Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer


1
School of Biological Sciences (SBS), Nanyang Technological University (NTU), 60 Nanyang Drive, Singapore 637551, Singapore
2
Institute of Molecular and Cell Biology (IMCB), A*STAR, Singapore 138673, Singapore
*
Authors to whom correspondence should be addressed.
Non-Coding RNA 2021, 7(1), 19; https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7010019
Submission received: 29 January 2021
/
Revised: 28 February 2021
/
Accepted: 5 March 2021
/
Published: 9 March 2021
(This article belongs to the Special Issue Post-transcriptional Regulation through Long Non-coding RNAs (lncRNAs))
Abstract
:It is becoming increasingly evident that the non-coding genome and transcriptome exert great influence over their coding counterparts through complex molecular interactions. Among non-coding RNAs (ncRNA), long non-coding RNAs (lncRNAs) in particular present increased potential to participate in dysregulation of post-transcriptional processes through both RNA and protein interactions. Since such processes can play key roles in contributing to cancer progression, it is desirable to continue expanding the search for lncRNAs impacting cancer through post-transcriptional mechanisms. The sheer diversity of mechanisms requires diverse resources and methods that have been developed and refined over the past decade. We provide an overview of computational resources as well as proven low-to-high throughput techniques to enable identification and characterisation of lncRNAs in their complex interactive contexts. As more cancer research strategies evolve to explore the non-coding genome and transcriptome, we anticipate this will provide a valuable primer and perspective of how these technologies have matured and will continue to evolve to assist researchers in elucidating post-transcriptional roles of lncRNAs in cancer.
Keywords:
lncRNA; cancer; post-transcription; RNA-binding; ribonucleoprotein; RNAi; interactome; prediction; database; CLIP1. Introduction
Transcription is at the forefront of the conversion of stable genomic information into reactive biochemical agents that form and modulate dynamic biological systems. This fundamental process relentlessly transcribes at least 62% of the human genome, resulting in a variety of non-coding RNA (ncRNAs) species that outnumbers the selection of more stable RNAs concerned with translation that accumulate in the cell such as ribosomal RNA (rRNA), transfer RNA (tRNA) and messenger RNA (mRNA) [1,2]. Far from being redundant transcriptional byproducts, ncRNAs can also act as pleiotropic reactive biochemical agents interacting with both RNAs and proteins and are first to propagate any genome level information changes to the biological network state that shapes cellular behaviour [3,4].
Diseased states such as cancer arise from cumulative corruption of the genomic source code, resulting in the opportunistic dysregulation of the conserved transcriptional [5], post-transcriptional [6], translational [7] and post-translational processes [8] that ultimately allow them to escape systemic control. Among the hundreds of thousands of genetic abnormalities that may occur, tremendous progress has been made in understanding how specific key “driver” mutations affect important protein coding genes (oncogenes and tumour suppressors) and influence the aforementioned processes to provide a selective advantage to cancerous cells to develop in a given tissue or microenvironment [9]. However, many mutations found in cancer also accumulate in the non-coding genome [10,11].
The large non-coding transcriptional contributions such as small non-coding RNA (sncRNA), enhancer RNA (eRNA) and long non-coding RNAs (lncRNA) have come under increased interest for cancer research. They feature prominently among the rapidly increasing list of non-coding regulatory elements vulnerable to mutations (promoters, enhancers) and contributing to dysregulating the critical processes aforementioned [12,13]. Although some of these ncRNAs, such as microRNAs (miRNAs, a subset of sncRNAs) have garnered plenty of research momentum [14], others such as lncRNAs and circular RNAs (circRNAs) are still burgeoning especially in the context of cancer biology [15].
Long non-coding RNAs (≥200 bp; lncRNAs) encompass the largest and perhaps most intriguing category of ncRNAs in cancer currently known, as they exhibit highly dynamic and tissue specific expression patterns [16], a trait shared with most oncogenes/tumour suppressors [17]. The majority of these transcripts are localised in the nucleus and transcribed by RNA Polymerase II (RNA Pol II). Long ncRNAs share similar characteristics to messenger RNAs (mRNAs), such as having a 5′-cap and 3′ poly-A tail. Alternatively, circular forms may assemble through non-coding splicing of exons and introns (circRNA), bolstering their resistance to degradation further [18,19]. Classification of the diversity of ncRNAs, especially lncRNAs, is a work in progress. GENCODE has classified lncRNA annotation on the basis of their genomic context, however this offers little indication of their diverse functional potential aside from identifying possible antisense lncRNAs [20,21,22]. Interestingly, the length of an RNA has been found to correlate with their propensity to interact with other biochemical molecules, such as proteins [23]. Hence, the “longer” spectrum of ncRNAs may well be more prone to assume varied roles in regulatory processes through cross-molecular interactions. Indeed, many have already been found to play a pivotal role in those exploited by cancers, such as development and differentiation of cells [24,25]. In fact, since the discovery of MALAT1 (Metastasis-associated lung adenocarcinoma transcript 1) in the early 2000s [26], dysregulation of more than a dozen other lncRNAs, such as H19 [27], XIST [28,29], and HOTAIR [30,31] have steadily been found to be associated with cancer progression and drug resistance [32,33,34]. There is also evidence of circRNAs playing a role in cancer progression [35] and chemotherapy resistance along with biomarker potential [36]. With approximately 16,000 lncRNA genes (28,000 transcripts) identified in Gencode 27, this may well represent a potential “goldmine” of hidden tumour suppressor/oncogenic targets [37].
A major challenge remains in uncovering these lncRNAs and revealing their functional roles in cancer-exploited regulatory processes. This is principally due to the novelty of the field, which is further complicated by their multifunctional interactive potential. At least four major mechanisms have been suggested to mediate their effects: (1) Act as signals to regulate transcription, (2) as decoys recruiting binding partners away from their other targets, (3) as guides directing the targeting of a ribonucleoprotein complex, for example; (4) as scaffolds bringing together multiple biomolecules together [38,39]. Such mechanisms are directly susceptible to propagating abnormalities in lncRNA expression or sequence typical of cancer to the post-transcriptional regulatory networks [40]. Mechanisms 2 and 3 are known to influence one of the most prominent post-transcriptional regulatory pathways: miRNA mediated RNA interference (RNAi) [41]. Effectively, lncRNAs may act as either target or ordnance for the RNA-induced silencing complex (RISC) that perturb stability of various RNAs including lncRNAs themselves [42]. Additionally, lncRNAs may interact with RNAs directly as antisense lncRNAs, such as KRT7-AS, which promotes gastric cancer progression [43]. In summary, aberrant lncRNAs may exert substantial influence over post-transcriptional dysregulation in cancer through RISC dependent and independent mechanisms mediated via their interactions with RNA-binding proteins (RBPs) or RNAs [44]. Identifying and characterising these interactive mechanisms utilising appropriate approaches is therefore critical to overcoming the aforementioned challenge in elucidating their roles in major regulatory processes such as post-transcription.
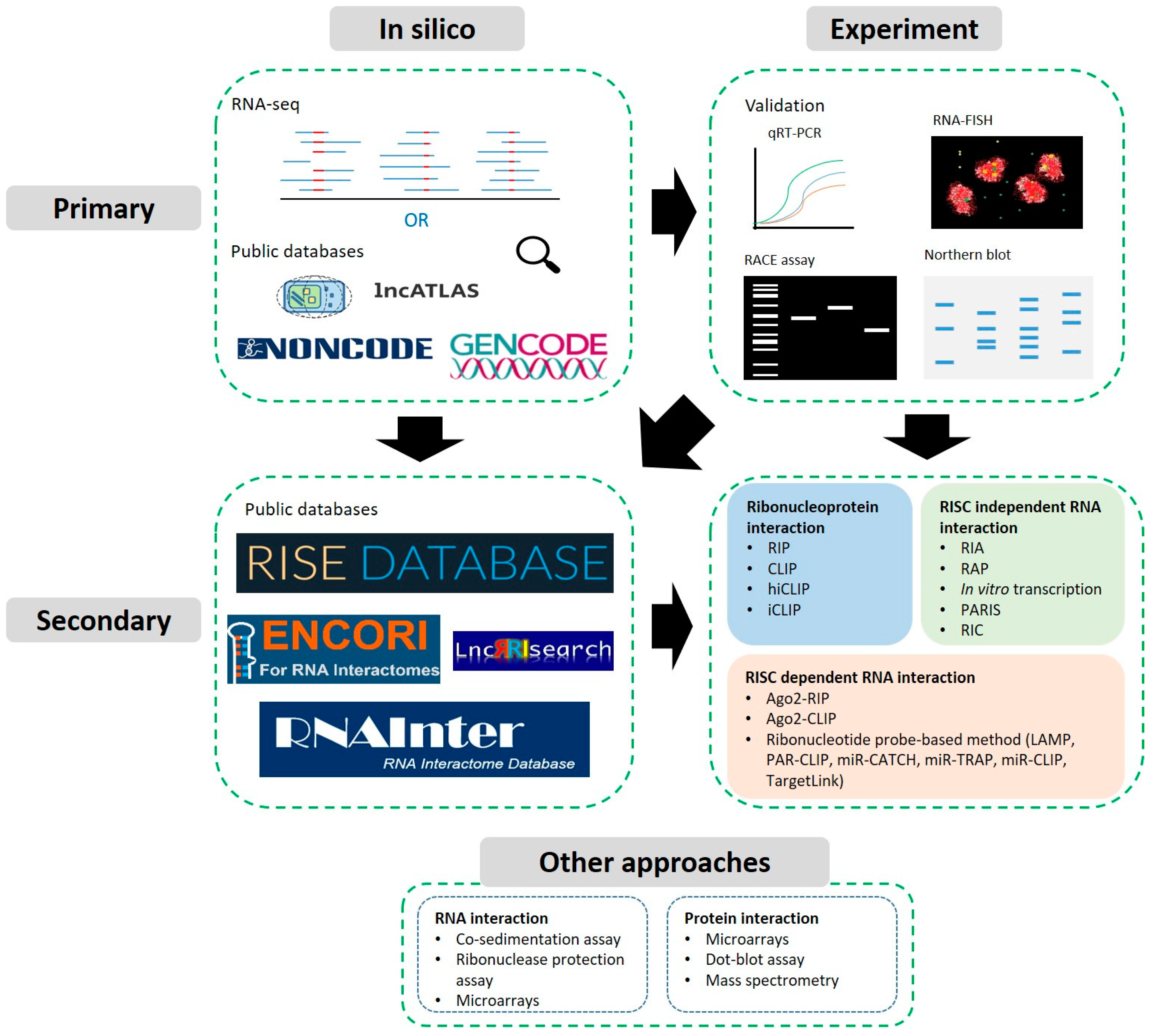
In this review, we will provide a survey of the most useful tools and techniques developed to help place lncRNAs on the post-transcriptional interactome map and reveal their roots to cancer. In the first part, we will provide an overview of such resources valued for primary identification and characterisation of lncRNAs, especially those capable of highlighting cancer relevant contextualisation. In the second part, we will cover how more advanced resources have been developed to help characterise how lncRNAs may interact with RNAs and proteins (Figure 1). Ultimately, we hope this will serve as a useful primer for new cancer research strategies interested in identifying and validating further lncRNAs as oncogenic/tumour suppressor-like players by mechanistically uncovering their post-transcriptional roles.
2. Identification and Primary Characterisation
2.1. Predictions, Identification from High-Throughput Data and Databases
Most cancer research strategies begin by identifying potential candidate genes/loci involved in the dysregulatory state under study. Screening for such candidates usually involves the intersection of high-throughput screening experiments, especially next-generation (NGS) or third-generation sequencing [45]. A number of RNA-sequencing techniques can directly provide valuable information on identity, expression and/or stability of RNAs including lncRNAs. These include capturing a sample of the total transcriptome via standard or single cell RNA-sequencing (RNA-seq/scRNA-seq); capturing nascent transcription using tagged nucleosides or analogs through Global or Precision Run-On sequencing (GRO-seq/PRO-seq; [46,47]) as well as Bru-seq/Bru-Chase-seq [48]; capturing full-length transcripts through Cap-analysis gene expression sequencing (CAGE-seq) and nano-cap analysis of gene expression (nanoCAGE+CAGEscan; [49]) or Oxford Nanopore native RNA sequencing [50]. Provided such sequencing datasets do not undergo selective library preparations these can allow the identification of lncRNAs directly by sequence. Other scenarios, such as Chromatin immunoprecipitation (ChIP) followed by high-throughput DNA sequencing or Assay for Transposase-Accessible Chromatin using sequencing (ChIP-seq or ATAC-seq) may lead towards selecting a putative non-coding region of the cancer genome [51,52]. Depending on the model system, experimental designs and resources, it will be worth considering whether performing additional sequencing experiments is really necessary to fulfil research objectives considering the large amounts of publicly available second-generation data already available.
Regardless of which experimental strategies have been implemented, orthogonal lines of evidence will always fall back on a specific locus or multiple loci of the non-coding genome. From this point, it is possible to infer the identity of potential linear lncRNAs by cross-referencing the loci coordinates with several large databases dedicated to cataloguing lncRNAs or predicting the coding potential of the region. If the coding potential has not already been evaluated, numerous machine-learning tools are available to perform this computation de novo. CPAT, FEELnc and PLEK may be particularly suitable for working with human cancer datasets as extensively evaluated alongside numerous other solutions in [53]. Further comparative reviews of the features of such tools can also be found in [54].
In the case of circular RNAs, most achieve their circular conformation via “backsplicing”. This refers to the covalent linkages between a downstream 3’ and an upstream 5’splice sites, which results in a reversal of exon sequences relative to the annotated transcript [55]. This unique mechanism can be exploited for their identification and therefore a number of tools have been developed to perform this on RNA-seq datasets, which are extensively evaluated and reviewed in [56,57]. CIRI and KNIFE are among some of the tools that showed robust performance even among background noise [58,59]. CIRI2 has also recently been released offering significant performance improvements over CIRI [60].
In the vast majority of cases—especially for cancer studies based on the human genome—a wealth of sequencing data, pre-generated predictions and annotations relevant for targeted loci are available from a number of public and restricted access databases. Aside from familiar initiatives such as RefSeq, Ensembl and FANTOM [61], many more specialised resources dedicated to allocating lncRNA identity and valuable annotations have emerged—some of which are tailored to cancer research, such as Lnc2Cancer or CSCD. Beyond simply determining whether the loci of interest is transcribed as a lncRNA, many of the resources presented offer insights into transcript localisation, expression as well as gene conservation, mutation and links to diseases, such as cancer. Much of the information is integrated from other public databases and projects such as ClinVar [62], COSMIC (Catalogue of Somatic Mutations in Cancer) [63], TCGA (The Cancer Genome Atlas) [64], 1000 Genomes Project (IGSR) [65], (G)ENCODE (Encyclopedia Of DNA Elements) [66,67], GEO (Gene Expression Omnibus) [68], dbSNP [69], UniProt [70], HPA (Human Protein Atlas) [71], GTEx (Genotype-Tissue Expression) [72], HBM2 (Human Body Map 2.0 GEO Dataset GSE30611), FANTOM (Functional Annotation of the Mammalian Genome) [73], CCLE (Cancer Cell Line Encyclopedia) [74], Disease Ontology [75], GO (Gene Ontology) [76], MeSH (Medical Subject Headings) [77] and TARGET (Therapeutically Applicable Research to Generate Effective Treatments) [78]. Additionally, many circRNA specialised databases integrate information and predictions from miRNA centric databases, since circRNAs often act miRNA sponges. These include Starbase [79], TargetScan [80], doRiNA [81], miRcode [82], miRTarBase [83], HMDD [84], OncomiRDB [85], dbDEMC [86] and miRecords [87]. Although most resources will require usage of the web interface, some offer more advanced programmatic access such as NONCODE [88]. All lncRNA and circRNA resources are summarized in Table 1 and Table 2 respectively.
While these databases offer an increasingly large and reliable set of annotations, they remain putative until validated in the specific cancer model system under study using the experimental approaches covered below.
2.2. Experimental Approaches: Validation of Expression, Localisation & Structure
Before any advanced experiments take place, it is usually preferable to validate basic characteristics of the target lncRNAs of interest. We will therefore briefly highlight some key primary techniques valued for lncRNA characterisation as well as potential limitations in their application.
Northern blotting has long been applied for analysing expression of specific RNAs, enabling relative quantification, determination of sizes and providing an assessment of the RNA quality [124,125]. Modern protocols allow reduced chemical usage and good specificity and this core method still remains a vital tool for primary characterisation of lncRNAs [126]. In addition, this technique is still one of the most direct methods for demonstrating the circular configuration of circRNAs. Furthermore, the method is often integrated with more advanced procedures to investigate ribonucleoprotein complexes [127].
Providing the target RNA may be reverse transcribed, RT-qPCR (reverse transcriptase polymerase chain reaction) may offer a more convenient high sensitivity assay. The exponential nature of qPCR, however, requires careful consideration of confounding factors such as genomic DNA contamination and appropriate selection of reference genes [128]. The latter can still be quite problematic when the system of study features aberrant expression of multiple genes including house-keeping genes commonly chosen as references. The recent availability of large pan-cancer datasets may be able to help overcome this problem [129].
Another valuable technique, especially for characterising unknown isoforms of lncRNAs suspected of undergoing splicing is rapid amplification of cDNA ends (RACE), which produces full length sequences of RNA transcripts. RACE utilises reverse transcription with a 5′ or 3′ primer of a known sequence of the RNA of interest to produce a cDNA copy, this is then followed up with PCR amplification. The product can then be coupled with high-throughput screening in a technique called RACE-Seq to characterize the RACE fragments [130,131].
RNA fluorescence in situ hybridization (RNA-FISH) is the reference technique for visually detecting and determining the distribution of any type of RNA in cellular compartments as well as cells that express the RNA of interest. This technique uses fluorescently labelled probes specific for the target RNA [132,133]. While this technique has been crucial in uncovering the mechanism of several lncRNAs [134,135], the high amounts of repetitive elements in lncRNAs increases the challenge of detecting a legitimate lncRNA signal. This may result in the probe binding to high-abundance, off-target RNAs instead of the intended lncRNA. Furthermore, lncRNA signals in the nucleus appear as “bright blobs”, which can be difficult to differentiate from non-specific background signals [136].
SHAPE (Selective 2′-hydroxyl acylation analysed by primer extension) involves the use of reagents, such as N-methylisatoic anhydride (NMIA) and 1-methyl-7-nitroisatoic anhydride (1M7) that react with the 2′-hydroxyl group of the RNA backbone, forming ribose 2′-O-adducts [137]. Adduct formation is dependent on nucleotide flexibility and is quantified at nucleotide resolution by performing RT and comparing the product against a control [138]. This can be further coupled with mutational profiling (MaP), which accounts for the occasional incorporation of noncomplementary nucleotides or deletions caused by reverse transcriptase enzymes, to generate SHAPE profiles where mutations are counted and facilitate the identification of RNA secondary structure formation at nucleotide resolution [139,140]. With valuable evidence supporting the expression, localisation and possible structure of the target lncRNAs, the next step is to estimate and conclusively identify what biomolecules may be interacting with it.
3. Secondary Characterisation: Predicting and Detecting Interactions
3.1. Predictions and Databases
As mentioned in the introduction, there are four types of molecular mechanisms suggested to mediate lncRNA effects through versatile interactions with DNA, RNA and protein molecules [141]. All three types of interactions have been studied and modelled. However, the interactions with the most direct effects on post-transcription are expected to involve only RNA and protein. For more information on predicting lncRNA:DNA interaction potential, we invite the reader to consult recent developments in this relatively new field [142,143,144].
In-silico prediction of RNA-RNA and RNA-protein interactions are active areas of research recently boosted by machine learning techniques that have grown in strength and numbers over the past decade, feeding on the wealth of accumulating experimental data [145,146]. A significant amount of time and resources have been invested into developing specialised databases and algorithms that predict potential interacting partners of particular lncRNA candidates, some of which have required supercomputer scale processing [147,148]. Exploring how some of these resources can help guide or supplement experimental approaches should therefore form a valuable addition to the secondary characterisation strategy for lncRNA candidates.
RNA:RNA interaction prediction stands to be the most well investigated in large part due to the strong overlap with the small RNA/miRNA field. For instance, lncRNAs acting as ceRNAs can be predicted through their interactions with miRNAs for which a multitude of databases and tools already exist to predict their general propensity to bind certain RNA sequences (Table 3); also see [149]. However, some lncRNA:RNA specific prediction tools have been developed too. Recent evaluation of a dozen such tools has shown that the real-life performance is still fairly average [53]. Tools such as ASSA or RIblast [150,151] that incorporate other sequence information, such as length and GC content and provide useful statistical outputs may be most relevant for real human datasets but cannot be solely relied on for confident inference.
RNA:Protein prediction requires a different approach. Notably, network and correlation based predictions [152] have gained popularity owing to the large increase in available expression data, allowing indirect inference of lncRNA:Protein relationships. RNA:RNA prediction can also benefit from this approach and should be used to complement predictions [153,154]. As such, most resources presented are databases integrating multiple sources of evidence from orthogonal experiments in repositories such as GEO [68], ArrayExpress [155], ENCODE [67], TCGA [64] and the SRA (Sequence Read Archive) [156] as well as pre-calculated prediction and annotation databases such as as LncRNADisease [94], MNDR (Mammal ncRNA-Disease Repository) [97], eDGAR (Database of Disease-Gene Associations with annotated Relationships among genes) [157], circRNADisease [114], RAIN (RNA-protein Association and Interaction Networks) [158], RAID (RNA Association Interaction Database) [159], NPInter [160] and RISE (RNA Interactome from Sequencing Experiments) [161] (Table 3). For a comprehensive review of tools and databases dedicated to miRNA specific predictions please refer to [149,162].

{kind=link}
Table 3.
Databases integrating computational and experimental sources for predicting lncRNA interactions. Databases are listed detailing the types of interactions they cover. A link to the hosting website is provided followed by the latest known version as well as the most recent publication describing the database. Most databases rely on a primary experimental (EXP) or computational (CPU) source, which are briefly explained. Any additional sources are also summarised.
Table 3.
Databases integrating computational and experimental sources for predicting lncRNA interactions. Databases are listed detailing the types of interactions they cover. A link to the hosting website is provided followed by the latest known version as well as the most recent publication describing the database. Most databases rely on a primary experimental (EXP) or computational (CPU) source, which are briefly explained. Any additional sources are also summarised.
| Database | Link | Interaction Type | Primary Source | Additional Sources |
|---|---|---|---|---|
| NPInter v4 (2019) [160] | http://bigdata.ibp.ac.cn/npinter4 (accessed on 8 March 2021) | miRNA-RNA; ncRNA-DNA; ncRNA-Protein; circRNA | EXP: Re-processing and integration of experimental data (GEO; ENCODE; RISE) | CPU: miRNA binding (miRanda, TargetScan); Disease association (LncRNADisease, MNDR, eDGAR and circRNADisease)EXP: Literature mining |
| lncRRIsearch (2019) [163] | http://rtools.cbrc.jp/LncRRIsearch/ (accessed on 8 March 2021) | lncRNA-mRNA | CPU: RIBlast | EXP: Tissue expression |
| DIANA-LncBase v3 (2020) [164] | https://diana.e-ce.uth.gr/lncbasev3 (accessed on 8 March 2021) | miRNA-lncRNA | EXP: Re-processing and integration of experimental data (miRNA, AGO2-CLIP-Seq and CLIP-Seq) | CPU: Correlation with lncRNA expression |
| SPONGEdb v1 (2021) [165] | https://exbio.wzw.tum.de/sponge/home (accessed on 8 March 2021) | miRNA-lncRNA | CPU: DIANA-LncBase, TargetScan, miRcode, miRTarBase | EXP: TCGA expression |
| LnCeVar v1 (2020) [166] | http://www.bio-bigdata.net/LnCeVar/ (accessed on 8 March 2021) | miRNA-lncRNA | EXP: SNP and mutation data from TCGA, COSMIC, 1000 Genomes Project | CPU: Integration from miRanda, mirBase, miRTarBase, TargetScan |
| miRSponge v1 (2015) [167] | http://bio-bigdata.hrbmu.edu.cn/miRSponge/ (accessed on 8 March 2021) | miRNA-lncRNA miRNA-circRNA | EXP: Manual curation from literature | CPU: Integration from TarBase, miRTarBase, miRanda, miRecord |
| starBase/ENCORI v2 (2014/2021) [79] | http://starbase.sysu.edu.cn/ (accessed on 8 March 2021) | miRNA-ncRNA; RBP-RNA;RNA-RNA | EXP: Re-processing and integration of experimental data (CLIP-Seq & variations) | CPU: Correlation of RBP somatic mutation with diseases EXP: Pan-Cancer networks from expression profiles (TCGA) |
| RAID v3/RNAInter (2020) [168] | http://www.rna-society.org/raid/ (accessed on 8 March 2021) | RNA-Protein; RNA-RNA; RNA-Histone; RNA-Drug | EXP/CPU: Integration of literature sources and 35 databases. | EXP: Methylation, localisation and editing data from other databases. |
| RISE (2018) [161] | http://0-rise-life-tsinghua-edu-cn.brum.beds.ac.uk/index.html (accessed on 8 March 2021) | RNA-RNA | EXP: Integration from sequencing based studies | CPU: Integration with several other databases (RAIN, RAID, NPInter) |
| LncRNA2Target v2 (2019) [169] | http://123.59.132.21/lncrna2target/ (accessed on 8 March 2021) | lncRNA-RNA | EXP: Manual extraction of interaction associations from literature | EXP: Re-processing of lncRNA perturbation RNA-Seq datasets |
| LncExpDB (2020) [170] | https://bigd.big.ac.cn/lncexpdb/interactions (accessed on 8 March 2021) | lncRNA-mRNA | CPU: Co-expression network analysis and prediction | EXP: Expression extracted from public repositories (GEO, SRA and ArrayExpress) |
| LncACTdb v2 (2019) [171] | http://www.bio-bigdata.net/LncACTdb (accessed on 8 March 2021) | miRNA-lncRNA-mRNAmiRNA-circRNA | EXP: Manual curation from literature | CPU: Predictions from networks and integration with Pan-Cancer data (TCGA) |
These tools and databases should help prioritise the types of hypotheses and experiments planned for experimental validation in the cancer model system of choice utilising several of the numerous techniques covered in the rest of this article.
3.2. Sequencing Compatible Approaches
With the advent of next-generation sequencing, multiple high-throughput techniques have been developed which allow for increased screening capabilities to discover novel lncRNA interactions in cancer. One of the primary mechanisms through which lncRNAs have been documented to dysregulate cancer post-transcription is through their participation in ribonucleoprotein (RNP) complexes [44,172]. Considering the importance of RBPs, we will first introduce the RIP and CLIP methods, which have been more recently adapted to RNA-Seq. We will then focus on how related methods have been specifically tailored for capturing RNA interactions involving the RNA-induced silencing complex (RISC). Finally, methods for exploring RISC independent RNA interactions will be presented.
3.2.1. Ribonucleoprotein Complex Interaction Detection
LncRNAs often interact with RNA-binding proteins (RBPs), such as the interaction between HOTAIR and EZH2 [173]. Several knock-on effects can result, such as competing with other mRNAs/lncRNAs for RBP binding and/or increasing the ceRNA potential of the lncRNA. In the MACC1-AS1 to PTBP1 interaction, such effects have significant consequences on breast cancer tumorigenesis [174]. RNA immunoprecipitation (RIP) is one of the first techniques employed to identify such RNAs bound to specific RBPs. RIP involves cell or tissue lysis, followed by immunoprecipitation of native RNA-protein complexes with a specific antibody against the target protein. As these complexes are not stabilized by covalent crosslinking, extra precaution must be taken during washing to remove nonspecific RNA while maintaining the RNA–protein interactions. This limitation makes detection of RNAs with low binding affinity to the protein of interest difficult. In addition, unstably bound RBPs may dissociate from their RNA targets and re-associate with other RNAs under harsh conditions [175,176].
Nevertheless, the RIP techniques have been successfully used over the years revealing relevant interactions in the context of cancer. For example, RIP was used by Tripathi et al. to investigate the interaction between lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and the serine/arginine (SR) splicing factors [177]. MALAT1 is known to be overexpressed in breast, pancreas, lung, colon and prostate carcinomas [178], in addition, it is associated with metastasis and poor survival in non-small cell lung cancer patients [26]. SR splicing factors can influence the alternative splicing (AS) events of many pre-mRNAs in a concentration and phosphorylation-dependent manner, but its cellular mechanism was unknown [179,180,181]. In the study, MALAT1 was found to interact with SRSF1 and regulate cellular levels of its phosphorylated forms, which modulated AS events downstream. Further exploration of MALAT1 interactions via RIP-Seq by Wang et al. was similarly fruitful. Their study revealed that MALAT1 also binds to EZH2 [182]. EZH2 is overexpressed in endometrium, prostate and breast cancers [183]. In prostate cancer patients, EZH2 is associated with increased cell proliferation, invasiveness and metastasis [184,185]. As EZH2 had been shown to interact with several lncRNAs, such as HOTAIR [173] and PCAT-1 [186], it was unclear which lncRNA was important for EZH2-driven prostate cancer progression. Ultimately, knockdown experiments demonstrated that EZH2-MALAT1 association played a significant role in cancer progression [182], thus representing a new alternative target for treating prostate cancer.
To overcome the low specificity of RIP, crosslinking and immunoprecipitation (CLIP) was developed by Ule et al. [187]. CLIP involves the usage of ultraviolet (UV) light to form covalent bonds between RBPs and their direct binding RNAs. An advantage in itself since UV does not crosslink proteins to each other, significantly improving its specificity. In CLIP-Seq, after crosslinking, RNA is fragmented, purified and prepared for sequencing [188]. This has led CLIP-Seq to be accepted as a gold standard for identification of endogenous RNA–protein interactions [189].
Since the development of CLIP-Seq, there have been major advancements in CLIP methods that further increase specificity. The first is the development of hybrid CLIP (hiCLIP), which enables identification of RNA duplexes bound to RBPs. This is achieved by ligating the two RNA strands with an additional RNA adaptor, following that, the RNA duplexes are immunoprecipitated and sequenced. This method was used to identify mRNA–mRNA and mRNA–lncRNA duplexes bound by Staufen 1 [190]. The second major improvement to CLIP is individual nucleotide CLIP (iCLIP), which maps RBP binding sites at nucleotide resolution. A limitation of CLIP is that cDNAs prematurely truncate before the crosslinked nucleotide [191]. However, iCLIP exploits this limitation through the addition of a second adaptor to the 3′ end of cDNA after reverse transcription via circularization [192]. This enables prematurely truncated cDNA at the crosslinked nucleotide to be amplified and therefore improves sensitivity.
3.2.2. RISC Dependent RNA Interactions
The active miRNA research field has led to the development and improvement of a number of methods for establishing which RNAs are being targeted by miRNAs. Thus, establishing whether candidate lncRNAs are involved in RNAi mediated regulation can provide valuable insight into their function. In the first case, lncRNAs targeted by a RISC that is loaded with a complementary miRNA or siRNA may act as decoys or competitive endogenous RNAs (ceRNAs). Circ-lncRNAs may be particularly ideal as ceRNAs due totheir increased stability [38,107]. Colloquially these lncRNAs or circ-lncRNAs are said to “sponge” away interference from targets with other cellular functions, such as mRNAs [193]. Alternatively, lncRNAs may act as the precursors to miRNAs or siRNAs—a further processing step that may be mediated by other RNA binding proteins, such as HuR. These opposing roles can be determined primarily by whether the lncRNA co-occupies the RISC with suspected RNA targets/loads by capturing the ribonucleic or protein part of the complex.
As a first approach, it is possible to identify RNA–RNA interactions on the basis of co-occupation of the RISC complexes isolated via RIP or CLIP based techniques introduced earlier. Ago2-RIP-Seq and Ago-HITS-CLIP (also called Ago-CLIP-Seq) in particular focus on applying RIP-Seq and CLIP-Seq respectively to AGO2, pulling down all miRNAs and possible targets in a single experiment [194,195]. Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) is another popular variant of CLIP-Seq that uses photoactivatable nucleoside analogues, such as 4-thiouridine (4SU) to crosslink RISC proteins, such as AGO2 or TNRC6 to the labelled RNAs [121,196]. A particular result of this method is the T to C transitions that occur at the crosslinking sites that can be used to enhance downstream analyses. This technique has been widely cited and implemented including in cancer research [197].
An alternative approach involves utilising a modified ribonucleotide probe to bait and capture any complementary RNAs when they are loaded in the RISC complex. This can be helpful to identify which miRNA/siRNAs are being sponged by a lncRNA acting as ceRNA (competitive endogenous RNA). Additionally, if a lncRNA is suspected of being processed into a miRNA/siRNA, a probe mimicking the lncRNA-derived miRNA/siRNA can be prepared to enable target identification. The first method to apply this concept employed biotinylated miRNA mimic probes to capture their targets in vivo [198]. An in vitro version employing digoxigenin instead has also shown similar performance and is known as the “labelled miRNA pull-down” (LAMP) assay system [199]. Many elaborate modifications have been devised to enhance the probes with interesting properties to better capture RNA-RNA duplexes.
A major type of enhancement to the original biotinylated approach has been the inclusion of photoactivatable tags or analogues into the probes (similar to PAR-CLIP). For example, miR-TRAP incorporates a psoralen analogue allowing photoactivatable crosslinking to targets, and more stringent purification [200]. The original miR-TRAP method has also recently been paired with RNA-Seq in PCP-Seq [201]. PCP-Seq was validated in A549 cancer cell lines.
Other crosslinking technologies incorporated into probes also alter the tag used for isolation procedures. “Photoclicking” a process borrowed from bioorthogonal protein chemistry uses tetrazole-ene or dibenzocyclooctyne (DBCO) [202]. DBCO-tagged mimic probes in particular have been reported to confer increased miRNA-RISC loading affinity and can be isolated via azide-immobilised magnetic beads [203]. In the PA-miRNA method, the biotin tag normally used for isolation is attached via a photo-cleavable linker. It is unclear whether this provides a particular advantage in identifying complementary targets, but the modification is claimed to allow the probe to be used as a photoactivatable source of miRNA [204]. TargetLink is a tagless method that utilises a Locked Nucleic Acid (LNA)-based probe for capturing crosslinked miRNA-mRNA complexes and was tested on a human colorectal cancer cell line yielding 12 target genes for miR-21 [205].
Similar approaches can be further enhanced by combining with the technologies used for RNA baiting. Such is the case for miR-CATCH which targets a single mRNA (or lncRNA) using a biotinylated DNA probe and crosslinked RISC ribonucleoprotein complexes to detect all miRNAs targetors [206,207]. miR-CLIP instead focuses on a single miRNA-like probe containing psoralen and biotin groups to capture the “targetome” after subsequent Ago2 immunoprecipitation and streptavidin purification followed by RNA sequencing [208]. Both techniques have shown promise in cancer research, miR-CATCH has been applied to MSLN mRNA, which is overexpressed in Malignant Pleural Mesothelioma and miR-CLIP was validated in Hela cells revealing the lncRNA H19 as a target of miR-106a [209].
Nucleoside analogues, such as diazirine and aryldiazirine, have shown promising results as a means of crosslinking RNA-RNA molecules and post-crosslink tagging has been developed [210,211,212]. Crosslinking chemistry is an active area of research that promises to deliver many more options that may give rise to further variations of RNA pull down methods [213]. Most interestingly, diazirine has even been encoded as an unnatural amino acid, which may open up new interesting possibilities for protein mediated interaction capture [214].
3.2.3. RISC Independent RNA Interactions
Although RISC independent lncRNA–RNA interactions may be less well known, they have been shown to regulate important biological processes, such as somatic tissue differentiation via post-transcriptional mechanisms [215] and cancer cell growth [216]. Interestingly, the extent of base-pairing could determine if the LncRNA–mRNA interactions positively or negatively regulate gene expression. LncRNAs associated with mRNAs through partial base-pairing have been found to promote mRNA decay [217] while more complete base-pairing protects the mRNA from degradation [218].
Many RISC independent RNA–RNA interactions may involve the participation of an RBP other than AGO2. Therefore, a similar RIP or CLIP-based approach targeting other known ribonucleoproteins may allow identification of other proximal RNAs interacting with the lncRNA of interest. Additionally, CLASH (cross-linking ligation and sequencing of hybrids), a modified version of iCLIP, is another technique that allows for identification of RNA–RNA interactions by using a tagged “bait” protein [219]. After UV crosslinking of RNA–protein interactions, the bait is pulled down and RNA is recovered and sequenced. When a particular RBP is not targeted MARIO (Mapping RNA interactome in vivo) allows EZ-link biotinylation of the protein [220]. A biotinylated RNA-linker is then ligated in a similar fashion to hiCLIP allowing RNA–RNA interactions mediated by other proteins to be captured and sequenced. However, without an RNA binding protein to mediate the interaction, other solutions are required.
Given that lncRNA expression tends to be lower than mRNAs, the levels of endogenous lncRNA must be considered otherwise there might be insufficient material being pulled down. One way to overcome this technical limitation is to perform in vitro transcribed biotin tagged mimics of the lncRNA prior to pull down. Not only does this ensure sufficient lncRNA for the pull down but can also improve specificity rather than relying on antisense DNA probe binding. This technique was used in characterising the function of antisense lncRNA of MACC1 in gastric cancer [221]. In that study, bioinformatics predictions suggested that MACC1-AS1 contained a binding site for MACC1 mRNA and the interaction between the two RNAs was later validated via qRT-PCR.
RIA-seq (RNA interactome analysis and sequencing) allows for mapping of transcriptome-wide RNA-RNA interactions before selectively probing for your lncRNAs of interest [215]. In brief, cells are fixed with 1% glutaraldehyde before lysis. The RNA are then sonicated to a size range of 100 to 500 nucleotides before addition of antisense DNA probes. The probes are biotinylated and target specific regions of the lncRNA of interest. Thereafter, streptavidin binding captures the beads–biotin-probes–RNA complexes. The RNA is then eluted and qRT–PCR is used to detect enriched transcripts. Alternatively, high-throughput sequencing can be used though sufficient read depth is required to detect interaction. This technique was used to discover a novel mechanism of lncRNA-mRNA interaction in colorectal cancer. The cytoplasmic lncRNA SNHG5 was found to interact with and stabilise their target mRNAs by protecting them from degradation by STAU1. As such, it promotes colorectal cancer cell survival [222]. However, the specificity of RIA-Seq depends largely on the probe design.
Finally, ribonucleoprotein agnostic methods exist to perform transcriptome wide identification of all RNA complexes without specific baits or probes. PARIS (Psoralen Analysis of RNA Interactions and Structures), for example, combines psoralen crosslinking with proximity ligation to identify interactions and structural information of all RNAs [223]. In brief, live cells are UV crosslinked and lysed before RNA is extracted and fragmented. PAGE gel electrophoresis is then used to purify RNA where only RNA duplexes are obtained. The RNA duplexes then undergo proximity ligation followed by photo-decrosslinking before the RNA is prepared for sequencing. This technique allows for identification of long-range RNA structures ranging from 200 to over 1000 nt [224]. Apart from detecting just intramolecular interactions and structures, PARIS has been reported to also identify and refine RNA–RNA interactions to near base pair resolution. In addition, unlike other techniques that require specific RNA baits, PARIS allows for identification of native base-pairing interactions through cross-linking of live cells.
Another similar technique is LIGR-seq (LIGation of interacting RNA followed by high-throughput sequencing) which uses a psoralen derivative aminomethyltrioxalen (AMT) that intercalates into the RNA for UV crosslinking [225,226]. circRNA ligase is used for proximity ligation of RNA before sequencing. Unlike the PARIS protocol, enrichment of RNA complexes occurs through RNase R digestion of uncrosslinked RNAs. SPLASH (Sequencing of Psoralen crosslinked, Ligated, and Selected Hybrids) might be seen as a more robust variation of PARIS and LIGR-seq as it utilises biotin-labelled psoralen for enrichment of crosslinked RNA using streptavidin beads [227].
RIC-Seq (RNA in situ conformation sequencing) has also recently entered this arena of whole RNA-interactome and secondary structure mapping [228]. Similar to SPLASH, in situ proximity ligation of RNA complexes is applied and biotin enables pulldown of crosslinked RNA. However, it substitutes psoralen with pCp thereby labelling the 3′ end of RNA [229] instead of staggered pyrimidines on opposite strands [230]. This step would seem to give RIC-Seq an edge over psoralen-based techniques as it appears to more effectively enrich RNA complexes allowing for detection of lowly expressed RNA [228].
Fundamentally, all these RNA–RNA interactome methods apply proximity ligation with key differences at the RNA-complex isolation steps. Interpreting the results from these largescale experiments is challenging especially considering the current bioinformatic tools are still somewhat underdeveloped. Effectively, analyses borrow tools traditionally used for HiC. However, they have all shown promise in being able to identify lncRNA interactions. For example, MALAT1 was found to interact with NEAT1 through analysis of RIC-Seq data [228]. Nevertheless, the aforementioned methods (summarised in Table 4) are providing valuable results that are being compiled into databases such as RISE and can be re-analysed using updated analytical tools when available to continue improving our understanding of RNA–RNA interactomes and structure. Such results can ultimately be followed by more precise experiments to reveal the regulatory effects of the lncRNA interaction.
3.3. Other Approaches and Biochemical Assays
Whether potential candidate lncRNA and interacting partners have been predicted using bioinformatic tools or via high-throughput sequencing techniques, it is also possible to apply other low- or medium-throughput technologies to characterise or further validate possible interactions.
3.3.1. Protein Interaction Assays
Microarrays provide an alternative method to Next-Generation Sequencing (NGS) for lncRNA-protein interaction studies. This relatively inexpensive method is able to provide information in a couple of hours. However, it has limitations in identifying novel RNA targets. It could be used to quantify and identify either annotated RNA targets or RBPs in ribonucleoprotein (RNP) complexes. RNP immunoprecipitation–microarray (RIP-Chip) has been applied successfully in detecting several QKI-5-binding lncRNAs, especially lnc10 that regulates the apoptosis of germ cells during their development [231]. In this method, the crosslinking reaction may be omitted during RIP as cell extracts will be used to identify RBPs. However, the crosslinking step can give results with high backgrounds and introduce sequence biases. Briefly, cell extracts are used for immunoprecipitation against the protein of interest and then washed extensively, following which the RNP is eluted and dissociated into RNA and protein [232]. Besides this, protein microarrays have also been widely used to detect RBPs that interact with certain lncRNA. Here, lncRNAs are transcribed in vitro and labelled with Cy5 dye, then labelled lncRNAs are incubated with a protein microarray [233]. Protein microarrays have been able to detect the interaction between TINCR RNA and STAU1 protein [215]. Aberrant expression of TINCR RNA is implicated in the progression of many cancers. TINCR RNA overexpression in epithelial ovarian cancer has been reported to correlate with tumour size, metastasis and survival rates in the patients. By silencing TINCR, FGF2 expression is downregulated and leads to the inhibition of epithelial ovarian cancer progression [234].
Dot-blot assay is widely used to study lncRNA–protein interaction and is especially useful in mapping the protein binding region in lncRNA. In this assay, lncRNAs of interest are biotinylated and transcribed in vitro, followed by in vitro RNA-protein binding via incubation of the biotinylated lncRNA with recombinant protein. The bound lncRNA is partially digested by RNase to allow only a small fragment attached to the protein. The lncRNA–protein complexes are subjected to proteinase K treatment to dissociate the complexes. Subsequently, the lncRNA is purified and hybridized to nylon or PVDF membranes spotted with 54–60 mer antisense DNA oligonucleotides tiled along the lncRNA of interest [235,236]. The hybridized membrane is washed and visualized by the detection of streptavidin-HRP signals. This assay has successfully identified the motif sequence of BCAR4 bound by SNIP1 and PNUTS, which is located at positions 235–288 and 991–1044 in BCAR4 [236]. In a tumour microenvironment study, positions 355–414 and 1298–1353 of lncRNA CamK-A are bound and protected by PNCK and IκBα, which is important in tumour progression [237].
Mass spectrometry (MS) is commonly used to characterize various proteins that are associated with lncRNAs, following pull-down of the lncRNA of interest. It can identify and quantify molecules in complex mixtures based on their mass and charge. However, the quantification accuracy may not be correct due to the difference in mass spectrometric responses. To overcome this issue, stable-isotope labelling has been applied before proceeding with MS. The stable-isotope labelling by amino acids in cell culture (SILAC) has been shown to simplify the quantification and remove false-positive results [238]. This labelling is performed by simply growing two cell populations in two different mediums containing either light or heavy amino acids. Then, the cells are mixed, and proteins extracted for MS analysis. This method has been used to identify several proteins that are specifically enriched and found to interact with Xist to mediate transcriptional silencing [239]. Moreover, aberrant expression of Xist is associated with tumour progression and metastasis in multiple cancers. Knockdown of Xist in colorectal cancer has been proven to inhibit cell proliferation, invasion, and epithelial-mesenchymal transition (EMT) [240]. Larger tumour size and advanced stage of tumour are significantly correlated with high expression of Xist. Hence, Xist expression is used to predict the prognosis and survival of colorectal cancer patients [241].
3.3.2. RNA Interaction Assays
Co-sedimentation assays can be used whereby RNA is extracted from cells and fractionated using sucrose or glycerol gradients. The RNAs found in the different fractions are examined by Northern blot. RNAs found in the same gradient fractions are thought to interact with each other [242,243] though it does not directly demonstrate interaction. A more robust experiment would be the electrophoretic mobility shift assay (EMSA), which involves studying interaction of RNA fragments by observing rate of migration of the samples during gel electrophoresis [244]. If the lncRNA–mRNA interacts, the complex would have a larger molecular mass compared to separate strands of RNA. Therefore, the complex would migrate slower on the gel compared to non-paired RNAs. Samples can be extracted from cells or synthesised in vitro. Synthesising of RNA fragments could potentially demonstrate interaction between specific regions of the lncRNA-mRNA complexes. However, these techniques can only screen for a given set of molecules.
Ribonuclease protection assays (RPA) can also be used to detect these sense-antisense RNA duplexes. RPA involves isolation of total RNA followed by RNase and DNase digestion [245,246,247]. Duplexed RNA should be protected from digestion and will be detected by PCR and gel electrophoresis or qRT-PCR. This technique was used to demonstrate interaction between PDCD4-AS1 and PDCD4 mRNA in breast cancer [248].
Microarrays as mentioned earlier have been used to identify the alternative splicing (AS) events regulated by MALAT1 for example. PolyA+ RNA isolated from MALAT1-antisense oligo treated and control HeLa cells were isolated and prepared into labelled cDNA. This was hybridized to a custom AS microarray. The GenASAP algorithm was then used to estimate the percent exon inclusion. Semiquantitative RT-PCR using primers specific for exons flanking the AS events was performed to validate the microarray predictions. This assay revealed that MALAT1 depleted cells have changes in AS of B-MYB and MGEA6 pre-mRNAs [177].
4. Closing Remarks
Throughout this review, we have introduced both computational resources and experimental methods to perform primary and secondary characterisations of lncRNAs to ascertain their potential roles in post-transcriptional regulation and cancer. Primary characterisation establishes basic ground truths relating expression, localisation and relative importance in a model system of choice under normal or perturbed conditions. Secondary characterisation particularly focuses on identifying the interacting RNAs and RBPs that the lncRNAs may influence and importantly deregulate in cancer states due to aberrant expression or non-coding mutations affecting their binding. The RIP and CLIP technologies in particular have been well adapted to next-generation sequencing allowing these newer methods to become reference options for performing high-throughput screening of RNA/Protein or RNA/RNA interactions. Furthermore, a host of improvements to the cross-linking biochemistry have been incorporated and enabled progressive advances in specificity and reliability. It will be important to continue generating these types of experiments and complementing the growing databases dedicated to cataloguing and integrating this information with other valuable sources as presented in Section 2.1, which are already useful starting points for orienting experimental strategies for more novel lncRNAs. The appearance of disease and cancer specialised lncRNA databases and metadatabases will prove highly valuable in integrating the diverse interaction sources and placing them in relevant cancer contexts to identify interesting regulatory patterns or correlations in diseased states.
Some of the findings concerning lncRNA functions in cancer may have direct applications for therapies involving Antisense oligonucleotides (ASOs) for example. ASOs are DNA:RNA chimeras that direct RNase H to degrade target RNAs [249] such as target lncRNAs associated with cancer. During preclinical trials, they were able to target MALAT1 in vivo, resulting in reduced metastasis [250]. Additionally, ASOs can operate through other mechanisms, such as steric blocking of TF binding and modulating splicing [251]. Unfortunately, RNA molecules like lncRNAs are able to form multiple conformations given their intrinsic flexibility [252]. This makes predicting their structure a challenge which could impede the success of targeted regulation of their expression.
As non-coding RNA continues to take on importance in influencing fundamental processes such as post-transcriptional regulation it will be interesting to integrate this knowledge with findings relating to other novel post-transcriptional regulatory mechanisms such as RNA modifications. This field has also benefited from the adoption of CLIP technologies to perform epitranscriptomic studies on some of the hundreds of modifications that are likely to affect RNA stability, structure, localisation and interactions—lncRNAs included [253,254,255]. All of these CLIP-based sequencing methods will continue evolving with the maturation of third-generation sequencing, which is already enabling native RNA sequencing including RNA modification detection and structural footprinting [256,257]. In the near future, it may well be possible to capture RNA interactomes, methylomes and structuromes in single experiments to reveal a more complete landscape of the post-transcriptional regulatory mechanisms susceptible to exploitation by cancers.
Funding
This work is supported by the National Research Foundation (NRF) Singapore, under its Singapore NRF Fellowship (NRF-NRFF2018-04). In addition, J.M. Carter, D.A. Ang and N. Sim are funded by the Nanyang Assistant Professorship (NAP) Start-up-grant to Y.L., from the Nanyang Technological University (NTU), Singapore.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors thank present and past colleagues for inspiring discussions and Aswin Doekhie for critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef] [Green Version]
- 6 Non-coding RNA characterization. Nature 2019. [CrossRef]
- Fabbri, M.; Girnita, L.; Varani, G.; Calin, G.A. Decrypting noncoding RNA interactions, structures, and functional networks. Genome Res. 2019, 29, 1377–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, S.; Lin, S.; Li, Y.; Zhao, W.; Mills, G.B.; Sahni, N. Functional variomics and network perturbation: Connecting genotype to phenotype in cancer. Nat. Rev. Genet. 2017, 18, 395–410. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jewer, M.; Findlay, S.D.; Postovit, L.-M. Post-transcriptional regulation in cancer progression: Microenvironmental control of alternative splicing and translation. J. Cell Commun. Signal. 2012, 6, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaklavas, C.; Blume, S.W.; Grizzle, W.E. Translational Dysregulation in Cancer: Molecular Insights and Potential Clinical Applications in Biomarker Development. Front. Oncol. 2017, 7, 158. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.-J.; Feng, Y.-H.; Gu, B.-H.; Li, Y.-M.; Chen, H. The post-translational modification, SUMOylation, and cancer (Review). Int. J. Oncol. 2018, 52, 1081–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, M. Rate, molecular spectrum, and consequences of human mutation. Proc. Natl. Acad. Sci. USA 2010, 107, 961–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, C.L.; Cenik, C.; Reuter, J.A.; Kiss, G.; Pande, V.S.; Snyder, M.P.; Greenleaf, W.J. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat. Genet. 2016, 48, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Khurana, E.; Fu, Y.; Chakravarty, D.; Demichelis, F.; Rubin, M.A.; Gerstein, M. Role of non-coding sequence variants in cancer. Nat. Rev. Genet. 2016, 17, 93–108. [Google Scholar] [CrossRef]
- Zhou, S.; Treloar, A.E.; Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discov. 2016, 6, 1215–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, H.; Weng, Z. Decoding the non-coding genome: Opportunities and challenges of genomic and epigenomic consortium data. Curr. Opin. Syst. Biol. 2018, 11, 82–90. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Gloss, B.S.; Dinger, M.E. The specificity of long noncoding RNA expression. Biochim. Biophys. Acta 2016, 1859, 16–22. [Google Scholar] [CrossRef]
- Haigis, K.M.; Cichowski, K.; Elledge, S.J. Tissue-specificity in cancer: The rule, not the exception. Science 2019, 363, 1150–1151. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Zuo, Y.; Wang, J.; Zhang, M.Q.; Malhotra, A.; Mayeda, A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006, 34, e63. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- St; St. Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Wenric, S.; ElGuendi, S.; Caberg, J.-H.; Bezzaou, W.; Fasquelle, C.; Charloteaux, B.; Karim, L.; Hennuy, B.; Frères, P.; Collignon, J.; et al. Transcriptome-wide analysis of natural antisense transcripts shows their potential role in breast cancer. Sci. Rep. 2017, 7, 17452. [Google Scholar] [CrossRef] [Green Version]
- Sanchez de Groot, N.; Armaos, A.; Graña-Montes, R.; Alriquet, M.; Calloni, G.; Vabulas, R.M.; Tartaglia, G.G. RNA structure drives interaction with proteins. Nat. Commun. 2019, 10, 3246. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Diederichs, S.; Wang, W.; Böing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar] [CrossRef] [Green Version]
- Matouk, I.J.; DeGroot, N.; Mezan, S.; Ayesh, S.; Abu-lail, R.; Hochberg, A.; Galun, E. The H19 non-coding RNA is essential for human tumor growth. PLoS ONE 2007, 2, e845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Sirchia, S.M.; Tabano, S.; Monti, L.; Recalcati, M.P.; Gariboldi, M.; Grati, F.R.; Porta, G.; Finelli, P.; Radice, P.; Miozzo, M. Misbehaviour of XIST RNA in breast cancer cells. PLoS ONE 2009, 4, e5559. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Tang, S.; Li, M.; Feng, A.; Qin, L.; Liu, Z.; Wang, X. The role of long noncoding RNA HOTAIR in the acquired multidrug resistance to imatinib in chronic myeloid leukemia cells. Hematology 2017, 22, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Yang, Y.A.; Zhang, A.; Fong, K.-W.; Kim, J.; Song, B.; Li, S.; Zhao, J.C.; Yu, J. LncRNA HOTAIR enhances ER signaling and confers tamoxifen resistance in breast cancer. Oncogene 2016, 35, 2746–2755. [Google Scholar] [CrossRef] [Green Version]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. Adv. Exp. Med. Biol. 2017, 1008, 1–46. [Google Scholar] [CrossRef]
- Wang, W.-T.; Han, C.; Sun, Y.-M.; Chen, T.-Q.; Chen, Y.-Q. Noncoding RNAs in cancer therapy resistance and targeted drug development. J. Hematol. Oncol. 2019, 12, 55. [Google Scholar] [CrossRef]
- Chi, Y.; Wang, D.; Wang, J.; Yu, W.; Yang, J. Long Non-Coding RNA in the Pathogenesis of Cancers. Cells 2019, 8, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Du, W.W.; Wu, N.; Yang, W.; Awan, F.M.; Fang, L.; Ma, J.; Li, X.; Zeng, Y.; Yang, Z.; et al. A circular RNA promotes tumorigenesis by inducing c-myc nuclear translocation. Cell Death Differ. 2017, 24, 1609–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, M.; Xiao, Y.; Ma, J.; Tang, Y.; Tian, B.; Zhang, Y.; Li, X.; Wu, Z.; Yang, D.; Zhou, Y.; et al. Circular RNAs in Cancer: Emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol. Cancer 2019, 18, 90. [Google Scholar] [CrossRef]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Matyunina, L.V.; Walker, L.D.; Chen, W.; Xiao, H.; Benigno, B.B.; Wu, R.; McDonald, J.F. Evidence for the importance of post-transcriptional regulatory changes in ovarian cancer progression and the contribution of miRNAs. Sci. Rep. 2017, 7, 8171. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.-H.; Abdelmohsen, K.; Gorospe, M. Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 2014, 34, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Song, J.H.; Cheng, Y.; Abraham, J.M.; Ibrahim, S.; Sun, Z.; Ke, X.; Meltzer, S.J. Long non-coding antisense RNA KRT7-AS is activated in gastric cancers and supports cancer cell progression by increasing KRT7 expression. Oncogene 2016, 35, 4927–4936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, R.-Z.; Luo, D.-X.; Mo, Y.-Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis. 2019, 6, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. J. Hum. Genet. 2020, 65, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardini, A. Global Run-On Sequencing (GRO-Seq). Methods Mol. Biol. 2017, 1468, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahat, D.B.; Kwak, H.; Booth, G.T.; Jonkers, I.H.; Danko, C.G.; Patel, R.K.; Waters, C.T.; Munson, K.; Core, L.J.; Lis, J.T. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 2016, 11, 1455–1476. [Google Scholar] [CrossRef]
- Paulsen, M.T.; Veloso, A.; Prasad, J.; Bedi, K.; Ljungman, E.A.; Magnuson, B.; Wilson, T.E.; Ljungman, M. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods 2014, 67, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Plessy, C.; Bertin, N.; Takahashi, H.; Simone, R.; Salimullah, M.; Lassmann, T.; Vitezic, M.; Severin, J.; Olivarius, S.; Lazarevic, D.; et al. Linking promoters to functional transcripts in small samples with nanoCAGE and CAGEscan. Nat. Methods 2010, 7, 528–534. [Google Scholar] [CrossRef] [Green Version]
- Workman, R.E.; Tang, A.D.; Tang, P.S.; Jain, M.; Tyson, J.R.; Razaghi, R.; Zuzarte, P.C.; Gilpatrick, T.; Payne, A.; Quick, J.; et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat. Methods 2019, 16, 1297–1305. [Google Scholar] [CrossRef]
- Raha, D.; Hong, M.; Snyder, M. ChIP-Seq: A method for global identification of regulatory elements in the genome. Curr. Protoc. Mol. Biol. 2010, 21. [Google Scholar] [CrossRef]
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962. [Google Scholar] [CrossRef] [Green Version]
- Antonov, I.V.; Mazurov, E.; Borodovsky, M.; Medvedeva, Y.A. Prediction of lncRNAs and their interactions with nucleic acids: Benchmarking bioinformatics tools. Brief. Bioinform. 2019, 20, 551–564. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Liu, C. The computational approaches of lncRNA identification based on coding potential: Status quo and challenges. Comput. Struct. Biotechnol. J. 2020, 18, 3666–3677. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, W.; Guo, M.; Zou, Q. A comprehensive overview and evaluation of circular RNA detection tools. PLoS Comput. Biol. 2017, 13, e1005420. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Salzman, J. Detecting circular RNAs: Bioinformatic and experimental challenges. Nat. Rev. Genet. 2016, 17, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Zhang, J.; Zhao, F. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018, 19, 803–810. [Google Scholar] [CrossRef]
- Zou, D.; Ma, L.; Yu, J.; Zhang, Z. Biological databases for human research. Genom. Proteom. Bioinform. 2015, 13, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tassé, A.-M.; Flicek, P. The international Genome sample resource (IGSR): A worldwide collection of genome variation incorporating the 1000 Genomes Project data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ENCODE Project Consortium; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Geo NCBI-GEO. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/geo/ (accessed on 29 January 2021).
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar]
- The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [CrossRef] [Green Version]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ (accessed on 29 January 2021).
- GTEx Portal. Available online: https://gtexportal.org/home/ (accessed on 29 January 2021).
- Lizio, M.; Abugessaisa, I.; Noguchi, S.; Kondo, A.; Hasegawa, A.; Hon, C.C.; de Hoon, M.; Severin, J.; Oki, S.; Hayashizaki, Y.; et al. Update of the FANTOM web resource: Expansion to provide additional transcriptome atlases. Nucleic Acids Res. 2019, 47, D752–D758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broad Institute Cancer Cell Line Encyclopedia (CCLE). Available online: https://portals.broadinstitute.org/ccle (accessed on 29 January 2021).
- Schriml, L.M.; Mitraka, E.; Munro, J.; Tauber, B.; Schor, M.; Nickle, L.; Felix, V.; Jeng, L.; Bearer, C.; Lichenstein, R.; et al. Human Disease Ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Res. 2019, 47, D955–D962. [Google Scholar] [CrossRef] [Green Version]
- Gene Ontology Consortium the Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- NCBI-MeSH. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/mesh/ (accessed on 29 January 2021).
- GenomeOC Therapeutically Applicable Research to Generate Effective Treatments. Available online: https://ocg.cancer.gov/programs/target (accessed on 29 January 2021).
- Li, J.-H.; Liu, S.; Zhou, H.; Qu, L.-H.; Yang, J.-H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef]
- Blin, K.; Dieterich, C.; Wurmus, R.; Rajewsky, N.; Landthaler, M.; Akalin, A. DoRiNA 2.0--upgrading the doRiNA database of RNA interactions in post-transcriptional regulation. Nucleic Acids Res. 2015, 43, D160–D167. [Google Scholar] [CrossRef] [Green Version]
- Jeggari, A.; Marks, D.S.; Larsson, E. miRcode: A map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Lin, Y.-C.-D.; Li, J.; Huang, K.-Y.; Shrestha, S.; Hong, H.-C.; Tang, Y.; Chen, Y.-G.; Jin, C.-N.; Yu, Y.; et al. miRTarBase 2020: Updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020, 48, D148–D154. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.; Shi, J.; Gao, Y.; Cui, C.; Zhang, S.; Li, J.; Zhou, Y.; Cui, Q. HMDD v3.0: A database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019, 47, D1013–D1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Gu, J.; Wang, T.; Ding, Z. OncomiRDB: A database for the experimentally verified oncogenic and tumor-suppressive microRNAs. Bioinformatics 2014, 30, 2237–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Wu, L.; Wang, A.; Tang, W.; Zhao, Y.; Zhao, H.; Teschendorff, A.E. dbDEMC 2.0: Updated database of differentially expressed miRNAs in human cancers. Nucleic Acids Res. 2017, 45, D812–D818. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009, 37, D105–D110. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, J.; Li, Y.; Song, T.; Wu, Y.; Fang, S.; Bu, D.; Li, H.; Sun, L.; Pei, D.; et al. NONCODEV6: An updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res. 2021, 49, D165–D171. [Google Scholar] [CrossRef]
- Volders, P.-J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Li, A.; Zou, D.; Xu, X.; Xia, L.; Yu, J.; Bajic, V.B.; Zhang, Z. LncRNAWiki: Harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 2015, 43, D187–D192. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D128–D134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Shang, S.; Guo, S.; Li, X.; Zhou, H.; Liu, H.; Sun, Y.; Wang, J.; Wang, P.; Zhi, H.; et al. Lnc2Cancer 3.0: An updated resource for experimentally supported lncRNA/circRNA cancer associations and web tools based on RNA-seq and scRNA-seq data. Nucleic Acids Res. 2021, 49, D1251–D1258. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: An updated database of long non-coding RNA-associated diseases. Nucleic Acids Res. 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Wang, Z.; Pan, T.; Sahni, N.; Jin, X.; Wang, G.; Li, J.; Zheng, X.; Zhang, Y.; et al. LncMAP: Pan-cancer atlas of long noncoding RNA-mediated transcriptional network perturbations. Nucleic Acids Res. 2018, 46, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Cui, T.; Zheng, B.; Wang, N.; Luo, J.; Yang, B.; Du, M.; Cheng, J.; Dou, Y.; Wang, D. MNDR v3.0: Mammal ncRNA-disease repository with increased coverage and annotation. Nucleic Acids Res. 2021, 49, D160–D164. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Liu, W.; Zhang, Q.; Guo, A.-Y. lncRNASNP2: An updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018, 46, D276–D280. [Google Scholar] [CrossRef]
- Chan, W.-L.; Huang, H.-D.; Chang, J.-G. lncRNAMap: A map of putative regulatory functions in the long non-coding transcriptome. Comput. Biol. Chem. 2014, 50, 41–49. [Google Scholar] [CrossRef]
- Zhao, H.; Shi, J.; Zhang, Y.; Xie, A.; Yu, L.; Zhang, C.; Lei, J.; Xu, H.; Leng, Z.; Li, T.; et al. LncTarD: A manually-curated database of experimentally-supported functional lncRNA-target regulations in human diseases. Nucleic Acids Res. 2020, 48, D118–D126. [Google Scholar] [CrossRef]
- Zhou, B.; Zhao, H.; Yu, J.; Guo, C.; Dou, X.; Song, F.; Hu, G.; Cao, Z.; Qu, Y.; Yang, Y.; et al. EVLncRNAs: A manually curated database for long non-coding RNAs validated by low-throughput experiments. Nucleic Acids Res. 2018, 46, D100–D105. [Google Scholar] [CrossRef] [Green Version]
- Lv, D.; Xu, K.; Jin, X.; Li, J.; Shi, Y.; Zhang, M.; Jin, X.; Li, Y.; Xu, J.; Li, X. LncSpA: LncRNA Spatial Atlas of Expression across Normal and Cancer Tissues. Cancer Res. 2020, 80, 2067–2071. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Ji, P.; Zhao, F. CircAtlas: An integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020, 21, 101. [Google Scholar] [CrossRef]
- Chen, X.; Han, P.; Zhou, T.; Guo, X.; Song, X.; Li, Y. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016, 6, 34985. [Google Scholar] [CrossRef]
- Meng, X.; Hu, D.; Zhang, P.; Chen, Q.; Chen, M. CircFunBase: A database for functional circular RNAs. Database 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Glažar, P.; Papavasileiou, P.; Rajewsky, N. circBase: A database for circular RNAs. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [Green Version]
- Maass, P.G.; Glažar, P.; Memczak, S.; Dittmar, G.; Hollfinger, I.; Schreyer, L.; Sauer, A.V.; Toka, O.; Aiuti, A.; Luft, F.C.; et al. A map of human circular RNAs in clinically relevant tissues. J. Mol. Med. 2017, 95, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, Q.; Shen, J.; Yang, B.B.; Ding, X. Circbank: A comprehensive database for circRNA with standard nomenclature. RNA Biol. 2019, 16, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Ma, X.-K.; Li, G.-W.; Yang, L. CIRCpedia v2: An Updated Database for Comprehensive Circular RNA Annotation and Expression Comparison. Genom. Proteom. Bioinform. 2018, 16, 226–233. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, K.; Wu, F.; Wang, W.; Zhang, K.; Hu, H.; Liu, Y.; Jiang, T. circRNA disease: A manually curated database of experimentally supported circRNA-disease associations. Cell Death Dis. 2018, 9, 475. [Google Scholar] [CrossRef]
- Fan, C.; Lei, X.; Fang, Z.; Jiang, Q.; Wu, F.-X. CircR2Disease: A manually curated database for experimentally supported circular RNAs associated with various diseases. Database 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Feng, J.; Lei, L.; Hu, J.; Xia, L.; Wang, J.; Xiang, Y.; Liu, L.; Zhong, S.; Han, L.; et al. Comprehensive characterization of tissue-specific circular RNAs in the human and mouse genomes. Brief. Bioinform. 2017, 18, 984–992. [Google Scholar] [CrossRef] [Green Version]
- Rophina, M.; Sharma, D.; Poojary, M.; Scaria, V. Circad: A comprehensive manually curated resource of circular RNA associated with diseases. Database 2020, 2020. [Google Scholar] [CrossRef]
- Zhao, M.; Qu, H. circVAR database: Genome-wide archive of genetic variants for human circular RNAs. BMC Genom. 2020, 21, 750. [Google Scholar] [CrossRef]
- Xia, S.; Feng, J.; Chen, K.; Ma, Y.; Gong, J.; Cai, F.; Jin, Y.; Gao, Y.; Xia, L.; Chang, H.; et al. CSCD: A database for cancer-specific circular RNAs. Nucleic Acids Res. 2018, 46, D925–D929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosal, S.; Das, S.; Sen, R.; Basak, P.; Chakrabarti, J. Circ2Traits: A comprehensive database for circular RNA potentially associated with disease and traits. Front. Genet. 2013, 4, 283. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Yao, D.; Zhang, L.; Zheng, M.; Sun, X.; Lu, Y.; Liu, P. Circ2Disease: A manually curated database of experimentally validated circRNAs in human disease. Sci. Rep. 2018, 8, 11018. [Google Scholar] [CrossRef] [Green Version]
- Dudekula, D.B.; Panda, A.C.; Grammatikakis, I.; De, S.; Abdelmohsen, K.; Gorospe, M. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016, 13, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kevil, C.G.; Walsh, L.; Laroux, F.S.; Kalogeris, T.; Grisham, M.B.; Alexander, J.S. An improved, rapid Northern protocol. Biochem. Biophys. Res. Commun. 1997, 238, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Feng, Y.; Hu, Z.; Zhang, Y.; Yuan, C.-X.; Xu, X.; Zhang, L. Detection of Long Noncoding RNA Expression by Nonradioactive Northern Blots. In Long Non-Coding RNAs: Methods and Protocols; Feng, Y., Zhang, L., Eds.; Springer: New York, NY, USA, 2016; pp. 177–188. ISBN 9781493933785. [Google Scholar]
- Streit, S.; Michalski, C.W.; Erkan, M.; Kleeff, J.; Friess, H. Northern blot analysis for detection and quantification of RNA in pancreatic cancer cells and tissues. Nat. Protoc. 2009, 4, 37–43. [Google Scholar] [CrossRef]
- Zang, S.; Lin, R.-J. Northwestern Blot Analysis: Detecting RNA–Protein Interaction After Gel Separation of Protein Mixture. In RNA-Protein Complexes and Interactions: Methods and Protocols; Lin, R.-J., Ed.; Springer: New York, NY, USA, 2016; pp. 111–125. ISBN 9781493935918. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Choi, S.; Oh, J.; Lee, S.-G.; Choi, S.Y.; Kim, K.K.; Park, C. Conventionally used reference genes are not outstanding for normalization of gene expression in human cancer research. BMC Bioinform. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivarius, S.; Plessy, C.; Carninci, P. High-throughput verification of transcriptional starting sites by Deep-RACE. Biotechniques 2009, 46, 130–132. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, J.; Uszczynska-Ratajczak, B.; Santoyo-Lopez, J.; Gonzalez, J.M.; Tapanari, E.; Mudge, J.M.; Steward, C.A.; Wilming, L.; Tanzer, A.; Howald, C.; et al. Extension of human lncRNA transcripts by RACE coupled with long-read high-throughput sequencing (RACE-Seq). Nat. Commun. 2016, 7, 12339. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.R.; Orjalo, A.V.; Semaan, S.J.; Johansson, H.E. Simultaneous Detection of Nuclear and Cytoplasmic RNA Variants Utilizing Stellaris® RNA Fluorescence In Situ Hybridization in Adherent Cells. In In Situ Hybridization Protocols; Nielsen, B.S., Ed.; Springer: New York, NY, USA, 2014; pp. 189–199. ISBN 9781493914593. [Google Scholar]
- Orjalo, A.V.; Johansson, H.E. Stellaris® RNA Fluorescence In Situ Hybridization for the Simultaneous Detection of Immature and Mature Long Noncod-ing RNAs in Adherent Cells. In Long Non-Coding RNAs: Methods and Protocols; Feng, Y., Zhang, L., Eds.; Springer: New York, NY, USA, 2016; pp. 119–134. ISBN 9781493933785. [Google Scholar]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Maamar, H.; Cabili, M.N.; Rinn, J.; Raj, A. linc-HOXA1 is a noncoding RNA that represses Hoxa1 transcription in cis. Genes Dev. 2013, 27, 1260–1271. [Google Scholar] [CrossRef] [Green Version]
- Dunagin, M.; Cabili, M.N.; Rinn, J.; Raj, A. Visualization of lncRNA by Single-Molecule Fluorescence In Situ Hybridization. In Nuclear Bodies and Noncoding RNAs: Methods and Protocols; Nakagawa, S., Hirose, T., Eds.; Springer: New York, NY, USA, 2015; pp. 3–19. ISBN 9781493922536. [Google Scholar]
- Mortimer, S.A.; Weeks, K.M. A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J. Am. Chem. Soc. 2007, 129, 4144–4145. [Google Scholar] [CrossRef] [PubMed]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- Smola, M.J.; Rice, G.M.; Busan, S.; Siegfried, N.A.; Weeks, K.M. Selective 2’-hydroxyl acylation analyzed by primer extension and mutational profiling (SHAPE-MaP) for direct, versatile and accurate RNA structure analysis. Nat. Protoc. 2015, 10, 1643–1669. [Google Scholar] [CrossRef]
- Schmidt, K.; Weidmann, C.A.; Hilimire, T.A.; Yee, E.; Hatfield, B.M.; Schneekloth, J.S., Jr.; Weeks, K.M.; Novina, C.D. Targeting the Oncogenic Long Non-coding RNA SLNCR1 by Blocking Its Sequence-Specific Binding to the Androgen Receptor. Cell Rep. 2020, 30, 541–554.e5. [Google Scholar] [CrossRef] [Green Version]
- Shields, E.J.; Petracovici, A.F.; Bonasio, R. lncRedibly versatile: Biochemical and biological functions of long noncoding RNAs. Biochem. J. 2019, 476, 1083–1104. [Google Scholar] [CrossRef]
- Mishra, K.; Kanduri, C. Understanding Long Noncoding RNA and Chromatin Interactions: What We Know So Far. Noncoding RNA 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Long, Y.; Kwoh, C.K. Deep learning based DNA:RNA triplex forming potential prediction. BMC Bioinform. 2020, 21, 522. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C.; Hänzelmann, S.; Sentürk Cetin, N.; Frank, S.; Zajzon, B.; Derks, J.-P.; Akhade, V.S.; Ahuja, G.; Kanduri, C.; Grummt, I.; et al. Detection of RNA-DNA binding sites in long noncoding RNAs. Nucleic Acids Res. 2019, 47, e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, T.; Al-Absi, H.R.H.; Schmeier, S. Deep Learning in LncRNAome: Contribution, Challenges, and Perspectives. Noncoding RNA 2020, 6. [Google Scholar] [CrossRef]
- Iwakiri, J.; Hamada, M.; Asai, K. Bioinformatics tools for lncRNA research. Biochim. Biophys. Acta 2016, 1859, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Terai, G.; Iwakiri, J.; Kameda, T.; Hamada, M.; Asai, K. Comprehensive prediction of lncRNA-RNA interactions in human transcriptome. BMC Genom. 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakiri, J.; Terai, G.; Hamada, M. Computational prediction of lncRNA-mRNA interactionsby integrating tissue specificity in human transcriptome. Biol. Direct 2017, 12, 15. [Google Scholar] [CrossRef] [Green Version]
- Mar-Aguilar, F.; Rodríguez-Padilla, C.; Reséndez-Pérez, D. Web-based tools for microRNAs involved in human cancer (Review). Oncol. Lett. 2016, 11, 3563–3570. [Google Scholar] [CrossRef] [Green Version]
- Antonov, I.; Marakhonov, A.; Zamkova, M.; Medvedeva, Y. ASSA: Fast identification of statistically significant interactions between long RNAs. J. Bioinform. Comput. Biol. 2018, 16, 1840001. [Google Scholar] [CrossRef]
- Fukunaga, T.; Hamada, M. RIblast: An ultrafast RNA-RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics 2017, 33, 2666–2674. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Le, T.D.; Liu, L.; Li, J. Inferring and analyzing module-specific lncRNA-mRNA causal regulatory networks in human cancer. Brief. Bioinform. 2019, 20, 1403–1419. [Google Scholar] [CrossRef]
- Pyfrom, S.C.; Luo, H.; Payton, J.E. PLAIDOH: A novel method for functional prediction of long non-coding RNAs identifies cancer-specific LncRNA activities. BMC Genom. 2019, 20, 137. [Google Scholar] [CrossRef]
- Gawronski, A.R.; Uhl, M.; Zhang, Y.; Lin, Y.-Y.; Niknafs, Y.S.; Ramnarine, V.R.; Malik, R.; Feng, F.; Chinnaiyan, A.M.; Collins, C.C.; et al. MechRNA: Prediction of lncRNA mechanisms from RNA-RNA and RNA-protein interactions. Bioinformatics 2018, 34, 3101–3110. [Google Scholar] [CrossRef] [Green Version]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef]
- NCBI-SRA. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/sra (accessed on 29 January 2021).
- Babbi, G.; Martelli, P.L.; Profiti, G.; Bovo, S.; Savojardo, C.; Casadio, R. eDGAR: A database of Disease-Gene Associations with annotated Relationships among genes. BMC Genom. 2017, 18, 554. [Google Scholar] [CrossRef]
- Junge, A.; Refsgaard, J.C.; Garde, C.; Pan, X.; Santos, A.; Alkan, F.; Anthon, C.; von Mering, C.; Workman, C.T.; Jensen, L.J.; et al. RAIN: RNA-protein Association and Interaction Networks. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, Y.; Zhao, Y.; Li, C.; Zhang, L.; Huang, H.; Li, Y.; Liu, L.; Hou, P.; Cui, T.; Tan, P.; et al. RAID v2.0: An updated resource of RNA-associated interactions across organisms. Nucleic Acids Res. 2017, 45, D115–D118. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Chen, X.; Xue, H.; Tang, Y.; Zhang, P.; Kang, Q.; Hao, Y.; Chen, R.; Zhao, Y.; He, S. NPInter v4.0: An integrated database of ncRNA interactions. Nucleic Acids Res. 2020, 48, D160–D165. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Shao, D.; Xu, K.; Lu, Z.; Lu, Z.J.; Yang, Y.T.; Zhang, Q.C. RISE: A database of RNA interactome from sequencing experiments. Nucleic Acids Res. 2018, 46, D194–D201. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the development of miRNA bioinformatics tools. Brief. Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, T.; Iwakiri, J.; Ono, Y.; Hamada, M. LncRRIsearch: A Web Server for lncRNA-RNA Interaction Prediction Integrated With Tissue-Specific Expression and Subcellular Localization Data. Front. Genet. 2019, 10, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagkouni, D.; Paraskevopoulou, M.D.; Tastsoglou, S.; Skoufos, G.; Karavangeli, A.; Pierros, V.; Zacharopoulou, E.; Hatzigeorgiou, A.G. DIANA-LncBase v3: Indexing experimentally supported miRNA targets on non-coding transcripts. Nucleic Acids Res. 2020, 48, D101–D110. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Pachl, E.; Hartung, M.; Stiegler, V.; Baumbach, J.; Schulz, M.H.; List, M. SPONGEdb: A pan-cancer resource for competing endogenous RNA interactions. NAR Cancer 2021, 3. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Gao, Y.; Guo, Q.; Ning, S.; Zhang, Y.; Shang, S.; Wang, J.; Wang, Y.; Zhi, H.; et al. LnCeVar: A comprehensive database of genomic variations that disturb ceRNA network regulation. Nucleic Acids Res. 2020, 48, D111–D117. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhi, H.; Zhang, Y.; Liu, Y.; Zhang, J.; Gao, Y.; Guo, M.; Ning, S.; Li, X. miRSponge: A manually curated database for experimentally supported miRNA sponges and ceRNAs. Database 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Liu, T.; Cui, T.; Wang, Z.; Zhang, Y.; Tan, P.; Huang, Y.; Yu, J.; Wang, D. RNAInter in 2020: RNA interactome repository with increased coverage and annotation. Nucleic Acids Res. 2020, 48, D189–D197. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, P.; Tian, R.; Wang, S.; Guo, Q.; Luo, M.; Zhou, W.; Liu, G.; Jiang, H.; Jiang, Q. LncRNA2Target v2.0: A comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019, 47, D140–D144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, L.; Jiang, S.; Li, Q.; Feng, C.; Du, Q.; Zou, D.; Xiao, J.; Zhang, Z.; Ma, L. LncExpDB: An expression database of human long non-coding RNAs. Nucleic Acids Res. 2021, 49, D962–D968. [Google Scholar] [CrossRef]
- Wang, P.; Li, X.; Gao, Y.; Guo, Q.; Wang, Y.; Fang, Y.; Ma, X.; Zhi, H.; Zhou, D.; Shen, W.; et al. LncACTdb 2.0: An updated database of experimentally supported ceRNA interactions curated from low- and high-throughput experiments. Nucleic Acids Res. 2019, 47, D121–D127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhang, Y.; Lu, J. The roles of long noncoding RNAs in breast cancer metastasis. Cell Death Dis. 2020, 11, 749. [Google Scholar] [CrossRef]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Chen, S.; Li, W.; Chen, W.; Gu, W. LncRNA MACC1-AS1 sponges multiple miRNAs and RNA-binding protein PTBP1. Oncogenesis 2019, 8, 73. [Google Scholar] [CrossRef]
- Mili, S.; Steitz, J.A. Evidence for reassociation of RNA-binding proteins after cell lysis: Implications for the interpretation of immunoprecipitation analyses. RNA 2004, 10, 1692–1694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, K.J.; Yario, T.A.; Steitz, J.A. Association of Argonaute proteins and microRNAs can occur after cell lysis. RNA 2012, 18, 1581–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.; Maeda, S.; Liu, C.; Karin, M.; Edgington, T.S. A large noncoding RNA is a marker for murine hepatocellular carcinomas and a spectrum of human carcinomas. Oncogene 2007, 26, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamm, S. Regulation of Alternative Splicing by Reversible Protein Phosphorylation. J. Biol. Chem. 2008, 283, 1223–1227. [Google Scholar] [CrossRef] [Green Version]
- Calarco, J.A.; Superina, S.; O’Hanlon, D.; Gabut, M.; Raj, B.; Pan, Q.; Skalska, U.; Clarke, L.; Gelinas, D.; van der Kooy, D.; et al. Regulation of vertebrate nervous system alternative splicing and development by an SR-related protein. Cell 2009, 138, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, C.F.; Lejeune, F.; Stévenin, J. Broad Specificity of SR (Serine/Arginine) Proteins in the Regulation of Alternative Splicing of Pre-Messenger RNA. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2004; Volume 78, pp. 37–88. [Google Scholar]
- Wang, D.; Ding, L.; Wang, L.; Zhao, Y.; Sun, Z.; Karnes, R.J.; Zhang, J.; Huang, H. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget 2015, 6, 41045–41055. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Bryant, R.J.; Cross, N.A.; Eaton, C.L.; Hamdy, F.C.; Cunliffe, V.T. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate 2007, 67, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Saramäki, O.R.; Tammela, T.L.J.; Martikainen, P.M.; Vessella, R.L.; Visakorpi, T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer 2006, 45, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D.; et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Haberman, N.; Huppertz, I.; Attig, J.; König, J.; Wang, Z.; Hauer, C.; Hentze, M.W.; Kulozik, A.E.; Le Hir, H.; Curk, T.; et al. Insights into the design and interpretation of iCLIP experiments. Genome Biol. 2017, 18, 7. [Google Scholar] [CrossRef] [Green Version]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Vigilante, A.; Darbo, E.; Zirra, A.; Militti, C.; D’Ambrogio, A.; Luscombe, N.M.; Ule, J. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 2015, 519, 491–494. [Google Scholar] [CrossRef] [Green Version]
- Urlaub, H.; Hartmuth, K.; Lührmann, R. A two-tracked approach to analyze RNA-protein crosslinking sites in native, nonlabeled small nuclear ribonucleoprotein particles. Methods 2002, 26, 170–181. [Google Scholar] [CrossRef]
- König, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.-Q.; Li, Z.-X.; Zhou, G.-Q.; Lin, L.; Zhang, L.-L.; Lv, J.-W.; Huang, X.-D.; Liu, R.-Q.; Chen, F.; He, X.-J.; et al. Long Noncoding RNA FAM225A Promotes Nasopharyngeal Carcinoma Tumorigenesis and Metastasis by Acting as ceRNA to Sponge miR-590-3p/miR-1275 and Upregulate ITGB3. Cancer Res. 2019, 79, 4612–4626. [Google Scholar] [CrossRef]
- Petri, R.; Jakobsson, J. Identifying miRNA Targets Using AGO-RIPseq. In mRNA Decay: Methods and Protocols; Lamandé, S.R., Ed.; Springer: New York, NY, USA, 2018; pp. 131–140. ISBN 9781493975402. [Google Scholar]
- Chi, S.W.; Zang, J.B.; Mele, A.; Darnell, R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 2009, 460, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Pollum, M.; Jockusch, S.; Crespo-Hernández, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.P.; Rajapakshe, K.I.; Bader, D.A.; Cerne, J.Z.; Smith, E.A.; Coarfa, C.; Hartig, S.M.; McGuire, S.E. The Landscape of microRNA Targeting in Prostate Cancer Defined by AGO-PAR-CLIP. Neoplasia 2016, 18, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Orom, U.A.; Lund, A.H. Isolation of microRNA targets using biotinylated synthetic microRNAs. Methods 2007, 43, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.-J.; Yang, H.-J.; Tsai, H.-J. Labeled microRNA pull-down assay system: An experimental approach for high-throughput identification of microRNA-target mRNAs. Nucleic Acids Res. 2009, 37, e77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baigude, H.; Li, Z.; Zhou, Y.; Rana, T.M. miR-TRAP: A benchtop chemical biology strategy to identify microRNA targets. Angew. Chem. Int. Ed. 2012, 51, 5880–5883. [Google Scholar] [CrossRef]
- Su, Z.; Ganbold, T.; Baigude, H. Analysis and Identification of Tumorigenic Targets of MicroRNA in Cancer Cells by Photoreactive Chemical Probes. Int. J. Mol. Sci. 2020, 21, 1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Huang, L.; Xiao, X.; Chen, Y.; Wang, X.; Zhou, Z.; Zhang, C.; Zhang, Y. Photoclickable MicroRNA for the Intracellular Target Identification of MicroRNAs. J. Am. Chem. Soc. 2016, 138, 15943–15949. [Google Scholar] [CrossRef]
- Zhang, P.; Fu, H.; Du, S.; Wang, F.; Yang, J.; Cai, W.; Liu, D. Click RNA for Rapid Capture and Identification of Intracellular MicroRNA Targets. Anal. Chem. 2019, 91, 15740–15747. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Y.; Li, J.; Zhang, Y. A photoactivatable microRNA probe for identification of microRNA targets and light-controlled suppression of microRNA target expression. Chem. Commun. 2020, 56, 627–630. [Google Scholar] [CrossRef]
- Xu, Y.; Chen, Y.; Li, D.; Liu, Q.; Xuan, Z.; Li, W.-H. TargetLink, a new method for identifying the endogenous target set of a specific microRNA in intact living cells. RNA Biol. 2017, 14, 259–274. [Google Scholar] [CrossRef] [Green Version]
- Vencken, S.; Hassan, T.; McElvaney, N.G.; Smith, S.G.J.; Greene, C.M. miR-CATCH: MicroRNA Capture Affinity Technology. In RNA Interference: Challenges and Therapeutic Opportunities; Sioud, M., Ed.; Springer: New York, NY, USA, 2015; pp. 365–373. ISBN 9781493915385. [Google Scholar]
- Hassan, T.; Smith, S.G.J.; Gaughan, K.; Oglesby, I.K.; O’Neill, S.; McElvaney, N.G.; Greene, C.M. Isolation and identification of cell-specific microRNAs targeting a messenger RNA using a biotinylated anti-sense oligonucleotide capture affinity technique. Nucleic Acids Res. 2013, 41, e71. [Google Scholar] [CrossRef] [Green Version]
- Imig, J.; Brunschweiger, A.; Brümmer, A.; Guennewig, B.; Mittal, N.; Kishore, S.; Tsikrika, P.; Gerber, A.P.; Zavolan, M.; Hall, J. miR-CLIP capture of a miRNA targetome uncovers a lincRNA H19-miR-106a interaction. Nat. Chem. Biol. 2015, 11, 107–114. [Google Scholar] [CrossRef]
- De Santi, C.; Vencken, S.; Blake, J.; Haase, B.; Benes, V.; Gemignani, F.; Landi, S.; Greene, C.M. Identification of MiR-21-5p as a Functional Regulator of Mesothelin Expression Using MicroRNA Capture Affinity Coupled with Next Generation Sequencing. PLoS ONE 2017, 12, e0170999. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Ueno, Y. Diazirine-containing RNA photo-cross-linking probes for capturing microRNA targets. J. Org. Chem. 2014, 79, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Minami, K.; Akao, Y.; Ueno, Y. Labeling of target mRNAs using a photo-reactive microRNA probe. Chem. Commun. 2016, 52, 6720–6722. [Google Scholar] [CrossRef]
- Nakamoto, K.; Akao, Y.; Ueno, Y. Diazirine-containing tag-free RNA probes for efficient RISC-loading and photoaffinity labeling of microRNA targets. Bioorg. Med. Chem. Lett. 2018, 28, 2906–2909. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, B.; Pollum, M.; Crespo-Hernández, C.E. Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases. Photochem. Photobiol. 2019, 95, 33–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziuba, D.; Hoffmann, J.-E.; Hentze, M.W.; Schultz, C. A Genetically Encoded Diazirine Analogue for RNA-Protein Photo-crosslinking. Chembiochem 2020, 21, 88–93. [Google Scholar] [CrossRef]
- Kretz, M.; Siprashvili, Z.; Chu, C.; Webster, D.E.; Zehnder, A.; Qu, K.; Lee, C.S.; Flockhart, R.J.; Groff, A.F.; Chow, J.; et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature 2013, 493, 231–235. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Panda, A.C.; Kang, M.-J.; Guo, R.; Kim, J.; Grammatikakis, I.; Yoon, J.-H.; Dudekula, D.B.; Noh, J.H.; Yang, X.; et al. 7SL RNA represses p53 translation by competing with HuR. Nucleic Acids Res. 2014, 42, 10099–10111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3’ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faghihi, M.A.; Modarresi, F.; Khalil, A.M.; Wood, D.E.; Sahagan, B.G.; Morgan, T.E.; Finch, C.E.; St Laurent, G., 3rd; Kenny, P.J.; Wahlestedt, C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat. Med. 2008, 14, 723–730. [Google Scholar] [CrossRef] [Green Version]
- Helwak, A.; Tollervey, D. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH). Nat. Protoc. 2014, 9, 711–728. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.C.; Cao, X.; Yu, P.; Xiao, S.; Lu, J.; Biase, F.H.; Sridhar, B.; Huang, N.; Zhang, K.; Zhong, S. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 2016, 7, 12023. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Liu, Y.; Lin, L.; Huang, Q.; He, W.; Zhang, S.; Dong, S.; Wen, Z.; Rao, J.; Liao, W.; et al. The lncRNA MACC1-AS1 promotes gastric cancer cell metabolic plasticity via AMPK/Lin28 mediated mRNA stability of MACC1. Mol. Cancer 2018, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Damas, N.D.; Marcatti, M.; Côme, C.; Christensen, L.L.; Nielsen, M.M.; Baumgartner, R.; Gylling, H.M.; Maglieri, G.; Rundsten, C.F.; Seemann, S.E.; et al. SNHG5 promotes colorectal cancer cell survival by counteracting STAU1-mediated mRNA destabilization. Nat. Commun. 2016, 7, 13875. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Gong, J.; Zhang, Q.C. PARIS: Psoralen Analysis of RNA Interactions and Structures with High Throughput and Resolution. Methods Mol. Biol. 2018, 1649, 59–84. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, E.; Sterne-Weiler, T.; O’Hanlon, D.; Blencowe, B.J. Global Mapping of Human RNA-RNA Interactions. Mol. Cell 2016, 62, 618–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvet, J.P.; Pederson, T. Photochemical cross-linking of secondary structure in HeLa cell heterogeneous nuclear RNA in situ 1. Nucleic Acids Res. 1979, 6, 1993–2001. [Google Scholar] [CrossRef] [Green Version]
- Aw, J.G.A.; Shen, Y.; Wilm, A.; Sun, M.; Lim, X.N.; Boon, K.-L.; Tapsin, S.; Chan, Y.-S.; Tan, C.-P.; Sim, A.Y.L.; et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 2016, 62, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-seq for global in situ profiling of RNA-RNA spatial interactions. Nature 2020, 582, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Szostak, J.W. A simple method for 3’-labeling of RNA. Nucleic Acids Res. 1996, 24, 4360–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimino, G.D.; Gamper, H.B.; Isaacs, S.T.; Hearst, J.E. Psoralens as photoactive probes of nucleic acid structure and function: Organic chemistry, photochemistry, and biochemistry. Annu. Rev. Biochem. 1985, 54, 1151–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhong, S.; Luo, Y.; Zou, D.; Li, M.; Li, Y.; Lu, Y.; Miao, S.; Wang, L.; Song, W. A long noncoding RNA binding to QKI-5 regulates germ cell apoptosis via p38 MAPK signaling pathway. Cell Death Dis. 2019, 10, 699. [Google Scholar] [CrossRef]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302–307. [Google Scholar] [CrossRef]
- Siprashvili, Z.; Webster, D.E.; Kretz, M.; Johnston, D.; Rinn, J.L.; Chang, H.Y.; Khavari, P.A. Identification of proteins binding coding and non-coding human RNAs using protein microarrays. BMC Genom. 2012, 13, 633. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wang, Y.; Xu, Y.; He, X.; Li, Y. Silencing the long noncoding RNA, TINCR, a molecular sponge of miR-335, inhibits the malignant phenotype of epithelial ovarian cancer via FGF2 suppression. Int. J. Oncol. 2019, 55, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Tian, G.; Cheung, H.-H.; Wei, W.; Lee, T.-L. Gas5 is an essential lncRNA regulator for self-renewal and pluripotency of mouse embryonic stem cells and induced pluripotent stem cells. Stem Cell Res. Ther. 2018, 9, 71. [Google Scholar] [CrossRef]
- Xing, Z.; Lin, A.; Li, C.; Liang, K.; Wang, S.; Liu, Y.; Park, P.K.; Qin, L.; Wei, Y.; Hawke, D.H.; et al. lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 2014, 159, 1110–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, L.-J.; Ju, H.-Q.; Liu, G.-P.; Tian, T.; Ma, G.-L.; Lu, Y.-X.; Liu, Z.-X.; Pan, R.-L.; Li, R.-H.; Piao, H.-L.; et al. LncRNA CamK-A Regulates Ca2+-Signaling-Mediated Tumor Microenvironment Remodeling. Mol. Cell 2018, 72, 71–83. [Google Scholar] [CrossRef] [Green Version]
- Mann, M. Functional and quantitative proteomics using SILAC. Nat. Rev. Mol. Cell Biol. 2006, 7, 952–958. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.-L.; Chen, L.-Z.; Lu, Y.-X.; Zhang, D.-S.; Zeng, Z.-L.; Pan, Z.-Z.; Huang, P.; Wang, F.-H.; Li, Y.-H.; Ju, H.-Q.; et al. Long noncoding RNA XIST expedites metastasis and modulates epithelial-mesenchymal transition in colorectal cancer. Cell Death Dis. 2017, 8, e3011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-T.; Pan, S.-X.; Wang, A.-H.; Kong, Q.-Y.; Jiang, K.-T.; Yu, Z.-B. Long Non-Coding RNA (lncRNA) X-Inactive Specific Transcript (XIST) Plays a Critical Role in Predicting Clinical Prognosis and Progression of Colorectal Cancer. Med. Sci. Monit. 2019, 25, 6429–6435. [Google Scholar] [CrossRef]
- Liang, X.-H.; Fournier, M.J. The helicase Has1p is required for snoRNA release from pre-rRNA. Mol. Cell. Biol. 2006, 26, 7437–7450. [Google Scholar] [CrossRef] [Green Version]
- Fayet-Lebaron, E.; Atzorn, V.; Henry, Y.; Kiss, T. 18S rRNA processing requires base pairings of snR30 H/ACA snoRNA to eukaryote-specific 18S sequences. EMBO J. 2009, 28, 1260–1270. [Google Scholar] [CrossRef]
- Bak, G.; Han, K.; Kim, K.-S.; Lee, Y. Electrophoretic mobility shift assay of RNA-RNA complexes. Methods Mol. Biol. 2015, 1240, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Blum, Y.; Verma, A.; Liu, Z.; Pramanik, K.; Leigh, N.R.; Chun, C.Z.; Samant, G.V.; Zhao, B.; Garnaas, M.K.; et al. A noncoding antisense RNA in tie-1 locus regulates tie-1 function in vivo. Blood 2010, 115, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Gilman, M. Ribonuclease protection assay. Curr. Protoc. Mol. Biol. 2001, 4. [Google Scholar] [CrossRef] [PubMed]
- Melton, D.A.; Krieg, P.A.; Rebagliati, M.R.; Maniatis, T.; Zinn, K.; Green, M.R. Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res. 1984, 12, 7035–7056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadaliha, M.; Gholamalamdari, O.; Tang, W.; Zhang, Y.; Petracovici, A.; Hao, Q.; Tariq, A.; Kim, T.G.; Holton, S.E.; Singh, D.K.; et al. A natural antisense lncRNA controls breast cancer progression by promoting tumor suppressor gene mRNA stability. PLoS Genet. 2018, 14, e1007802. [Google Scholar] [CrossRef] [PubMed]
- Ideue, T.; Hino, K.; Kitao, S.; Yokoi, T.; Hirose, T. Efficient oligonucleotide-mediated degradation of nuclear noncoding RNAs in mammalian cultured cells. RNA 2009, 15, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of mammary tumors and reduction in metastasis upon Malat1 lncRNA loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef] [Green Version]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef]
- Dethoff, E.A.; Chugh, J.; Mustoe, A.M.; Al-Hashimi, H.M. Functional complexity and regulation through RNA dynamics. Nature 2012, 482, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Carter, J.-M.; Emmett, W.; Mozos, I.R.; Kotter, A.; Helm, M.; Ule, J.; Hussain, S. FICC-Seq: A method for enzyme-specified profiling of methyl-5-uridine in cellular RNA. Nucleic Acids Res. 2019, 47, e113. [Google Scholar] [CrossRef]
- Linder, B.; Grozhik, A.V.; Olarerin-George, A.O.; Meydan, C.; Mason, C.E.; Jaffrey, S.R. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 2015, 12, 767–772. [Google Scholar] [CrossRef]
- Aw, J.G.A.; Lim, S.W.; Wang, J.X.; Lambert, F.R.P.; Tan, W.T.; Shen, Y.; Zhang, Y.; Kaewsapsak, P.; Li, C.; Ng, S.B.; et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Liu, H.; Begik, O.; Lucas, M.C.; Ramirez, J.M.; Mason, C.E.; Wiener, D.; Schwartz, S.; Mattick, J.S.; Smith, M.A.; Novoa, E.M. Accurate detection of m6A RNA modifications in native RNA sequences. Nat. Commun. 2019, 10, 4079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Workflow(s) for the detection and functional characterisation of a lncRNA of interest and its interacting partners. Primary approaches focus on identifying and assigning basic properties based on existing knowledge, predictions or biochemical experiments to validate expression or localisation for example. Secondary approaches focus on the identification of interactions with RNAs or RBPs utilising sequencing-based techniques, see Section 3.2. Further approaches may also be useful to validate high throughput or predictive results.
Figure 1.
Workflow(s) for the detection and functional characterisation of a lncRNA of interest and its interacting partners. Primary approaches focus on identifying and assigning basic properties based on existing knowledge, predictions or biochemical experiments to validate expression or localisation for example. Secondary approaches focus on the identification of interactions with RNAs or RBPs utilising sequencing-based techniques, see Section 3.2. Further approaches may also be useful to validate high throughput or predictive results.
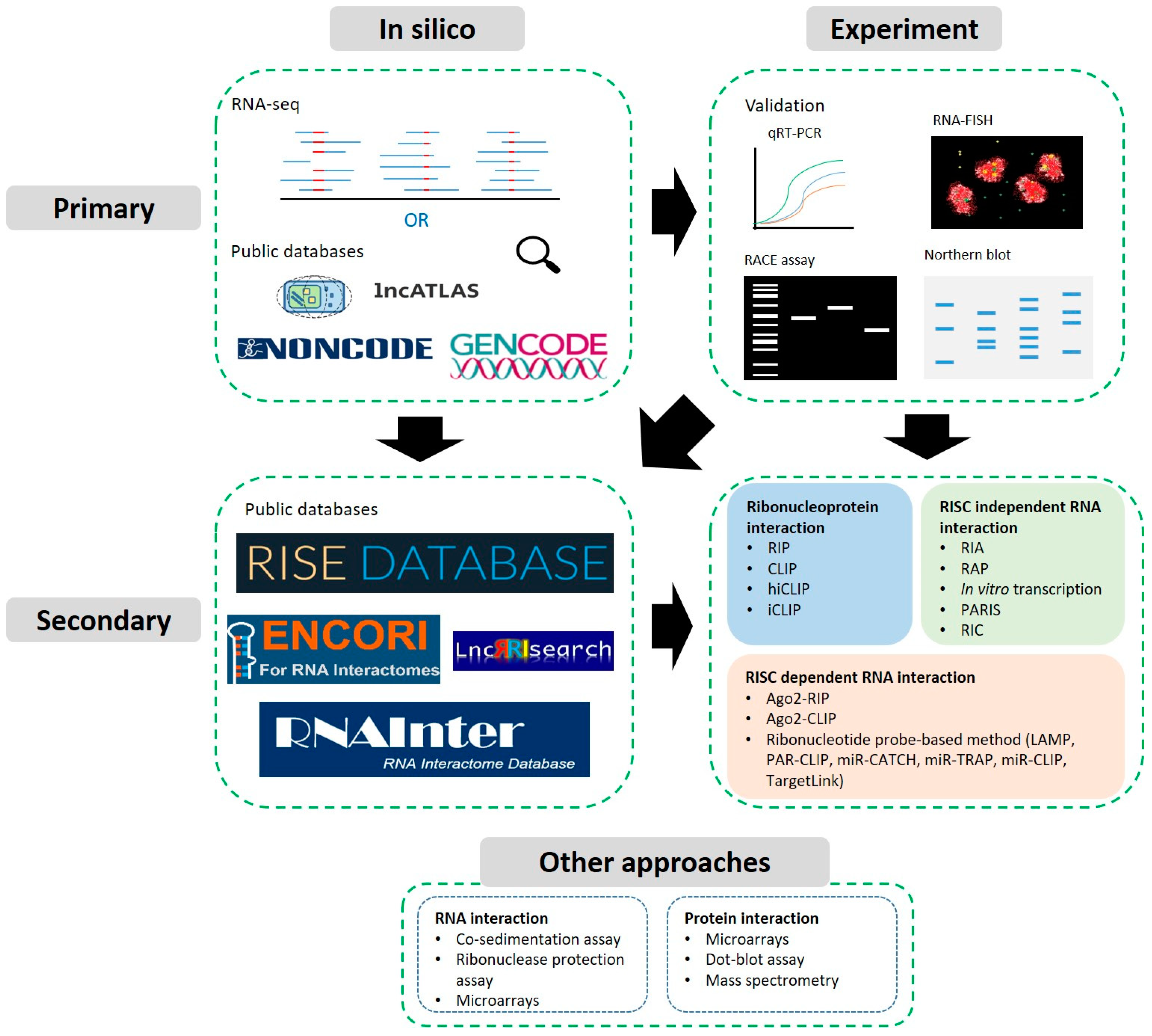
Table 1.
Databases for identifying lncRNAs and their basic properties or associations. Overview of active databases cataloguing various properties (sequence conservation, mutation, expression, localisation) or associations attributed to lncRNA genes or transcripts. A link to the hosting website is provided followed by the latest known version as well as the most recent publication describing the database.
Table 1.
Databases for identifying lncRNAs and their basic properties or associations. Overview of active databases cataloguing various properties (sequence conservation, mutation, expression, localisation) or associations attributed to lncRNA genes or transcripts. A link to the hosting website is provided followed by the latest known version as well as the most recent publication describing the database.
| Database/Version/Ref. | Link | Conservation | Mutations | Expression | Localisation | Associations |
|---|---|---|---|---|---|---|
| LNCiPedia v5 (2019) [89] | https://lncipedia.org/ (accessed on 8 March 2021) | H. sapiens, D. melanogaster, D. rerio, M. musculus, P. troglodytes | NA | NA | NA | Relevant references |
| lncATLAS (2017) [90] | https://lncatlas.crg.eu/ (accessed on 8 March 2021) | NA | NA | GENCODE | GENCODE | NA |
| NONCODE v6 (2020) [88] | http://www.noncode.org/ (accessed on 8 March 2021) | H. sapiens, M. musculus and 15 more | dbSNP | Human Body Map; NCBI GEO | NA | Gene Ontology |
| lncWiki/Book (2019) [91,92] | https://bigd.big.ac.cn/lncrnawiki/index.php/Main_Page (accessed on 8 March 2021) https://bigd.big.ac.cn/lncbook/index (accessed on 8 March 2021) | NA | ClinVar; COSMIC | HPA; GTEx; Methylation | NA | Gene Ontology; MeSH Ontology; miRNA Interaction Prediction; |
| Lnc2Cancer v3 (2020) [93] | http://bio-bigdata.hrbmu.edu.cn/lnc2cancer/ (accessed on 8 March 2021) | NA | NA | Literature Mining | lncATLAS | Expression Correlation; Survival; TF Motif; lncBook |
| LncRNADisease v2 (2019) [94] | http://www.rnanut.net/lncrnadisease/ (accessed on 8 March 2021) | NA | NA | NA | NA | Disease Ontology; MeSH Ontology; Predictive Associations |
| LncMAP v2 (2018) [95] | http://bio-bigdata.hrbmu.edu.cn/LncMAP/ (accessed on 8 March 2021) | NA | NA | NA | NA | Associations with: TF, Genes, Drugs, Survival |
| TANRIC v2 (2019) [96] | https://www.tanric.org (accessed on 8 March 2021) | NA | TCGA Somatic Mutations | TCGA | NA | TCGA/CCLE Correlations: Expression, Stage; Survival |
| MNDR v3.1 (2020) [97] | https://www.rna-society.org/mndr/ (accessed on 8 March 2021) | NA | NA | Mammalian | NA | Evidenced disease associations and Predictor |
| lncRNASNP v2 (2018) [98] | http://bioinfo.life.hust.edu.cn/lncRNASNP/#!/ (accessed on 8 March 2021) | NA | TCGA and COSMIC SNVs | NA | NA | miRNA binding & SNP effects; GWAS LD; Mutation effects |
| lncRNAMAP (2014) [99] | https://lncrnamap.mbc.nctu.edu.tw (accessed on 8 March 2021) | NA | NA | NCBI GEO | NA | miRNA and endo-siRNA predictors |
| LncTarD (2020) [100] | http://bio-bigdata.hrbmu.edu.cn/LncTarD/ (accessed on 8 March 2021) | NA | NA | NA | NA | Disease-related Target Prediction |
| EVLncRNAs (2017) [101] | http://biophy.dzu.edu.cn/EVLncRNAs/ (accessed on 8 March 2021) | NA | NA | NA | NA | Manually curated disease association |
| LncSPA (2020) [102] | http://bio-bigdata.hrbmu.edu.cn/LncSpA/ (accessed on 8 March 2021) | NA | NA | GTEx, HPA, HBM2, FANTOM, TCGA, TARGET | NA | Expression in diseased tissues |
Table 2.
Databases for identifying circRNAs and their basic properties or associations. The table summarises for each database what species the data are based on, as well as data sources, integrations or predictions of circRNA genes or transcripts. A link to the hosting website is provided followed by the latest known version as well as the most recent publication describing the database. IRES and MRE correspond to Internal Ribosome Entry Sites and miRNA Response Elements. See Section 3.2 for more information on CLIP and PAR-CLIP techniques.
Table 2.
Databases for identifying circRNAs and their basic properties or associations. The table summarises for each database what species the data are based on, as well as data sources, integrations or predictions of circRNA genes or transcripts. A link to the hosting website is provided followed by the latest known version as well as the most recent publication describing the database. IRES and MRE correspond to Internal Ribosome Entry Sites and miRNA Response Elements. See Section 3.2 for more information on CLIP and PAR-CLIP techniques.
| Database/Version/Ref. | Link | Species | Data Sources | Integrations | Predictions |
|---|---|---|---|---|---|
| CircAtlas (2020) [103] | http://159.226.67.237:8080/new/index.php (accessed on 8 March 2021) | H. sapiens, M. mulatta, M. musculus, R norvegicus, S. scrofa and G gallus | 1070 RNA-seq samples across 6 species | Integrates circR2Disease and circRNADIsease for disease associations | Co-expression network; Functional inference from GO/KEGG; RBP and miRNA binding |
| circRNAdb (2016) [104] | http://reprod.njmu.edu.cn/cgi-bin/circrnadb/circRNADb.php (accessed on 8 March 2021) | H. sapiens | Literature and RNA-seq dataset | UniProt | Protein domains, post-translational modifications, half-lifes |
| CircFunBase (2019) [105] | http://bis.zju.edu.cn/CircFunBase/ (accessed on 8 March 2021) | H. sapiens, M. musculus + 13 more. | Literature search | CircInteractome (CLIP data), miRBase | miRNA-circRNA interactions |
| circBase (2017) [106] | http://www.circbase.org/ (accessed on 8 March 2021) | H. sapiens, C. elegans, D. melanogaster, M. musculus, L. chalumnae, L. menadoensis | Various publications [18,107,108,109,110,111] | doRiNA | NA |
| Circbank (2019) [112] | http://www.circbank.cn/ (accessed on 8 March 2021) | M. musculus, R. norvegicus, D. melanogaster | circBase, miRBase | m6A literature, COSMIC somatic mutations | IRES, circRNA-miRNA prediction |
| CIRCpedia v2 (2018) [113] | https://www.picb.ac.cn/rnomics/circpedia/ (accessed on 8 March 2021) | H. sapiens, M. musculus, R. norvegicus, D. rerio, D. melanogaster, C | 180 RNA-seq samples across 6 species | NA | Putative circRNAs |
| CircRNADisease (2018) [114] | http://cgga.org.cn:9091/circRNADisease/ (accessed on 8 March 2021) | H. sapiens | Manual curation of 800 publications | NA | Association to diseases |
| CircR2Disease (2018) [115] | http://bioinfo.snnu.edu.cn/CircR2Disease/ (accessed on 8 March 2021) | H. sapiens | Manual curation of literature | NA | Association to diseases |
| TSCD (2017) [116] | http://gb.whu.edu.cn/tscd/ (accessed on 8 March 2021) | H. sapiens, M. musculus | ENCODE + NCBI GEO RNA-seq | Starbase, Gene Ontology | MRE, Protein binding sites |
| circad (2020) [117] | http://clingen.igib.res.in/circad/ (accessed on 8 March 2021) | H. sapiens, M. musculus, R. rattus | Manual curation of literature | NA | Asssociation to diseases |
| circVAR (2020) [118] | http://soft.bioinfo-minzhao.org/circvar/ (accessed on 8 March 2021) | H. sapiens | circBase, circNet, circRNAdb | 1000 Genomes, ClinVAR, GWASCatalog, ClinVAR, COSMIC | Association to diseases/cancer |
| CSCD (2018) [119] | http://gb.whu.edu.cn/cscd/ (accessed on 8 March 2021) | H. sapiens | 228 RNA-seq samples from ENCODE | Starbase | Cancer Association, MRE, RBP, ORFs |
| Circ2Traits (2013) [120] | http://gyanxet-beta.com/circdb/ (accessed on 8 March 2021) | H. sapiens | RNA-seq [107] | Starbase, TargetScan, miRCode, dbSNP, GWAS catalog, PAR-CLIP Data [121] | miRNA interactions |
| Circ2Disease (2018) [122] | http://bioinformatics.zju.edu.cn/Circ2Disease/index.html (accessed on 8 March 2021) | H. sapiens | Manual curation of literature | HMDD, OncomiRDB, miRTarBase, dbDEMC, miRecords | miRNA interactions |
| CircInteractome (2016) [123] | https://circinteractome.nia.nih.gov/ (accessed on 8 March 2021) | H. sapiens | circBase | Starbase, miRBase | IRES, RBP and miRNA binding sites |
Table 4.
Summary of sequencing approaches for facilitating the characterization of lncRNAs.
| Method | Specifications | Limitations | Requirements (Time/Special Resources) |
|---|---|---|---|
| RIP/RIP-seq [182] (tagged/endogenous RBP mediated RNA co-occupancy) | Characterization of native RNA-protein complexes without crosslinking; antibody enrichment | Low specificity; dependent on antibody availability | 3–4 d/IP compatible antibody; Autoradiograph facilities |
| CLIP/CLIP-seq [187] (tagged/endogenous RBP mediated RNA co-occupancy) | RNA-protein interaction sites via RNA-Protein UV crosslinking; antibody enrichment | 5′ and 3′ sites of RNA tags affected by cleavage and ligation biases; dependent on antibody availability | 5–8 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| hiCLIP [190] (tagged/endogenous RBP mediated RNA co-occupancy and RNA-duplexes) | RNA-protein interaction sites and RNA duplexes via UV crosslinking; antibody enrichment | May only capture highly expressed RNA species; dependent on antibody availability | 5 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| iCLIP [192] (tagged/endogenous RBP mediated RNA co-occupancy) | RNA-protein interaction sites at nucleotide resolution via UV crosslinking; antibody enrichment | miRNA-target interaction strength; dependent on antibody availability | 5 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| PAR-CLIP [121] (tagged/endogenous RBP mediated RNA co-occupancy) | RNA-protein interaction sites at nucleotide resolution; enhanced UV cross-linking and analysis choices; antibody enrichment | cultured cells only; 4-SU can induce cellular stress; dependent on antibody availability | 5 d/IP compatible antibody; UV Crosslinker; Autoradiograph facilities |
| Biotin-mimics/LAMP [199] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; Biotin enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence | 2 d/Streptavidin magnetic beads |
| miR-TRAP/PCP-seq [200,201] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions at nucleotide resolution; UVA crosslinking; Poly-A enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence | 2–3 d/UV Crosslinker |
| DBCO-tagged mimics [203] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; increased loading affinity; Click enrichment | Requires known miRNA sequence | 3 d/Azide-immobilized magnetic Beads |
| PA-miRNA [204] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; Photocleavable linker; Biotin enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence; linker is not easily acquired | 5 d/Solid phase synthesis; HPLC; Mass spectrometry; UV Crosslinker; Streptavidin magnetic beads |
| TargetLink [205] (tagged miRNA mimic probing RNA targets) | One or more miRNAs to many RNA targets; LNA+Biotin enrichment | Requires KO control; Requires known miRNA sequence | 6 d/UV Crosslinker; HPLC; Streptavidin magnetic beads |
| miR-CATCH [206] (tagged RNA mimic probing miRNA targetors) | One RNA to many miRNA interactions; RNA-RISC crosslinking by formaldehyde; Biotin enrichment | Delivered by transfection to cultured cells; Requires known RNA sequence | 3–4 d/Dynamag-2; FastPrep-24; Hybridization Oven; Streptavidin magnetic beads |
| miR-CLIP [208] (tagged miRNA mimic probing RNA targets) | One miRNA to many RNA interactions; RNA-RNA crosslinking by psoralen; Biotin enrichment | Delivered by transfection to cultured cells; Requires known miRNA sequence; Probe needs testing | 3–4 d/HPLC; UV Crosslinker; Streptavidin magnetic beads |
| CLASH [219] (tagged RBP mediated RNA-Protein/duplex capture) | RNA-protein interaction sites and RNA duplexes via UV crosslinking; IgG+Ni-NTA enrichment | Delivered by transfection to cultured cells; Tagged protein expression design may be challenging | 4–5 d/UV Crosslinker; Autoradiography facilities |
| MARIO [220] (endogenous RBP mediated RNA-duplex capture) | Global RNA-RNA interactions mediated by RBPs; RNA-Protein UV crosslinking; 2-step biotin enrichment; proximity ligation | Limited to RBP mediated interactions | 5 d/UV Crosslinker; Streptavidin magnetic beads |
| RIA-seq [215] (endogenous RNA-duplex capture) | One RNA to all RNA interactions; glutaraldehyde crosslinking; biotin enrichment | Limited to RBP mediated interactions; probe preparation may be challenging | 5 d/Streptavidin magnetic beads |
| PARIS [223] (endogenous RNA-duplex capture) | All to all RNA interactions; psoralen crosslinking of RNAs; 2D enrichment of crosslinked duplexes; proximity ligation | Possible AMT side effects; 2D gel setup may be challenging | 5 d/UV Crosslinker; SequaGel UreaGel System |
| LIGR-seq [225] (endogenous RNA-duplex capture) | All to all RNA interactions; psoralen crosslinking of RNAs; RNAseR enrichment of crosslinked duplexes; proximity ligation | Possible AMT side effects | 4 d/UV Crosslinker; RNAseR |
| SPLASH [227] (endogenous RNA-duplex capture) | All to all RNA interactions; psoralen crosslinking of RNAs; biotin enrichment of crosslinked duplexes; proximity ligation | Possible AMT side effects | 4 d/UV Crosslinker; Streptavidin magnetic beads |
| RIC-seq [228] (endogenous RBP mediated RNA-duplex capture) | Global RNA-RNA interactions mediated by RBPs; RNA-Protein formaldehyde crosslinking; biotin enrichment; in situ proximity ligation | Limited to RBP mediated interactions; cell permeabilization may need optimizing | 5 d/Streptavidin magnetic beads |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Carter, J.-M.; Ang, D.A.; Sim, N.; Budiman, A.; Li, Y. Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer. Non-Coding RNA 2021, 7, 19. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7010019
AMA Style
Carter J-M, Ang DA, Sim N, Budiman A, Li Y. Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer. Non-Coding RNA. 2021; 7(1):19. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7010019
Chicago/Turabian StyleCarter, Jean-Michel, Daniel Aron Ang, Nicholas Sim, Andrea Budiman, and Yinghui Li. 2021. "Approaches to Identify and Characterise the Post-Transcriptional Roles of lncRNAs in Cancer" Non-Coding RNA 7, no. 1: 19. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna7010019
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.