Comparison of Poly-A+ Selection and rRNA Depletion in Detection of lncRNA in Two Equine Tissues Using RNA-seq

 , , , , and
, , , , and 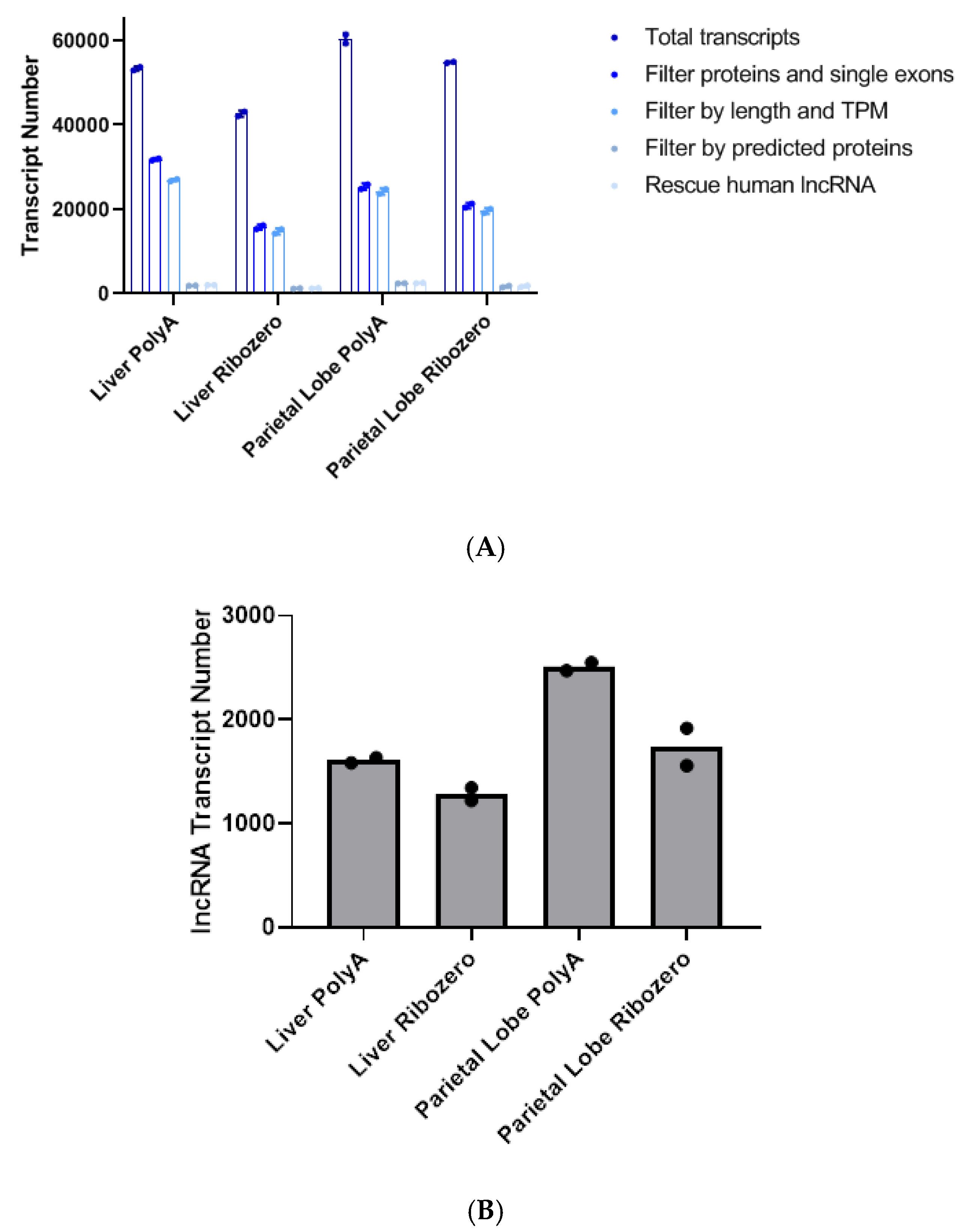{kind=link}
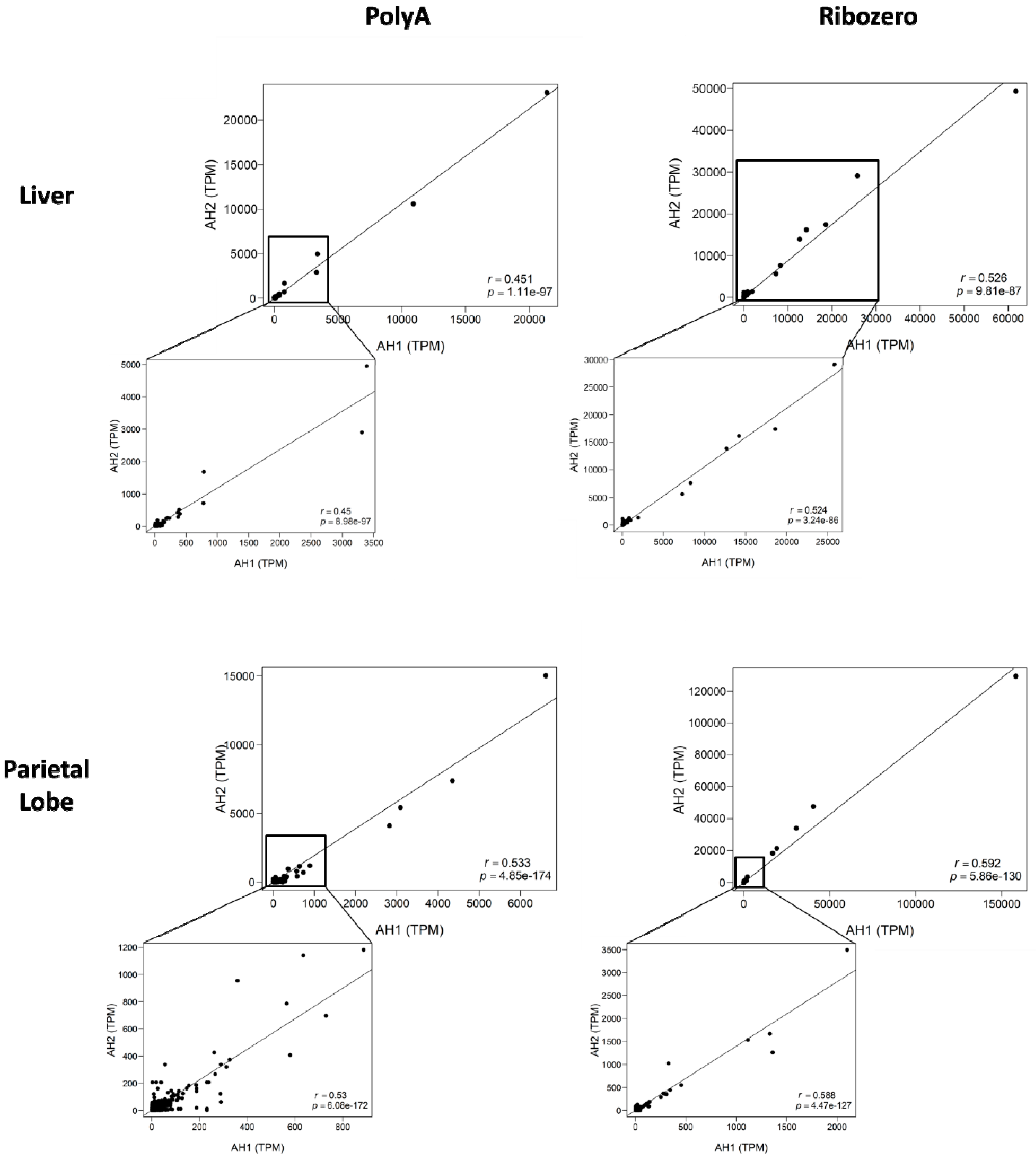{kind=link}
{kind=link}
{kind=link}
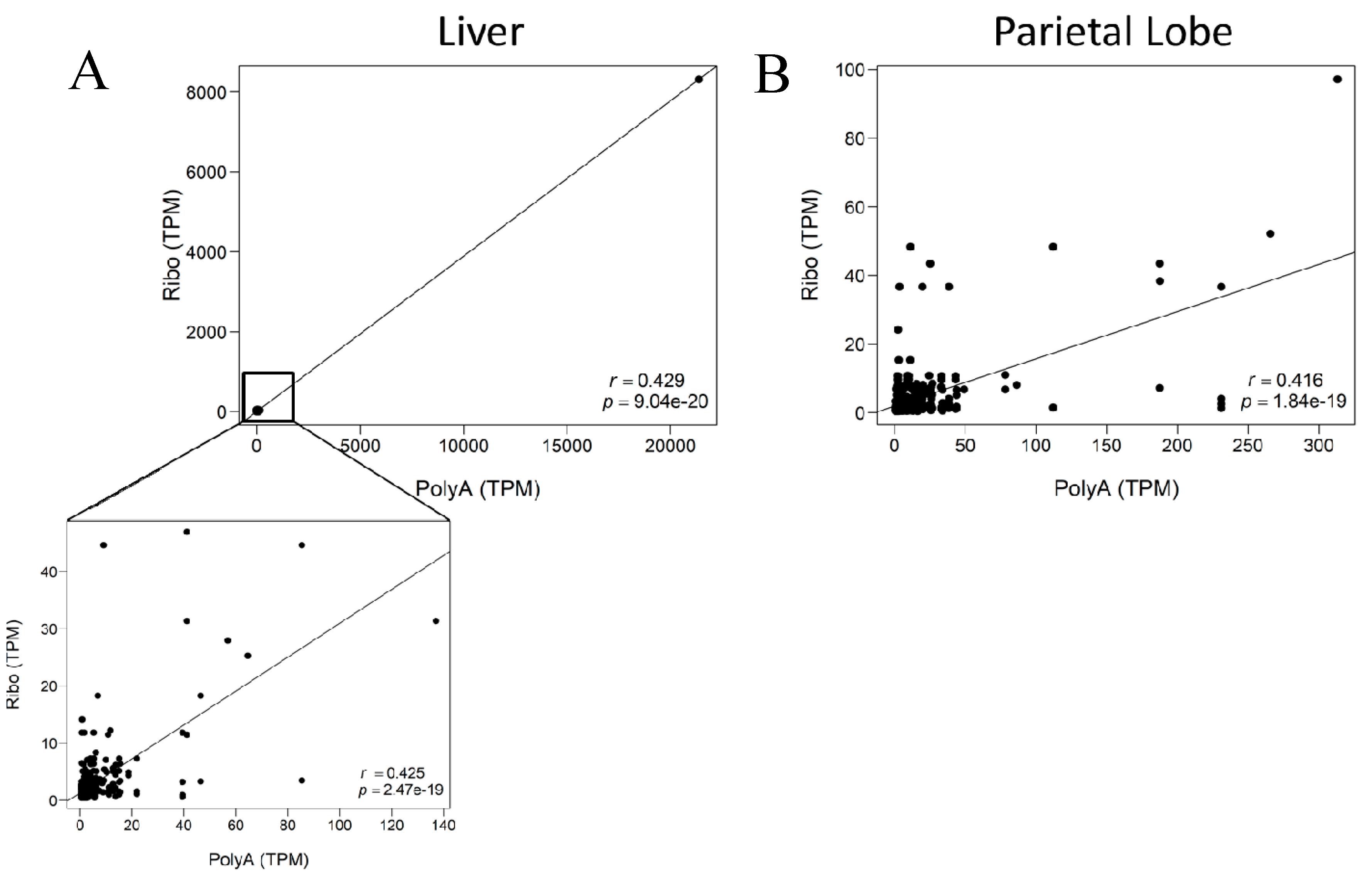{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Samples and Sequencing
4.2. lncRNA Identification
4.3. Analysis
4.4. qRT-PCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin Signature Reveals over a Thousand Highly Conserved Large Non-Coding RNAs in Mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef]
- Martens, J.A.; Laprade, L.; Winston, F. Intergenic Transcription Is Required to Repress the Saccharomyces Cerevisiae SER3 Gene. Nature 2004, 429, 571–574. [Google Scholar] [CrossRef]
- Blume, S.W.; Meng, Z.; Shrestha, K.; Snyder, R.C.; Emanuel, P.D. The 5′-Untranslated RNA of the Human Dhfr Minor Transcript Alters Transcription Pre-Initiation Complex Assembly at the Major (Core) Promoter. J. Cell. Biochem. 2003, 88, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Martignetti, J.A.; Brosius, J. BC200 RNA: A Neural RNA Polymerase III Product Encoded by a Monomeric Alu Element. Proc. Natl. Acad. Sci. USA 1993, 90, 11563–11567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redrup, L.; Branco, M.R.; Perdeaux, E.R.; Krueger, C.; Lewis, A.; Santos, F.; Nagano, T.; Cobb, B.S.; Fraser, P.; Reik, W. The Long Noncoding RNA Kcnq1ot1 Organises a Lineage-Specific Nuclear Domain for Epigenetic Gene Silencing. Development 2009, 136, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Dinger, M.; Amaral, P.; Mercer, T. Long Noncoding RNAs in Mouse Embryonic Stem Cell Pluripotency and Differentiation. Genome Res. 2008, 1433–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalil, A.M.; Faghihi, M.A.; Modarresi, F.; Brothers, S.P.; Wahlestedt, C. A Novel RNA Transcript with Antiapoptotic Function Is Silenced in Fragile X Syndrome. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Liang, W.-C.; Fu, W.-M.; Wong, C.-W.; Wang, Y.; Wang, W.-M.; Hu, G.-X.; Zhang, L.; Xiao, L.-J.; Wan, D.C.-C.; Zhang, J.-F.; et al. The LncRNA H19 Promotes Epithelial to Mesenchymal Transition by Functioning as MiRNA Sponges in Colorectal Cancer. Oncotarget 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yu, B.; Li, J.; Su, L.; Yan, M.; Zhu, Z.; Liu, B. Overexpression of LncRNA H19 Enhances Carcinogenesis and Metastasis of Gastric Cancer. Oncotarget 2014, 5, 2318–2329. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pitchiaya, S.; Cieślik, M.; Niknafs, Y.S.; Tien, J.C.Y.; Hosono, Y.; Iyer, M.K.; Yazdani, S.; Subramaniam, S.; Shukla, S.K.; et al. Analysis of the Androgen Receptor-Regulated LncRNA Landscape Identifies a Role for ARLNC1 in Prostate Cancer Progression. Nat. Genet. 2018, 50, 814–824. [Google Scholar] [CrossRef]
- Johnson, R. Long Non-Coding RNAs in Huntington’s Disease Neurodegeneration. Neurobiol. Dis. 2012, 46, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Spreafico, M.; Grillo, B.; Rusconi, F.; Battaglioli, E.; Venturin, M. Multiple Layers of CDK5R1 Regulation in Alzheimer’s Disease Implicate Long Non-Coding RNAs. Int. J. Mol. Sci. 2018, 19, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, E.Y.; Mansour, T.; Bellone, R.R.; Brown, C.T.; Mienaltowski, M.J.; Penedo, M.C.; Ross, P.J.; Valberg, S.J.; Murray, J.D.; Finno, C.J. Identification of Long Non-Coding RNA in the Horse Transcriptome. BMC Genom. 2017, 18, 511. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Gamini, R.; Zhang, B.; Von Schack, D. Evaluation of Two Main RNA-Seq Approaches for Gene Quantification in Clinical RNA Sequencing: PolyA+ Selection versus RRNA Depletion. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ulitsky, I.; Shkumatava, A.; Jan, C.H.; Sive, H.; Bartel, D.P. Conserved Function of LincRNAs in Vertebrate Embryonic Development Despite Rapid Sequence Evolution. Cell 2011, 147, 1537–1550. [Google Scholar] [CrossRef] [Green Version]
- Hezroni, H.; Koppstein, D.; Schwartz, M.G.; Avrutin, A.; Bartel, D.P.; Ulitsky, I. Principles of Long Noncoding RNA Evolution Derived from Direct Comparison of Transcriptomes in 17 Species. Cell Rep. 2015, 11, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Muret, K.; Désert, C.; Lagoutte, L.; Boutin, M.; Gondret, F.; Zerjal, T.; Lagarrigue, S. Long Noncoding RNAs in Lipid Metabolism: Literature Review and Conservation Analysis across Species. BMC Genom. 2019, 20, 882. [Google Scholar] [CrossRef] [Green Version]
- Wilusz, J.E.; Freier, S.M.; Spector, D.L. 3′ End Processing of Long Nuclear-Retained Non-Coding RNA Yields a TRNA-like Cytoplasmic RNA. Cell 2009, 135, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Kapranov, P.; Drenkow, J.; Dike, S.; Brubaker, S.; Patel, S.; Long, J.; Stern, D.; Tammana, H.; Helt, G.; et al. Transcriptional Maps of 10 Human Chromosomes at 5-Nucleotide Resolution. Science 2005, 308, 1149–1154. [Google Scholar] [CrossRef]
- Burns, E.N.; Bordbari, M.H.; Mienaltowski, M.J.; Affolter, V.K.; Barro, M.V.; Gianino, F.; Gianino, G.; Giulotto, E.; Kalbfleisch, T.S.; Katzman, S.A.; et al. Generation of an Equine Biobank to Be Used for Functional Annotation of Animal Genomes Project. Anim. Genet. 2018, 49, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Capomaccio, S.; Vitulo, N.; Verini-Supplizi, A.; Barcaccia, G.; Albiero, A.; D’Angelo, M.; Campagna, D.; Valle, G.; Felicetti, M.; Silvestrelli, M.; et al. RNA Sequencing of the Exercise Transcriptome in Equine Athletes. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A Triple Helix Stabilizes the 3′ Ends of Long Noncoding RNAs That Lack Poly(A) Tails. Genes Dev. 2012, 26, 2392–2407. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhao, S.; Sheng, Q.; Guo, M.; Lehmann, B.; Pietenpol, J.; Samuels, D.C.; Shyr, Y. RNAseq by Total RNA Library Identifies Additional RNAs Compared to Poly(A) RNA Library. BioMed Res. Int. 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sultan, M.; Amstislavskiy, V.; Risch, T.; Schuette, M.; Dökel, S.; Ralser, M.; Balzereit, D.; Lehrach, H.; Yaspo, M.L. Influence of RNA Extraction Methods and Library Selection Schemes on RNA-Seq Data. BMC Genom. 2014, 15, 675. [Google Scholar] [CrossRef] [Green Version]
- Cui, P.; Lin, Q.; Ding, F.; Xin, C.; Gong, W.; Zhang, L.; Geng, J.; Zhang, B.; Yu, X.; Yang, J.; et al. A Comparison between Ribo-Minus RNA-Sequencing and PolyA-Selected RNA-Sequencing. Genomics 2010, 96, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.P.; Chen, Y.; Takata, Y.; Tomida, M.W.; Lin, K.; Kirk, J.S.; Simper, M.S.; Mikulec, C.D.; Rundhaug, J.E.; Fischer, S.M.; et al. Systematic Evaluation of RNA-Seq Preparation Protocol Performance. BMC Genom. 2019, 20, 571. [Google Scholar] [CrossRef]
- Schuierer, S.; Carbone, W.; Knehr, J.; Petitjean, V.; Fernandez, A.; Sultan, M.; Roma, G. A Comprehensive Assessment of RNA-Seq Protocols for Degraded and Low-Quantity Samples. BMC Genom. 2017, 18, 442. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Yang, M.; Guo, H.; Yang, L.; Wu, J.; Li, R.; Liu, P.; Lian, Y.; Zheng, X.; Yan, J.; et al. Single-Cell RNA-Seq Profiling of Human Preimplantation Embryos and Embryonic Stem Cells. Nat. Struct. Mol. Biol. 2013, 20, 1131–1139. [Google Scholar] [CrossRef]
- Ingolia, N.T.; Lareau, L.F.; Weissman, J.S. Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity of Mammalian Proteomes. Cell 2011, 147, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. A Peptide Encoded by a Transcript Annotated as Long Noncoding RNA Enhances SERCA Activity in Muscle. Science 2016, 351, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Joshi, N.; Fass, J. Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files, Version 1.33 [Software]. 2011. Available online: https://github.com/najoshi/sickle (accessed on 6 April 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Kalbfleisch, T.S.; Rice, E.S.; DePriest, M.S.; Walenz, B.P.; Hestand, M.S.; Vermeesch, J.R.; O′Connell, B.L.; Fiddes, I.T.; Vershinina, A.O.; Saremi, N.F.; et al. Improved Reference Genome for the Domestic Horse Increases Assembly Contiguity and Composition. Commun. Biol. 2018, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G. GffCompare. Available online: http://0-ccb-jhu-edu.brum.beds.ac.uk/software/stringtie/gffcompare.shtml (accessed on 6 April 2020).
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr. 2018. Available online: https://cran.r-project.org/web/packages/dplyr/index.html (accessed on 6 April 2020).
- Sonnhammer, E.L.; Eddy, S.R.; Durbin, R. Pfam: A Comprehensive Database of Protein Domain Families Based on Seed Alignments. Proteins Struct. Funct. Bioinform. 1997, 28, 405–420. [Google Scholar] [CrossRef]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, 279–285. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.; McCarthy, D.; Smyth, G. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dahlgren, A.R.; Scott, E.Y.; Mansour, T.; Hales, E.N.; Ross, P.J.; Kalbfleisch, T.S.; MacLeod, J.N.; Petersen, J.L.; Bellone, R.R.; Finno, C.J. Comparison of Poly-A+ Selection and rRNA Depletion in Detection of lncRNA in Two Equine Tissues Using RNA-seq. Non-Coding RNA 2020, 6, 32. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6030032
Dahlgren AR, Scott EY, Mansour T, Hales EN, Ross PJ, Kalbfleisch TS, MacLeod JN, Petersen JL, Bellone RR, Finno CJ. Comparison of Poly-A+ Selection and rRNA Depletion in Detection of lncRNA in Two Equine Tissues Using RNA-seq. Non-Coding RNA. 2020; 6(3):32. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6030032
Chicago/Turabian StyleDahlgren, Anna R., Erica Y. Scott, Tamer Mansour, Erin N. Hales, Pablo J. Ross, Theodore S. Kalbfleisch, James N. MacLeod, Jessica L. Petersen, Rebecca R. Bellone, and Carrie J. Finno. 2020. "Comparison of Poly-A+ Selection and rRNA Depletion in Detection of lncRNA in Two Equine Tissues Using RNA-seq" Non-Coding RNA 6, no. 3: 32. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6030032