
Blood Transcriptome Analysis of Septic Patients Reveals a Long Non-Coding Alu-RNA in the Complement C5a Receptor 1 Gene

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Overview
2.2. External Bulk RNA-Seq Datasets
2.3. Whole Blood Model Induced with S. aureus
2.4. RNA Isolation for RNA-Seq
2.5. Illumina Sequencing
2.6. Ion S5 Stranded RNA Sequencing
2.7. Mapping of Transcripts
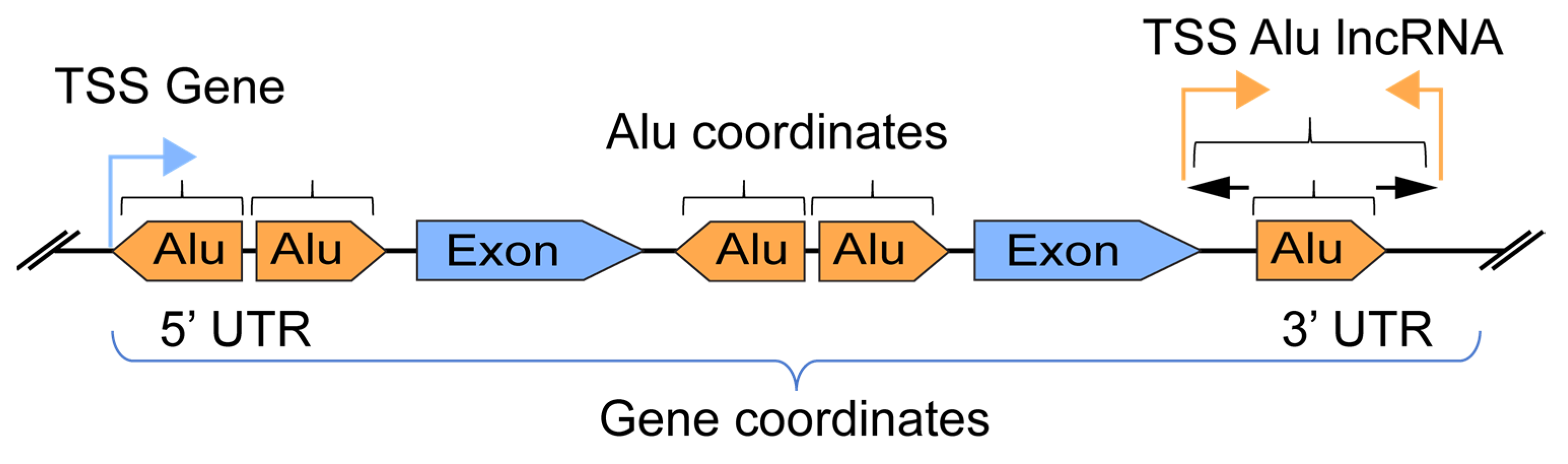
2.8. Alu Coordinates Intersection with Genes for Expression Analysis
2.9. Counting Alu Transcript Levels with the Homer Software
- Entity List: All Entities
- Interpretation: condition1 (Non-averaged)
- Distance Metric: Euclidean
- Minimum Distance: 0.009999999776482582
- Number of neighbours: 15
- Initialization Method: Spectral
- Created from Advanced Analysis operation: Clustering:
- Entity List: All Entities
- Interpretation: condition1 (Non-averaged)
- Experiment: 22j
- Clustering Algorithm: Hierarchical
- Clustered By: Normalized intensity values
- Clustered On: Entities and Conditions
- Similarity Measure: Euclidean
- Linkage Rule: Wards
- Cluster Within Conditions: No
3. Results
3.1. Alu Insertions in the Human Genome
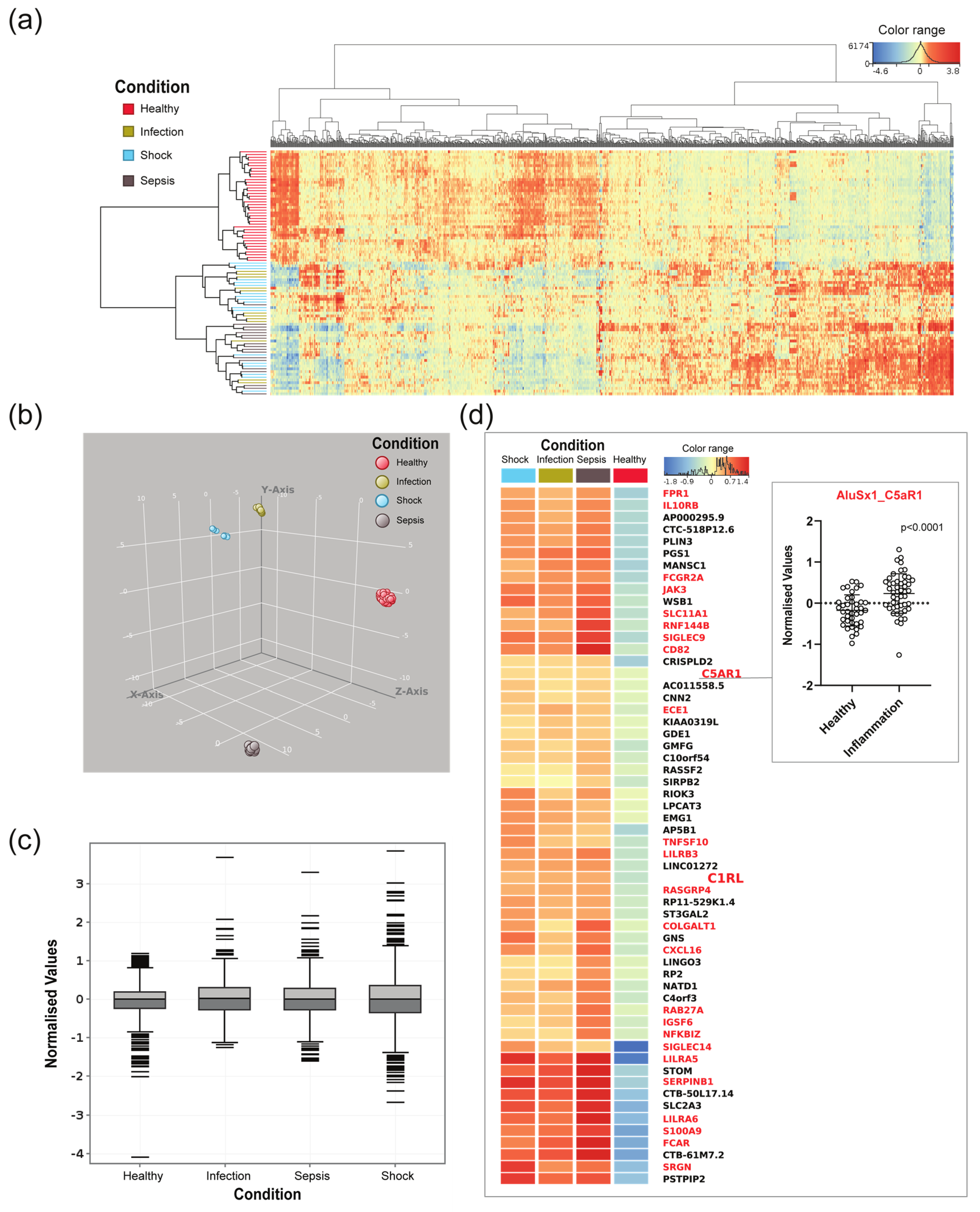
3.2. Alu-RNAs in Blood from Sepsis Patients and Healthy Controls
3.3. A Differentially Expressed Alu-Containing lncRNA Is Transcribed from the C5aR1 3′UTR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Batzer, M.A.; Deininger, P.L.; Hellmann-Blumberg, U.; Jurka, J.; Labuda, D.; Rubin, C.M.; Schmid, C.W.; Zietkiewicz, E.; Zuckerkandl, E. Standardized Nomenclature for Alu Repeats. J. Mol. Evol. 1996, 42, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu Elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-Mediated Retrotransposition of Marked Alu Sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Häsler, J.; Samuelsson, T.; Strub, K. Useful “Junk”: Alu RNAs in the Human Transcriptome. Cell. Mol. Life Sci. 2007, 64, 1793–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-O.; Gingeras, T.R.; Weng, Z. Genome-Wide Analysis of Polymerase III-Transcribed Alu Elements Suggests Cell-Type-Specific Enhancer Function. Genome Res. 2019, 29, 1402–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raha, D.; Wang, Z.; Moqtaderi, Z.; Wu, L.; Zhong, G.; Gerstein, M.; Struhl, K.; Snyder, M. Close Association of RNA Polymerase II and Many Transcription Factors with Pol III Genes. Proc. Natl. Acad. Sci. USA 2010, 107, 3639–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, K.L.; Latchman, D.S. HSV Infection Induces Increased Transcription of Alu Repeated Sequences by RNA Polymerase III. FEBS Lett. 1989, 258, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Z.; Wespiser, A.R.; Caffrey, D.R. The Domain Structure and Distribution of Alu Elements in Long Noncoding RNAs and MRNAs. RNA 2016, 22, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fort, V.; Khelifi, G.; Hussein, S.M.I. Long Non-Coding RNAs and Transposable Elements: A Functional Relationship. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118837. [Google Scholar] [CrossRef] [PubMed]
- Wight, M.; Werner, A. The Functions of Natural Antisense Transcripts. Essays Biochem. 2013, 54, 91–101. [Google Scholar] [PubMed]
- Khorkova, O.; Myers, A.J.; Hsiao, J.; Wahlestedt, C. Natural Antisense Transcripts. Hum. Mol. Genet. 2014, 23, R54–R63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinad, H.S.; Natasya, I.; Werner, A. Natural Antisense Transcripts at the Interface between Host Genome and Mobile Genetic Elements. Front. Microbiol. 2017, 8, 2292. [Google Scholar] [CrossRef]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long Non-Coding Antisense RNA Controls Uchl1 Translation through an Embedded SINEB2 Repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef] [PubMed]
- Zucchelli, S.; Cotella, D.; Takahashi, H.; Carrieri, C.; Cimatti, L.; Fasolo, F.; Jones, M.H.; Sblattero, D.; Sanges, R.; Santoro, C.; et al. SINEUPs: A New Class of Natural and Synthetic Antisense Long Non-Coding RNAs That Activate Translation. RNA Biol. 2015, 12, 771–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häsler, J.; Strub, K. Alu RNP and Alu RNA Regulate Translation Initiation in Vitro. Nucleic Acids Res. 2006, 34, 2374–2385. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Ivanova, E.; Gareau, C.; Scherrer, A.; Mazroui, R.; Strub, K. Direct Binding of the Alu Binding Protein Dimer SRP9/14 to 40S Ribosomal Subunits Promotes Stress Granule Formation and Is Regulated by Alu RNA. Nucleic Acids Res. 2014, 42, 11203–11217. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Molina, R.; Arzate-Mejía, R.G.; Ayala-Ortega, E.; Guerrero, G.; Meier, K.; Suaste-Olmos, F.; Recillas-Targa, F. An Intronic Alu Element Attenuates the Transcription of a Long Non-Coding RNA in Human Cell Lines. Front. Genet. 2020, 11, 928. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating Gene Expression through RNA Nuclear Retention. Cell 2005, 123, 249–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-L.; DeCerbo, J.N.; Carmichael, G.G. Alu Element-Mediated Gene Silencing. EMBO J. 2008, 27, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. LncRNAs Transactivate STAU1-Mediated MRNA Decay by Duplexing with 3′ UTRs via Alu Elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.; Bhattacharya, A.; Bhardwaj, V.; Jha, V.; Mandal, A.K.; Mukerji, M. Alu-MiRNA Interactions Modulate Transcript Isoform Diversity in Stress Response and Reveal Signatures of Positive Selection. Sci. Rep. 2016, 6, 32348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, N.; Roy-Engel, A.M.; Imataka, H.; Sonenberg, N.; Deininger, P. Shared Protein Components of SINE RNPs. J. Mol. Biol. 2002, 321, 423–432. [Google Scholar] [CrossRef]
- Chen, L.-L.; Yang, L. ALUternative Regulation for Gene Expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, M.; Han, D.; Boyd-Kirkup, J.; Yu, X.; Han, J.-D.J. Evolution of Alu Elements toward Enhancers. Cell Rep. 2014, 7, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lara, J.C.-F.; Arzate-Mejía, R.G.; Recillas-Targa, F. Enhancer RNAs: Insights Into Their Biological Role. Epigenet. Insights 2019, 12, 2516865719846093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, P.R.; Wells, A.D.; Li, X.C. Diversity and Emerging Roles of Enhancer RNA in Regulation of Gene Expression and Cell Fate. Front. Cell Dev. Biol. 2019, 7, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, M.V.; Sim, R.B. Complement in Health and Disease. Adv. Drug Deliv. Rev. 2011, 63, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollnes, T.E.; Huber-Lang, M. Complement in Sepsis-When Science Meets Clinics. FEBS Lett. 2020, 594, 2621–2632. [Google Scholar] [CrossRef]
- Walther, K.; Schulte, L.N. The Role of LncRNAs in Innate Immunity and Inflammation. RNA Biol. 2021, 18, 587–603. [Google Scholar] [CrossRef]
- Herwanto, V.; Tang, B.; Wang, Y.; Shojaei, M.; Nalos, M.; Shetty, A.; Lai, K.; McLean, A.S.; Schughart, K. Blood Transcriptome Analysis of Patients with Uncomplicated Bacterial Infection and Sepsis. BMC Res. Notes 2021, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Mishra, K.; Kanduri, C. Understanding Long Noncoding RNA and Chromatin Interactions: What We Know So Far. Noncoding RNA 2019, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Gil, N.; Ulitsky, I. Regulation of Gene Expression by Cis-Acting Long Non-Coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.W.; Brinkman, F.S.L.; Lynn, D.J. InnateDB: Systems Biology of Innate Immunity and beyond--Recent Updates and Continuing Curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef] [PubMed]
- Lizio, M.; Harshbarger, J.; Shimoji, H.; Severin, J.; Kasukawa, T.; Sahin, S.; Abugessaisa, I.; Fukuda, S.; Hori, F.; Ishikawa-Kato, S.; et al. Gateways to the FANTOM5 Promoter Level Mammalian Expression Atlas. Genome Biol. 2015, 16, 22. [Google Scholar] [CrossRef] [Green Version]
- Mollnes, T.E.; Brekke, O.-L.; Fung, M.; Fure, H.; Christiansen, D.; Bergseth, G.; Videm, V.; Lappegård, K.T.; Köhl, J.; Lambris, J.D. Essential Role of the C5a Receptor in E Coli-Induced Oxidative Burst and Phagocytosis Revealed by a Novel Lepirudin-Based Human Whole Blood Model of Inflammation. Blood 2002, 100, 1869–1877. [Google Scholar] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Neph, S.; Kuehn, M.S.; Reynolds, A.P.; Haugen, E.; Thurman, R.E.; Johnson, A.K.; Rynes, E.; Maurano, M.T.; Vierstra, J.; Thomas, S.; et al. BEDOPS: High-Performance Genomic Feature Operations. Bioinformatics 2012, 28, 1919–1920. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sun, H.; Zhang, Y.; Zhang, T.; Gong, J.; Wei, Y.; Duan, Y.-G.; Shu, M.; Yang, Y.; Wu, D.; et al. Dimensionality Reduction by UMAP Reinforces Sample Heterogeneity Analysis in Bulk Transcriptomic Data. Cell Rep. 2021, 36, 109442. [Google Scholar] [CrossRef]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap Analysis Gene Expression for High-Throughput Analysis of Transcriptional Starting Point and Identification of Promoter Usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantarella, S.; Carnevali, D.; Morselli, M.; Conti, A.; Pellegrini, M.; Montanini, B.; Dieci, G. Alu RNA Modulates the Expression of Cell Cycle Genes in Human Fibroblasts. Int. J. Mol. Sci. 2019, 20, 3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aune, T.M.; Tossberg, J.T.; Heinrich, R.M.; Porter, K.P.; Crooke, P.S., 3rd. Alu RNA Structural Features Modulate Immune Cell Activation and A-to-I Editing of Alu RNAs Is Diminished in Human Inflammatory Bowel Disease. Front. Immunol. 2022, 13, 818023. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Saville, L.; Gollen, B.; Veronesi, A.A.; Mohajerani, M.; Joseph, J.T.; Zovoilis, A. Increased Alu RNA Processing in Alzheimer Brains Is Linked to Gene Expression Changes. EMBO Rep. 2021, 22, e52255. [Google Scholar] [CrossRef] [PubMed]
- Mus, E.; Hof, P.R.; Tiedge, H. Dendritic BC200 RNA in Aging and in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2007, 104, 10679–10684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, T.; Kim, S.; Chowdhury, T.; Yu, H.J.; Kim, K.-M.; Kang, H.; Won, J.-K.; Park, S.-H.; Shin, J.H.; Park, C.-K. Genome-Wide Perturbations of Alu Expression and Alu-Associated Post-Transcriptional Regulations Distinguish Oligodendroglioma from Other Gliomas. Commun. Biol. 2022, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.-H.; Yeh, C.-T. A Gene Expression Restriction Network Mediated by Sense and Antisense Alu Sequences Located on Protein-Coding Messenger RNAs. BMC Genom. 2013, 14, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Name | Alu Subfamily | Genomic Location | Position in Gene | TSS | Alu Orientation | p-Value |
|---|---|---|---|---|---|---|
| ECE1 | AluSx1 | chr1:21218585-21218890 | 3′UTR | no | Sense | 4 × 10−12 |
| S100A9 | AluSp | chr1:153359680-153359988 | Intron | no | Inverted | 3 × 10−33 |
| FCGR2A | AluSz6 | chr1:161518618-161518922 | 3′UTR | yes 1 | Inverted | 2 × 10−23 |
| SRGN | AluSz | chr10:69091596-69091908 | Intron | no | Sense | 9 × 10−16 |
| CD82 | AluY | chr11:44619480-44619789 | 3′UTR | yes | Sense | 3 × 10−13 |
| C1RL | AluSc | chr12:7095120-7095413 | 3’UTR | no | Sense | 1 × 10−21 |
| AluSq | chr12:7097283-7097581 | Intron | no | Sense | 1 × 10−14 | |
| AluSq2 | chr12:7098913-7099254 | Intron | yes 1 | Inverted | 6 × 10−20 | |
| RAB27A | AluY | chr15:55203412-55203721 | 3′UTR | no | Sense | 1 × 10−13 |
| IGSF6 | AluSx | chr16:21639920-21640237 | 3′UTR | no | Sense | 9 × 10−8 |
| AluSx | chr16:21640468-21640796 | 3′UTR | yes | Inverted | 2 × 10−10 | |
| CXCL16 | AluSx3 | chr17:4733927-4734234 | 3′UTR | yes | Inverted | 2 × 10−12 |
| COLGALT1 | AluSz | chr19:17581909-17582208 | 3′UTR | yes | Inverted | 2 × 10−11 |
| JAK3 | AluY | chr19:17825619-17825921 | 3′UTR | yes | Inverted | 1 × 10−21 |
| AluSz | chr19:17826103-17826414 | 3′UTR | yes | Inverted | 1 × 10−17 | |
| AluSx | chr19:17836278-17836571 | Intron | no | Sense | 3 × 10−24 | |
| RASGRP4 | AluSg | chr19:38409458-38409752 | 3′UTR | no | Inverted | 4 × 10−17 |
| C5AR1 | AluSx1 | chr19:47321326-47321636 | 3′UTR | yes | Sense | 2 × 10−5 |
| SIGLEC9 | AluY | chr19:51131305-51131590 | Intron | no | Sense | 2 × 10−16 |
| AluSz | chr19:51131634-51131933 | Intron | no | Sense | 2 × 10−15 | |
| AluSx1 | chr19:51133290-51133609 | Intron | no | Sense | 7 × 10−19 | |
| AluSc | chr19:51134129-51134456 | Intron | no | Inverted | 4 × 10−20 | |
| SIGLEC14 | AluSx1 | chr19:51642741-51643045 | 3′UTR | yes | Inverted | 4 × 10−4 |
| FPR1 | AluSx1 | chr19:51745155-51745455 | 3′UTR | no | Sense | 3 × 10−20 |
| LILRB3 | AluJb | chr19:54215999-54216282 | 3′UTR | no | Sense | 1 × 10−21 |
| AluY | chr19:54216298-54216609 | 3′UTR | no | Sense | 2 × 10−21 | |
| AluJo | chr19:54216689-54216866 | 3′UTR | no | Sense | 1 × 10−20 | |
| LILRA6 | AluSx3 | chr19:54238010-54238335 | Intron | no | Sense | 3 × 10−19 |
| LILRA5 | AluSc | chr19:54307138-54307296 | 3′UTR | no | Inverted | 1 × 10−26 |
| FCAR | AluSx | chr19:54889964-54890253 | 3′UTR | no | Inverted | 4 × 10−23 |
| AluSg | chr19:54890415-54890727 | 3′UTR | yes | Inverted | 6 × 10−26 | |
| SLC11A1 | AluSz | chr2:218383482-218383779 | Intron | no | Inverted | 3 × 10−13 |
| AluSx1 | chr2:218385379-218385690 | Intron | no | Inverted | 2 × 10−8 | |
| AluSc | chr2:218388087-218388396 | Intron | yes | Sense | 3 × 10−13 | |
| AluSx3 | chr2:218388511-218388823 | Intron | no | Sense | 7 × 10−20 | |
| AluJb | chr2:218388830-218389102 | Intron | no | Sense | 6 × 10−19 | |
| AluJo | chr2:218389289-218389568 | Intron | no | Sense | 5 × 10−14 | |
| AluJo | chr2:218390899-218391030 | Intron | no | Sense | 4 × 10−14 | |
| AluSx1 | chr2:218391611-218391922 | Intron | yes | Inverted | 4 × 10−11 | |
| AluSg | chr2:218391923-218392055 | Intron | yes | Inverted | 2 × 10−11 | |
| AluSp | chr2:218392056-218392362 | Intron | yes | Inverted | 3 × 10−12 | |
| AluJo | chr2:218392363-218392634 | Intron | yes | Inverted | 4 × 10−13 | |
| AluSz6 | chr2:218395463-218395753 | 3′UTR | yes | Inverted | 2 × 10−17 | |
| IL10RB | AluSz | chr21:33296666-33296973 | 3′UTR | no | Sense | 8 × 10−25 |
| NFKBIZ | AluJo | chr3:101860180-101860475 | 3′UTR | yes 1 | Inverted | 2 × 10−12 |
| TNFSF10 | AluSc | chr3:172505725-172506023 | 3′UTR | yes | Sense | 1 × 10−14 |
| RNF144B | AluJo | chr6:18467641-18467939 | 3′UTR | yes | Inverted | 3 × 10−16 |
| SERPINB1 | AluSx | chr6:2832525-2832836 | 3′UTR | yes | Inverted | 2 × 10−21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emblem, Å.; Knutsen, E.; Jørgensen, T.E.; Fure, H.; Johansen, S.D.; Brekke, O.-L.; Mollnes, T.E.; Karlsen, B.O. Blood Transcriptome Analysis of Septic Patients Reveals a Long Non-Coding Alu-RNA in the Complement C5a Receptor 1 Gene. Non-Coding RNA 2022, 8, 24. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna8020024
Emblem Å, Knutsen E, Jørgensen TE, Fure H, Johansen SD, Brekke O-L, Mollnes TE, Karlsen BO. Blood Transcriptome Analysis of Septic Patients Reveals a Long Non-Coding Alu-RNA in the Complement C5a Receptor 1 Gene. Non-Coding RNA. 2022; 8(2):24. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna8020024
Chicago/Turabian StyleEmblem, Åse, Erik Knutsen, Tor Erik Jørgensen, Hilde Fure, Steinar Daae Johansen, Ole-Lars Brekke, Tom Eirik Mollnes, and Bård Ove Karlsen. 2022. "Blood Transcriptome Analysis of Septic Patients Reveals a Long Non-Coding Alu-RNA in the Complement C5a Receptor 1 Gene" Non-Coding RNA 8, no. 2: 24. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna8020024