.; Manickavinayaham, S.; Biswas, A.K.; Clary, R.W.; Premkumar, T.; Cole, F.; Johnson, D.G. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev. 2016, 30, 2500–2512. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, H.; Gong, M.; Gong, F. The chromatin remodeling protein BRG1 modulates BRCA1 response to UV irradiation by regulating ATR/ATM activation. Front. Oncol. 2013, 3, 7. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.T.; Yen, P.H. A novel nucleocytoplasmic shuttling sequence of DAZAP1, a testis-abundant RNA-binding protein. RNA 2006, 12, 1486–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goina, E.; Skoko, N.; Pagani, F. Binding of DAZAP1 and hnRNPA1/A2 to an exonic splicing silencer in a natural BRCA1 exon 18 mutant. Mol. Cell. Biol. 2008, 28, 3850–3860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloutier, A.; Shkreta, L.; Toutant, J.; Durand, M.; Thibault, P.; Chabot, B. hnRNP A1/A2 and Sam68 collaborate with SRSF10 to control the alternative splicing response to oxaliplatin-mediated DNA damage. Sci. Rep. 2018, 8, 2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, B.; Paunesku, T.; Protic, M.; Woloschak, G.E. Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int. J. Radiat. Biol. 2009, 85, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantha, R.W.; Alcivar, A.L.; Ma, J.; Cai, H.; Simhadri, S.; Ule, J.; Konig, J.; Xia, B. Requirement of heterogeneous nuclear ribonucleoprotein C for BRCA gene expression and homologous recombination. PLoS ONE 2013, 8, e61368. [Google Scholar] [CrossRef] [Green Version]
- Nazim, M.; Masuda, A.; Rahman, M.A.; Nasrin, F.; Takeda, J.I.; Ohe, K.; Ohkawara, B.; Ito, M.; Ohno, K. Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res. 2017, 45, 1455–1468. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taiana, E.; Ronchetti, D.; Todoerti, K.; Nobili, L.; Tassone, P.; Amodio, N.; Neri, A. LncRNA NEAT1 in Paraspeckles: A Structural Scaffold for Cellular DNA Damage Response Systems? Non-Coding RNA 2020, 6, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6030026
Taiana E, Ronchetti D, Todoerti K, Nobili L, Tassone P, Amodio N, Neri A. LncRNA NEAT1 in Paraspeckles: A Structural Scaffold for Cellular DNA Damage Response Systems? Non-Coding RNA. 2020; 6(3):26. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6030026
Chicago/Turabian StyleTaiana, Elisa, Domenica Ronchetti, Katia Todoerti, Lucia Nobili, Pierfrancesco Tassone, Nicola Amodio, and Antonino Neri. 2020. "LncRNA NEAT1 in Paraspeckles: A Structural Scaffold for Cellular DNA Damage Response Systems?" Non-Coding RNA 6, no. 3: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6030026