Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation


1
Department of Civil and Environmental Engineering, University of Connecticut, Unit 3037, 261 Glenbrook Rd., Storrs, CT 06269-3037, USA
2
Department of Chemistry, University of Connecticut, Unit 3060, 55 N. Eagleville Rd., Storrs, CT 06269-3037, USA
*
Author to whom correspondence should be addressed.
C 2018, 4(3), 40; https://0-doi-org.brum.beds.ac.uk/10.3390/c4030040
Submission received: 15 May 2018
/
Revised: 22 June 2018
/
Accepted: 28 June 2018
/
Published: 9 July 2018
Abstract
:Enhancing the performance of nanofibrous carbons requires the specific chemical functionalization of the surface, while limiting material degradation or causing other detrimental changes in the surface area and pore structures. We compare traditional oxidation protocols using HNO3, HNO3/H2SO4, and KMnO4 with the much less used oxidants RuO4 and OsO4, in tandem with secondary oxidants (such as KMnO4 or Oxone®), for their ability to form carboxylic acids on the surface of polyacrylonitrile-based activated carbon nanofiber membrane (ACNF) materials. While the traditional methods increased the carboxylic acid contents, they also destroyed the macrostructure of the ACNF, concomitant with the loss of up to 17 wt.% of the material. RuO4-mediated oxidations proved also to be too harsh. On the contrary, some of the OsO4-based protocols were characterized by very high mass yields; significant increase in carboxylic acid functionalization (6.3 µmol/mg) compared with the unmodified ACNF (1.7 µmol/mg), but with no concomitant loss of macrostructure, as measured by the retention of the Brunauer-Emmett-Teller (BET) surface area; and average pore width. While there was some reduction in micropore volume, the microporosity of the material remained high. The temperature-programmed desorption mass spectrometry (up to 1000 °C) indicated the presence of both single and adjacent carboxylic acid groups. We thus identified mild and highly effective reaction conditions for the functionalization of carbon nanomaterials without undue degradation of their physical properties.
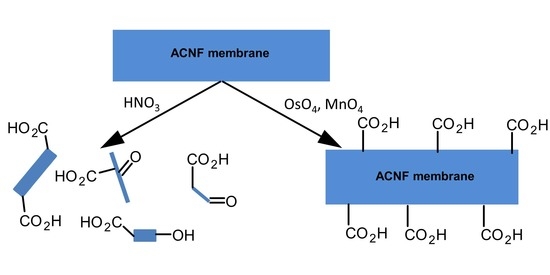
1. Introduction
Carbon nanomaterials with their tunable chemical, physical, electrical, and mechanical properties, are useful as adsorbents, sensors, filters, antimicrobial agents, or in energy storage devices [1]. For example, materials such as carbon nanotubes (CNT) [2,3,4], carbon nanofibers (CNF) [5,6,7], carbon cloth [1,8,9,10], or activated carbon nanofiber (ACNF) [11,12,13], have all been used for these purposes. The properties of nanocarbon adsorbents with short intra-particle diffusion distances, controllable pore sizes, and the option for high specific surface areas make them particularly suitable for contaminant binding [14]. However, as the dimensionality of the carbon materials get smaller and more structured (e.g., electrospun materials or woven clothes), it gets increasingly difficult to alter their surface chemistry without also damaging their overall structure.
In considering the use of carbon nanomaterials as adsorbents for environmental contaminants, tuning the surface chemistry is key to targeting specific contaminants. A variety of methods have been used to modify the surface of carbon nanomaterials, including plasma treatment, gaseous reactions, or wet chemical oxidations [15,16,17,18,19]. These oxidation steps produce a variety of oxygen-based surface functional groups, including carbonyl, carboxylic acid, or phenolic groups. Carboxylic acid groups, in particular, are most versatile precursors for the subsequent functionalizations in applications that require the alteration of conductivity of the carbonaceous materials [20], to attach sensors [21], or as a functional handle to attach other chemical moieties through (e.g., amidation reactions) [19,22]. The introduction of carboxylic acid groups was primarily achieved by oxidation strategies, using the oxidants nitric acid (HNO3) with and without the addition of sulfuric acid (H2SO4) [23,24,25,26,27,28], organic and inorganic peroxides (R–O–O–R) [29], ozone (O3) [17], or potassium permanganate (KMnO4) [30]. However, these oxidation reactions generally also lead to a loss of pore volume, reduced surface area, and an increase in the mean pore size, through the destruction of pore walls [25,31,32]. For instance, Rasheed et al. examined the oxidation of graphitized carbon nanofibers with HNO3 and generated oxidized materials with mass yields of less than 76% [18]; in other words, about a quarter of the material was lost in the form of soluble or volatile carbon products. Other researchers regularly observed the breakage of carbon nanotubes under the strongly oxidizing acid treatments [16,27]. Thus, applying these methods to carbon cloth or ACNF, the undesirable effects of fiber breakage and the destruction of the larger macrostructure are likely to be expected (and as we will also confirm experimentally).
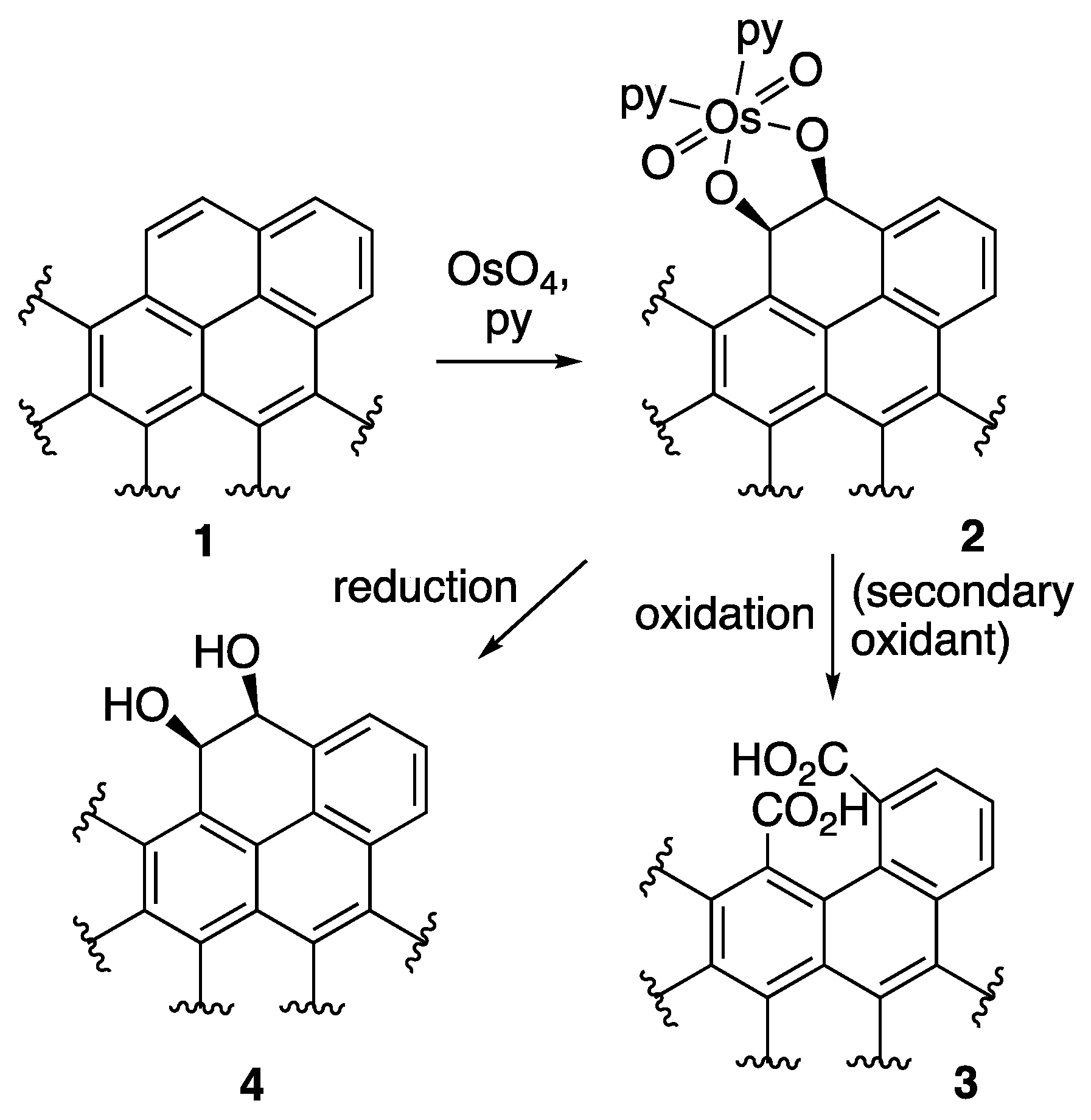
The introduction of specific oxygen functionalities through the oxidation processes of carbonaceous materials, while minimizing material loss and/or maintaining the carbon nanomaterial macrostructure is non-trivial, because these carbon sources are not readily oxidized; consequently, only few rationally designed nano-adsorbents were described [33]. The testing of alternative oxidants of larger selectivity may fundamentally provide more protection against dramatic changes in pore volume, surface area, or fiber dimensions [34]. For instance, osmium tetraoxide (OsO4) and ruthenium tetroxide (RuO4) are powerful and very selective oxidants, known to specifically functionalize only olefinic or pseudo-olefinic double bonds [35]. These oxidants have been used to open the ends of carbon nanotubes, introducing ketone functionalities [36]. Literature evidence suggests that, while RuO4 is capable of transforming a C=C double bond to a bis-ketone functionality in one step [37], OsO4 forms with a suitable double bond in a polycyclic structure 1 (Scheme 1) an intermediate osmate ester 2 (Scheme 1). This ester requires a secondary oxidation step to affect C–C bond cleavage, and the introduction of two aldehyde or carboxylic acid groups on the carbon framework, 3 (Scheme 1). Numerous secondary oxidants are an option. For instance, KMnO4, potassium peroxysulfate (KHSO5), Oxone® (2 KHSO5·KHSO4·K2SO4), potassium periodate (KIO4), or manganese dioxide (MnO2) could be used, allowing a fine-tuning of the process [35].
Furthermore, a reduction of the intermediate osmate ester provides a vic-cis-diol functionality in 2 (Scheme 1), which subsequently could also be specifically oxidized to generate either ketone/aldehyde or carboxylic acid functions. In general, the OsO4- and RuO4-mediated oxidation methods have the advantage of introducing well-defined functionalities on adjacent carbon atoms [35]. This is potentially beneficial for enhancing the sorbent interactions upon secondary functionalization, in cases where the chelate effects play a role.
We report here on the chemical oxidation of polyacrylonitrile (PAN)-based activated carbon nanofiber (ACNF) membranes, using a range of well-known and novel oxidation methods. The material is nonwoven in its macrostructure, with nanoscale fiber diameters and a well-defined carbon framework structure [38]. The objectives of this study are to assess the ability of a range of oxidants so as to introduce surface functional groups, with a particular focus on generating carboxylic acids, while at the same time minimizing changes to the surface area, pore distribution, and macrostructure. Thus, following each oxidation reaction, we compared the physical appearance, surface area, and pore size distribution using field emission scanning electron microscopy (FESEM) and physisorption experiments, respectively, and we probed the chemical functionalities present on the modified ACNF membranes using elemental analysis, FTIR spectroscopy, Boehm titrations, and temperature programmed desorption mass spectrometry (TPD-MS). The experiments suggest several novel options for generating carboxylic acids on carbon nanomaterials that minimize the physical damage to the material and that are therefore superior to traditional oxidation methods.
2. Materials and Methods
2.1. Materials
All reagents and solvents were certified as ACS grade. All aqueous solutions were prepared with 18.2 MΩ deionized water (H2O) (Millipore Integral treatment system). The concentration of H2SO4 was 95–98% and the concentration of HNO3 was 70% (Thermo Fisher Scientific, Waltham, MA, USA). KMnO4 and Oxone® were purchased from Acros (Thermo Fisher Scientific, Waltham, MA, USA), RuO2 from Sigma-Aldrich (St. Louis, MO, USA), and OsO4 from Pressure Chemical Company (Pittsburgh, PA, USA).
The filter assembly used was a Kontes Ultra-Ware all-glass funnel/support assembly with stainless steel support mesh, 47 mm diameter, 430 mm height, funnel capacity 300 mL, using Millipore type AP40 glass fiber filters, pore size 0.7 μm.
The polyacrylonitrile (PAN)-based activated carbon nanofiber (ACNF) membranes were fabricated from a 14 wt.% solution of polyacrylonitrile (PAN, with an average molecular weight of 150,000 amu, obtained from Scientific Polymer Products Inc., Ontario, NY, USA) in dimethylformamide (DMF) using the electrospinning, stabilization, pyrolysis, and activation steps similar to as described in the literature [38]. After electrospinning, the PAN mats were stabilized at 280 °C in a muffle furnace (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h, in an air atmosphere, where the temperature was increased at a rate of 1 °C/min. The stabilized mats were then heated with a ramp rate of 3 °C/min up to 600 °C and were then carbonized for 2 h under a N2 atmosphere. Finally, the carbonized mats were activated by injecting a water steam at a flow rate of 1 mL/min, while the temperature was increased at a rate of 5 °C/min up to 750 °C, where it was maintained for 1 h.
2.2. Oxidation Experiments
We chose four classic HNO3- and MnO4-based activated carbon oxidation conditions as controls to the much less explored OsO4- and RuO4-based oxidation methods evaluated here. The temperature, timing, and oxidant concentration of the reactions for the classic oxidations were based on the ranges found in the literature [18,23,27,28,39]. Our approach was meant to maintain certain stoichiometric ratios of carbon to oxidant, while allowing the different reaction times (e.g., OsO4 reacts more slowly and was therefore given more time) and temperatures typical for the different reagents. For the MnO4, OsO4, or RuO4 oxidations, the ACNF:oxidant ratios of were kept constant, and were estimated based on surface area and assuming that the bulk of the surface was aromatic carbon (carbon:oxidant = 106:1, or 106:4 for the higher concentration of MnO4). This molar ratio was based on preliminary data acquired by us and by the considerations that two surface carbons react with one equivalent of MnO4, OsO4, or RuO4. OsO4 was used as a two-stage stoichiometric oxidant (Scheme 1), as follows: OsO4 specifically adds to the double bond with the least resonance stabilization (i.e., at the edges or defects of the carbon framework). The initially formed osmate ester can be oxidatively cleaved with a secondary oxidant to generate two carboxylic acid functionalities in close vicinity to each other. We pursued this reaction with stoichiometric amounts of OsO4 in a step-wise fashion, rather than using catalytic quantities of OsO4 in the presence of the secondary oxidant, for a better comparison of the effects of the secondary oxidants on identically osmate ester-derivatized ACNFs, and to eliminate any kinetic effects that might slow the catalytic cycle. A similar scheme was followed for the sub-stoichiometric quantities of RuO4 generated in situ from RuO2 and aqueous sodium hypochlorite (NaOCl, bleach). The reaction of RuO4 with a double bond directly generates carboxylic acids rapidly in one step.
2.2.1. Control Method 1: HNO3 Oxidation
The ACNF (0.5 g) was placed into a 25 mL wide-mouth round bottom flask along with aq 10 M HNO3 (15 mL). In a well-ventilated fume hood, the suspension was heated to gentle reflux for 8 h, then allowed to cool to ambient temperature. The reaction mixture was filtered through a glass fiber filter and washed with a large amount of DI H2O (~5 L), until the pH of the filtrate was above six. The mass yield of the material dried at 80 °C was greater than 80%.
2.2.2. Control Method 2: HNO3/H2SO4 Oxidation
The ACNF mats (0.5 g) were placed into a loosely stoppered Erlenmeyer flask and a mixture of 13.5 M H2SO4 and 4 M HNO3 (40 mL) was added. The mixture was placed in a fume hood on a horizontal shaker and gently swirled at an ambient temperature for 24 h. The resulting suspension was filtered through a glass fiber filter assembly and the filter cake washed with DI H2O (~1.5 L), until the pH of the filtrate was above six. The mass yield of the modified ACNF dried at 80 °C was lower than 30%.
2.2.3. Low Concentration MnO4 Oxidation (MnO4-L)
The ACNF mats (0.5 g), KMnO4 (61 mg), and aq 0.5 M H2SO4 (50 mL) were placed into a tall beaker. The suspension was sonicated in a Fisher FS30D sonication bath (Thermo Fisher Scientific, Waltham, MA, USA) for 4 h, while the temperature rose to between 40 and 50 °C [18]. As the reaction progressed, the initially purple solution gradually became colorless. The product was recovered by filtration through a glass fiber filter assembly and the filter cake was transferred wet to another flask. A concentration of aq HCl (100 mL) was added and the suspension was warmed to 55 °C for 30 min (to dissolve the manganese oxides that had precipitated onto the ACFN). The final product was collected on a glass fiber filter assembly and DI H2O (~1.0 L) was used to wash the filter cake until the filtrate became colorless and the pH was above six. The mass yield of the modified ACNF dried at 80 °C was in excess of 95%.
2.2.4. High Concentration MnO4 Oxidation (MnO4-H)
The procedure as described for KMnO4-L, except that the ACNF (0.5 g) was reacted with KMnO4 (253 mg) in aq 0.5 M H2SO4 (50 mL). The mass yield of the modified ACNF dried at 80 °C was above 95%.
2.2.5. OsO4 + Oxone® Oxidation
CAUTION: OsO4 is volatile and toxic, handle in well-ventilated fume hood, only wearing appropriate personal protection equipment (nitrile gloves, goggles, and lab coat); consult the Material Safety Data Sheet.
In a well-ventilated fume hood, OsO4 (1.0 g) was dissolved in a volumetric flask in pyridine (50 mL). The ACNF (2.0 g) was suspended in a wide-mouthed flask in 30% pyridine/CHCl3 (180 mL) and 20 mL of the OsO4/pyridine solution was added. The stoppered flask was placed in a fume hood on a horizontal shaker for one week at an ambient temperature. The modified ACNF product was filtered through a glass fiber filter assembly, washed with CH2Cl2 (200 mL), and dried overnight in an oven at 80 °C. A band in the FTIR spectra of this sample at 830 cm−1 indicated the presence of the osmate esters [40,41]. The osmylated ACNF (1.0 g) was suspended in DMF (100 mL), and Oxone® (4.03 g) was added. The suspension was gently swirled at ambient conditions for 3 h. The product was recovered on a glass fiber filter assembly and washed with DI H2O (~300 mL). The product was dried in the oven at 80 °C to produce the final modified ACNF product, at greater than 95% mass yield.
2.2.6. OsO4 + MnO4-L Oxidation
Osmylated ACNF was synthesized, as described in Section 2.2.5. Osmylated ACNF (0.5 g), KMnO4 (61 mg), and aq H2SO4 (0.5 M, 50 mL) were placed in a loosely lidded beaker and suspended in a sonicator (Fisher FS30D) for 4 h. The temperature of the reaction mixture rose to 40 to 45 °C during the sonication. The product was recovered on a glass fiber filter, briefly rinsed with water, and then transferred to a beaker where the concentration of HCl (100 mL) was added. The suspension was warmed to 55 °C for 30 min. The final product was recovered on a glass fiber filter assembly and the filter cake was washed with DI H2O, until the filtrate solution became colorless (~1.0 L). The modified ACNF dried at 80 °C was obtained in mass yields above 95%.
2.2.7. OsO4 + MnO4-H Oxidation
The procedure as described in Section 2.2.6, except that the osmylated ACNF (0.5 g) was reacted with KMnO4 (253 mg) and aq H2SO4 (0.5 M, 50 mL) to obtain the modified ACNF dried at 80 °C in mass yields above 95%.
2.2.8. RuO4 Oxidation
RuO2 (12 mg) and NaOCl (6 wt.% aqueous solution, 180 mL) were stirred at ambient conditions for 60 min in a tall wide-mouthed flask with a lid, in a well-ventilated fume hood. In due process, the color of the solution changed from yellow to light greenish yellow. Then, the ACNF (1.0 g) was added and the reaction was gently agitated for 3 d. The final product was collected on a glass filter assembly and washed with DI H2O (200 mL). The product was dried in the oven at 80 °C and produced an approximate mass yield of a modified ACNF of 50%.
2.3. Physical Characterization of Oxidized ACNF Samples
The constitution of the modified ACNF membranes were assessed using a Field Emission Scanning Electron Microscopy (FESEM, JSM-6335F, 5 kV operation voltage, Jeol, Peabody, MA, USA). The mean fiber diameters were determined by averaging 100 randomly selected nanofibers using ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA).
The specific surface area and pore size distribution were measured with an ASAP 2020 Physisorption Analyzer (Micrometrics Instrument Corporation, Norcross, GA, USA). Prior to the measurements, the samples were degassed at 100 °C for 12 h; N2 sorption isotherms were measured at 77 K using N2. The density functional theory (DFT) assuming an infinite slit pore model was used in conjunction with N2 isotherm data to calculate micropore volumes and pore size distributions [42].
Samples for analysis by Fourier-transform infrared spectroscopy (FTIR, Nicolet Magna 560, Thermo Fisher Scientific, Waltham, MA, USA) were ground and well mixed with pure and dry KBr (Thermo Fisher Scientific, Waltham, MA, USA) powder (carbon loading w/w = 0.4%); pellets were prepared at 14,000 psi pressure. The FTIR spectra of the samples were acquired in transmission mode, with pure KBr as background spectrum.
2.4. Chemical Characterization of Oxidized ACNF Samples
Total carbon, hydrogen, and nitrogen (CHN) contents of 2 mg samples of the differently oxidized ACNFs were analyzed in accordance with an application note for carbon-based materials, using a Perkin Elmer® (Waltham, MA, USA) 2400 CHN analyzer [43]. Prior to analysis, the analyte samples were dried at 140 °C at normal pressure.
The metal contents (Mn, Ru, and Os) were measured following digestion and analysis, using ICP-MS. The digestion procedure followed Balcerzak et al., and the samples were stabilized with a solution of 5% HCl, 0.1% acetic acid, 0.076% thiourea, and 0.01% ascorbic acid [44,45]. The samples were analyzed on an Agilent 7700x inductively coupled plasma mass spectrometer (ICP-MS) with He collision cell (Agilent, Newark, DE, USA). The measured analyte masses/internal standards were 55Mn/45Sc, 101Ru/89Y, and 189Os/209Bi. The standards were purchased from Spex CertiPrep (Metuchen, NJ, USA). The recovery of spiked samples was at least 70%.
The temperature programmed desorption (TPD) profiles were obtained on ~50 mg ACNF samples ground into a Al2O3 crucible using a Netzsch TG 209 F1 Libra instrument (Netzsch, Burlington, MA, USA), coupled to the QMS 403 D Aëolos mass spectrometer (Netzsch, Burlington, MA, USA). The purge gas (flow rate of 20 mL/min) and protective gas (flow rate of 30 mL/min) was Ar; temperature ramp rate 20 °C/min starting at 27 °C to a final T of 1000 °C.
Boehm titrations of 250 mg ACFN samples suspended (and shaken) in water for 24 h at ambient conditions were used to determine the amount of surface active hydrogen groups [46]. We adopted a back-titration technique, as follows: Addition of excess of aqueous bases (either 25.00 mL of 0.05 M NaOH, 0.05 M Na2CO3, or 0.05 M NaHCO3), then back-titration with aqueous 0.05 M HCl. The quantity of NaHCO3 consumed was taken as equal to the amount of carboxylic acids, the equivalence difference between NaHCO3 and Na2CO3 was taken to reflect lactone groups, while the equivalence difference between Na2CO3 and NaOH was translated to the amount of phenol groups [47].
3. Results and Discussion
3.1. Physical Characterization of the Oxidation Products
We inspected the physical appearance of the ACNF membrane after the oxidations. In the case of the HNO3-based oxidations, the membranes disintegrated into powders, as perhaps also expected by the very large mass losses during the treatments (Table 1). We therefore did not include them in the more detailed surface area, pores, functional group, etc., investigations. We note that while we used concentrations reported in the literature, lower acid concentrations or shorter reaction times could perceivably have resulted in the maintenance of the membrane structures.
Oxidation with RuO4 resulted in the lowest yield, while surprisingly still maintaining the membrane structure. The other oxidation treatments resulted in near-quantitative yields and an excellent retention of the membrane structure. We expected the yield to be higher for the more specific oxidants that functionalize only olefinic (or pseudo-olefinic) double bonds or break open a carbon ring, rather than the harsh oxidants that non-selectively degrade nearly all carbon functionalities to CO, CO2, or small carbon acids.
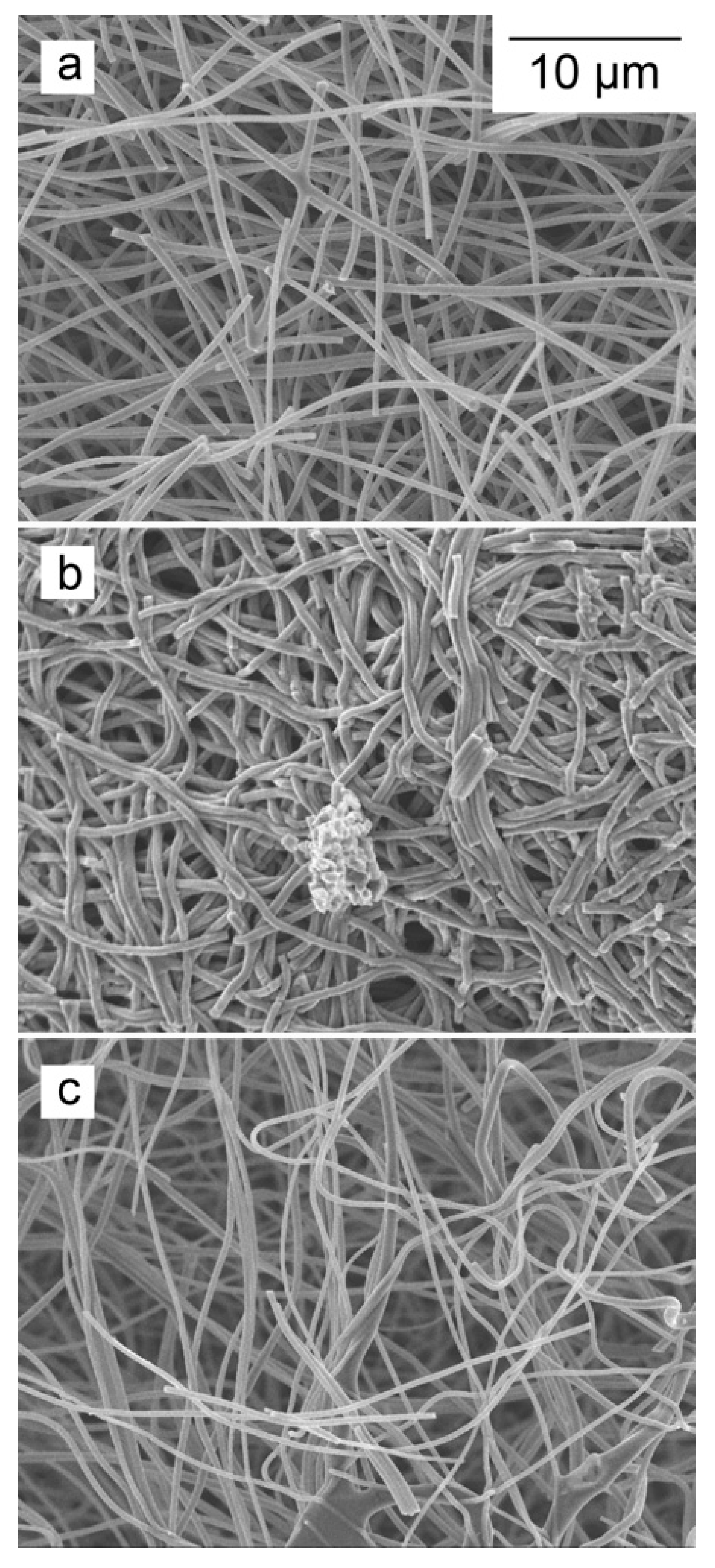
The differences in the degree of degradation also manifest themselves in the physical appearance of the fibers. The SEM image of the untreated ACNF possesses a homogeneous morphology of cylindrical fibers (Figure 1a). After the majority of oxidation treatments, a similar fiber structure was maintained (Figure 1 and Figure S1), but with different fiber diameters. The HNO3/H2SO4-treated samples produced many degraded fibers that collapsed on themselves (Figure 1b). Also, while we could examine small pieces of ACNF membrane material under the electron microscope in the HNO3 and HNO3/H2SO4 treatments, the material changed from a membrane sheet (50 mm × 50 mm) to small (<1 mm2) membrane fragments. Likely as a result of the combination of chemical treatment and the mechanical stress during the sample manipulations, some individual broken fibers and amorphous carbonaceous materials were also observed in the materials treated with MnO4 and RuO4. However, not nearly as many as were observed by us, or Rasheed et al. [18], resulting from the HNO3 oxidation of 60–130 nm graphitized carbon nanofibers. Most importantly, in the OsO4-based oxidation treatments, the nonwoven structure of the material was well maintained (Figure 1c), despite some modest lowering of the average fiber diameters.
Interestingly, there was a significant increase in the average fiber diameter in three cases. In part, the observed increase in the average fiber size after oxidation could be due to amorphous carbon accumulating on the surface, particularly as the largest increases were observed in the samples with the largest material degradation, but the exact nature of the increase is not readily discerned using the SEM images. The observed average fiber size increase after the MnO4-L treatment has also a high variability and might also be due to incomplete washing of the MnO2 off the fibers.
In all cases, the oxidations of ACNF resulted generally in a lower Brunauer-Emmett-Teller (BET) surface area and larger average pore width compared with the unmodified ACNF (Table 2), consistent with the notion of the destruction of (thin) pore walls. This is also evidenced in the increase in average pore width and the corresponding decrease in micropore volume and % microporosity for most of the oxidized materials. This type of oxidative degradation is well-documented, particularly for HNO3-based protocols [34]. Samples with the largest reduction in BET specific surface area, OsO4-Oxone®, OsO4-MnO4-L, and RuO4, all had 3–4-fold increases in the average pore width. Pore size distributions indicate that the oxidized samples that still maintained BET specific surface areas greater than 100 m2/g, generally shifted to one dominant larger size distribution and sometimes some much larger pores in the 100–1000 Å range, from four 0–20 Å peaks in the unmodified material (Figure S2). This is a potentially useful attribute for more homogeneous surface interactions with sorbates. The samples with the least change in the BET surface area, average pore width, and % microporosity were MnO4-L, MnO4-H, and OsO4-MnO4-H treated materials, which all had high yields and insignificant changes in the average fiber diameters (Table 1).
3.2. FTIR Characterization of the Oxidation Products
The characterization of ACNF using FTIR allowed the identification of the presence of a variety of oxygen groups on both the original and the treated materials (Figure 2).
The functional groups that are expected to be present on a carbon material activated with steam at high temperatures include ketones and aldehydes, carboxylic acid functionalities, phenolic groups, and ether groups [48,49]. In the case of the ACNF membrane, there is also a substantial amount of nitrogen present, about 7 mass % by elemental analysis (Table S1), likely in the form of nitrogen-based heterocycles (pyrrole, pyrazole, pyridine, etc., annulated to the carbon framework) [49,50]. The unmodified ACNF material showed two broad peaks centered around 1576 and 1265 cm−1, likely representing a combination of C=N, C–N, O–H, C–O, and N–H bonds. The presence of pyridinic C=N bonds (expected at 1590 cm−1) formed during the stabilization phase of the ACNF fabrication process was shown before [38]. There is also a visible shoulder on the second peak around 1100 cm−1, likely representing the C–O stretching or O–H bending modes of alcoholic, phenolic, ether, and carboxylic groups (carboxylic O–H groups are expected to show at around 1125 cm−1) [25,51,52,53,54]. A broad peak at around 3400 cm−1 represents the water O–H bonds potentially still present or the O–H stretching mode of the phenolic functional groups. A nearly invisible band at 2400 cm−1, visible in most samples is most likely an artefact stemming from residual CO2 in the analysis cavity.
Following oxidation treatments, the IR bands centered around 3400, 1580, and 1260 cm−1 remained, but several subtle shifts or the appearance of new peaks are noted. In the MnO4-L and MnO4-H (data not shown) oxidized material, a peak at around 580 cm−1 can be assigned to the MnOx stretching modes [55], providing direct evidence that not all MnO2 was removed from the ACNF membranes. In the RuO4 and OxO4-Oxone® oxidized materials, small bands were observed at around 850 and 830 cm−1, respectively, which could be representing the M=O vibration of Os and Ru esters [40]. This suggests that some intermediate metal esters are still present. On all oxidized materials, a more or less pronounced shoulder appeared on the first peak at around 1650–1750 cm−1. This is likely a carbonyl group in carboxylic acids (expected to be at 1710–1760 cm−1 in aliphatic carbon chains). The additional O–H groups formed during oxidation could have contributed to the slightly more intense bands at around 1200 cm−1. The presence of overlapping and broad peaks make a clear distinction and quantification of the oxygen functionalities using FTIR difficult.
While all oxidations resulted in a lower percent of carbon of the material, reduced from 75 to 65% or lower, the percent nitrogen increased in all but the OsO4-MnO4 oxidations (Table S1). This suggests a preferential attack on C=C bonds for most oxidations, but potentially, also an attack on some C=N bonds and a loss of N in the OsO4-MnO4 oxidations. However, given the overlap of the bands, this is difficult to discern using the FTIR data only. Some of our findings derived by FTIR could be supported by Boehm titrations, elemental analyses, and the TPD-MS data described below.
3.3. Specification of the Active Hydrogen Functionalities in the Oxidized ACNFs Using Boehm Titrations
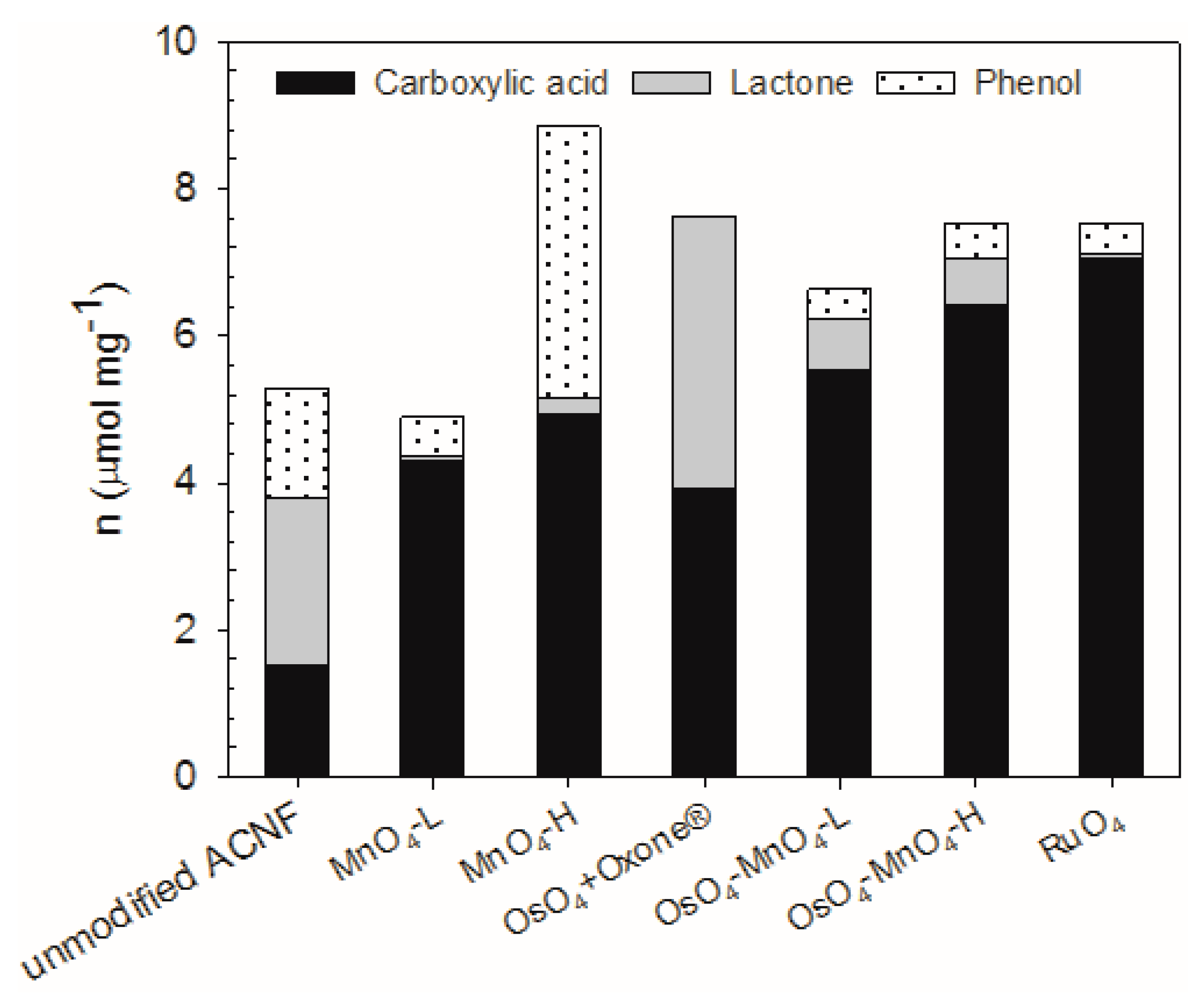
The oxidation treatments produced a change from −7 to +68% in the total oxygen groups on the surface of ACNF, as determined by the Boehm titrations (Figure 3).
In terms of specific oxygen groups, we achieved our goal of increasing the number of carboxylic acid groups (–CO2H) by the oxidations, from 158 to 365%, depending on the method chosen. The RuO4 treatment produced the most –CO2H groups, at 7.07 µmol/mg, which represent 94% of the total acidic hydrogens on that material and a ~5-fold increase in the –CO2H concentration compared with the unmodified ACNF. While the MnO4-L treatment resulted in a decrease in the total acidic oxygen groups by 7%, it still increased the –CO2H groups by almost 3-fold. The least effective oxidation to produce carboxylic acids was the OsO4-Oxone® reaction, but which nevertheless resulted in a 158% increase in –CO2H groups. Parallel to the –CO2H group increase, the number of lactone and phenol groups also decreased in many cases. This is in contrast to other simple oxidants such as ammonium persulfate, which may increase carboxylic acid groups, but more substantially increased phenolic groups on activated carbons [56]. The increased surface functionalization is even more apparent when considering surface area, where compared with the 2.52 µmol/m2 of the –CO2H groups on the unmodified ACNF, the OsO4-MnO4-H treatment resulted in almost a 10-fold increase in –CO2H groups. Although the oxidation reactions reduced the surface area and pore volumes, in several cases the functionalization per unit surface area was significantly increased.
The OsO4- and RuO4-mediated reactions were particularly successful in the introduction of –CO2H and lactone groups. The advantage of using OsO4 in the activation of the carbon structure, followed by a cleavage of the osmate ester by an oxidant (Scheme 1), becomes clear when comparing the outcome of the MnO4-L and MnO4-H reactions with the corresponding OsO4-MnO4-L and OsO4-MnO4-H reactions, respectively. The two-step oxidation introduces more –CO2H groups and less other functionalities. Travis et al. suggested Oxone® as an effective oxidant of osmate esters [57], but it also promotes the formation of esters, a known reactivity of this reagent [58]. While RuO4 introduced the highest yield of carboxylic acid groups, this oxidant is evidently very harsh and less selective than hoped for, as indicated by the small mass yields of these reactions. Evidently, much of the carbon was lost in the form of soluble forms of carbon or CO2. A high percentage of –CO2H groups were also observed in the RuO4 oxidation of other graphitized carbon materials [18].
3.4. TPD-MS Analyses of the Oxidized ACNFs
In temperature programmed desorption mass spectrometry (TPD-MS), a sample is heated at a predetermined rate and the temperature-dependent emissions of small molecule fragments are recorded by mass spectrometry, whereby channels for specific fragments are monitored in parallel. Thus, the TPD-MS data for CO and CO2 from an ambient temperature up to 1200 K were gathered. Carboxylic acids decarboxylate at relatively low temperatures (350–680 K), while lactones would decarboxylate and anhydrides decarbonylate/decarboxylate at higher temperatures (450–950 K and 620–900 K, respectively) [23,24,48,59]. Phenols, ethers, carbonyls, and quinones only start to decompose at relatively high temperatures (973–1253 K) [48]. Our analysis of different oxygen groups on the ACNF is mainly a comparison of the shape and position of TPD-MS peaks following different treatments. A deconvolution of the TPD-MS profiles into a series of smaller peaks corresponding to specific oxygen groups was, given the broadness of the signals, not attempted.
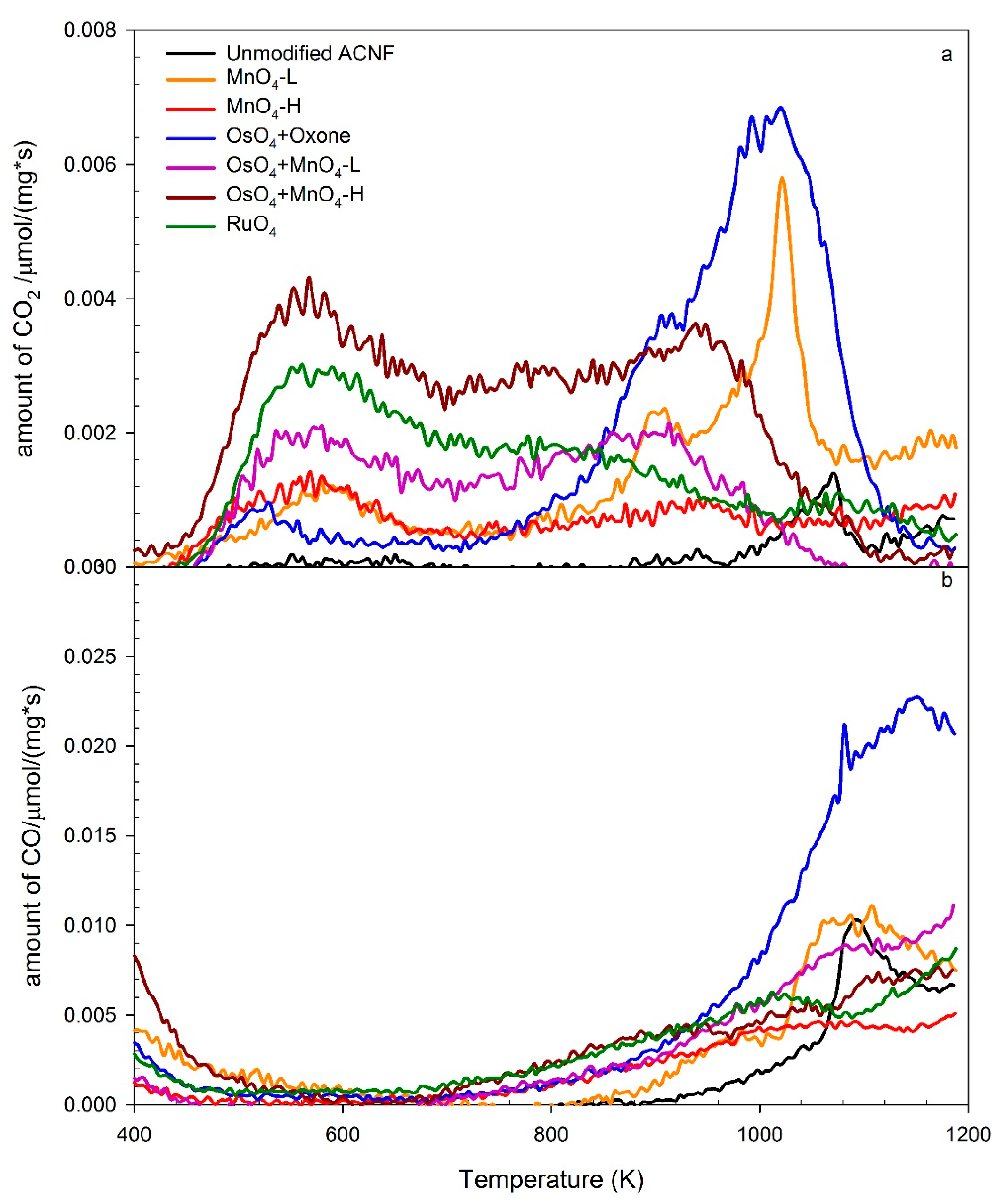
Based on the broad distribution of the CO2 evolution, the TPD-MS data suggest the presence of clearly different surface oxygen chemical environments on the different materials. All samples decarboxylated at the temperature range between 400–700 K, indicating the presence of significant amounts of –CO2H groups (Figure 4a). The ACNF treatments resulted in a substantial increase in CO2 peak area reflecting the increase of carboxylic acid functionalities, following the sequence of OsO4-MnO4-H > RuO4 > OsO4-MnO4-L > MnO4-H > MnO4-L > unmodified ACNF. The release of CO2 from OsO4-MnO4-H started at a lower temperature than others, perhaps an effect of the presence of adjacent functional groups. On the other hand, as the temperature increases, the carboxylic acid groups located on adjacent sites might dehydrate to form carboxylic acid anhydrides at lower temperatures, and ultimately eliminate CO2 and CO at higher temperatures.
The second dominant CO2 peak can attribute to lactones and carboxylic anhydrides [52]. While there are some distinct peaks in the CO2 trace, typically around 900–1000 K or around 1000–1100 K (Figure 4a), there is a broad peak in the CO trace, starting around 900 up to 1200 K (Figure 4b). It is notable that the second dominant CO2 peak of the samples with oxidants of MnO4-L, MnO4-H, OsO4-MnO4-L, and OsO4-MnO4-H rose earlier than the unmodified ACNF, samples oxidized by OsO4-Oxone® and RuO4, all MnO4–based primary or secondary oxidations, and OsO4-Oxone®, but not with the RuO4 oxidations, showed a distinct peak at around 900 K for CO2, and above 1000 K for CO, which we interpret as an indicator for the presence of carbonyl and quinones, which are known to produce CO at 900–1200 K [52]. The most significant difference between the samples was a peak around 1040–1080 K in the CO2 channel, most notable in the OsO4-Oxone® sample, and to a lesser extent in MnO4-L and unmodified ACNF. The corresponding CO trace showed a peak for those materials as well in a similar temperature range, starting at 1040 K and up to 1160K in the case of OsO4-Oxone®. Although this is outside the typical range for anhydrides, there is some overlap in CO and CO2 traces in this case, suggestive that the peaks originate from carboxylic acid anhydride functionalities.
A larger portion of the CO2 trace is likely due to lactones. In fact, both the TPD-MS data (peak position and shape) and Boehm titrations are a good match for lactones. The high content of carboxylic acid in RuO4 or OsO4-MnO4-H modified ACNF, as deduced from the TPD data, also largely correspond to the Boehm titration data. Others have also found good relative comparisons of oxygen groups when comparing Boehm titrations and TPD data, but quantitation can deviate because of the differences in accessibility of the oxygen group to the analytical tool [60]. In addition, the Boehm titration methods are limited to the identification of the acidic carboxyl acid, lactone, and phenol groups, and ignore the portions of oxygen functionalities, such as ethers and ketones. Furthermore, while the Boehm titration method provides the total amount of titratable carboxylic acids, it cannot provide any information about their relative placements.
4. Conclusions
The choice of the oxidation method to establish oxygen functionalities in general, and carboxylic acid groups in particular, on ACNF materials, has a dramatic effect on the chemical yield, the physical appearance, and the chemical composition of the functionalized material. The use of HNO3 (with or without the addition of H2SO4) proved to be the most destructive method, leading to a disintegration of the membranes. Considering a balance between limiting fiber damage, maximizing yield, maintaining pore distribution, and increasing the number of carboxylic acid groups, the OsO4 treatments that followed with high concentrations of the secondary oxidant MnO4 provided the best results. Notably, the prior activation of the carbonaceous material with OsO4 yielded much better results than using only an identical MnO4 treatment. This indicates that OsO4 was able to oxidize the double bonds that were inaccessible to KMnO4, but without causing broad structural damage. Overall, the Boehm titrations qualitatively and quantitatively showed that RuO4 produced the highest percentage of carboxylic acids among all of the oxidants, per unit mass of modified ACNF, but at the expense of the chemical yield. We thus also identified RuO4 as another promising oxidant. However, further optimization experiments to increase the yield of the modified ACNF would be required for this oxidation pathway to become a practical method.
The chemical oxidation protocols tested generated multiple different products suitable for, for example, sorbent or catalyst attachment. Given the known chemistry of OsO4 and RuO4, a benefit of these oxidants is the expected formation of carboxylic acid functionalities arranged as pairs on the carbon framework surface. When considering further functionalizing the surface in the future, the placement of adjacent secondary functional groups will increase the likelihood of generating (e.g., more selective chelating interactions).
In conclusion, alternative chemical oxidation pathways to establish specific surface oxygen groups on an ACNF material with generally graphitic carbon structures and relatively high nitrogen content was shown. The effectiveness of the OsO4- and RuO4-based oxidations on other carbon nanomaterials is worth considering in the future.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2311-5629/4/3/40/s1, Figure S1: SEM images of oxidized ACNF; Figure S2: Pore volume distribution of oxidized ACNF; Table S1: Elemental analysis of oxidized ACNF samples.
Author Contributions
All authors conceived and designed the experiments, analyzed the data, and wrote the paper. Y.H. and R.L. performed the experiments.
Funding
This work was supported by the U.S. National Science Foundation under Grant CBET 1438518 to Timothy M. Vadas and Christian Brückner.
Acknowledgments
Yi Han acknowledges support from the Chinese Scholarship Council. The SEM studies were performed using the facilities in the UConn/FEI Center for Advanced Microscopy and Materials Analysis (CAMMA). We thank Jeffrey R. McCutcheon and Basma Waisi, the University of Connecticut, for providing the ACNF material, and Eileen Meehan, the University of Connecticut, for preliminary experiments.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Mauter, M.S.; Elimelech, M. Environmental applications of carbon-based nanomaterials. Environ. Sci. Technol. 2008, 42, 5843–5859. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.P.; Lu, C.; Su, F. Sorption of divalent metal ions from aqueous solution by carbon nanotubes: A review. Sep. Purif. Technol. 2007, 58, 224–231. [Google Scholar] [CrossRef]
- Hummer, G.; Rasaiah, J.C.; Noworyta, J.P. Water conduction through the hydrophobic channel of a carbon nanotube. Nature 2001, 414, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Vecitis, C.D.; Schnoor, M.H.; Rahaman, M.S.; Schiffman, J.D.; Elimelech, M. Electrochemical multiwalled carbon nanotube filter for viral and bacterial removal and inactivation. Environ. Sci. Technol. 2011, 45, 3672–3679. [Google Scholar] [CrossRef] [PubMed]
- Al-Saleh, M.H.; Sundararaj, U. A review of vapor grown carbon nanofiber/polymer conductive composites. Carbon 2009, 47, 2–22. [Google Scholar] [CrossRef]
- Jang, J.; Bae, J. Carbon nanofiber/polypyrrole nanocable as toxic gas sensor. Sens. Actuators B Chem. 2007, 122, 7–13. [Google Scholar] [CrossRef]
- Mitchell, R.R.; Gallant, B.M.; Thompson, C.V.; Shao-Horn, Y. All-carbon-nanofiber electrodes for high-energy rechargeable Li-O2 batteries. Energy Environ. Sci. 2011, 4, 2952–2958. [Google Scholar] [CrossRef]
- Faur-Brasquet, C.; Kadirvelu, K.; Le Cloirec, P. Removal of metal ions from aqueous solution by adsorption onto activated carbon cloths: Adsorption competition with organic matter. Carbon 2002, 40, 2387–2392. [Google Scholar] [CrossRef]
- Gao, L.; Wang, X.; Xie, Z.; Song, W.; Wang, L.; Wu, X.; Qu, F.; Chen, D.; Shen, G. High-performance energy-storage devices based on WO3 nanowire arrays/carbon cloth integrated electrodes. J. Mater. Chem. A 2013, 1, 7167–7173. [Google Scholar] [CrossRef]
- Zhao, F.; Rahunen, N.; Varcoe, J.R.; Chandra, A.; Avignone-Rossa, C.; Thumser, A.E.; Slade, R.C.T. Activated carbon cloth as anode for sulfate removal in a microbial fuel cell. Environ. Sci. Technol. 2008, 42, 4971–4976. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Shiratori, N.; Lee, G.H.; Miyawaki, J.; Mochida, I.; Yoon, S.-H.; Jang, J. Activated carbon nanofiber produced from electrospun polyacrylonitrile nanofiber as a highly efficient formaldehyde adsorbent. Carbon 2010, 48, 4248–4255. [Google Scholar] [CrossRef]
- Fan, Z.; Yan, J.; Wei, T.; Zhi, L.; Ning, G.; Li, T.; Wei, F. Asymmetric supercapacitors based on graphene/MnO2 and activated carbon nanofiber electrodes with high power and energy density. Adv. Funct. Mater. 2011, 21, 2366–2375. [Google Scholar] [CrossRef]
- Kim, C.; Yang, K.S. Electrochemical properties of carbon nanofiber web as an electrode for supercapacitor prepared by electrospinning. Appl. Phys. Lett. 2003, 83, 1216–1218. [Google Scholar] [CrossRef]
- Qu, X.; Brame, J.; LI, Q.; Alvarez, P.J.J. Nanotechnology for a safe and sustainable water supply: Enabling integrated water treatment and reuse. Acc. Chem. Res. 2012, 46, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Dai, L.; Gao, M.; Huang, S.; Mau, A. Plasma activation of carbon nanotubes for chemical modification. J. Phys. Chem. B 2001, 105, 618–622. [Google Scholar] [CrossRef]
- Karousis, N.; Tagmatarchis, N.; Tasis, D. Current progress on the chemical modification of carbon nanotubes. Chem. Rev. 2010, 110, 5366–5397. [Google Scholar] [CrossRef] [PubMed]
- Morales-Lara, F.; Pérez-Mendoza, M.J.; Altmajer-Vaz, D.; García-Román, M.; Melguizo, M.; López-Garzón, F.J.; Domingo-García, M. Functionalization of multiwall carbon nanotubes by ozone at basic pH. Comparison with oxygen plasma and ozone in gas phase. J. Phys. Chem. C 2013, 117, 11647–11655. [Google Scholar] [CrossRef]
- Rasheed, A.; Howe, J.Y.; Dadmun, M.D.; Britt, P.F. The efficiency of the oxidation of carbon nanofibers with various oxidizing agents. Carbon 2007, 45, 1072–1080. [Google Scholar] [CrossRef]
- Saito, T.; Matsushige, K.; Tanaka, K. Chemical treatment and modification of multi-walled carbon nanotubes. Phys. B Condens. Matter 2002, 323, 280–283. [Google Scholar] [CrossRef]
- Kumar, S.; Rath, T.; Mahaling, R.N.; Reddy, C.S.; Das, C.K.; Pandey, K.N.; Srivastava, R.B.; Yadaw, S.B. Study on mechanical, morphological and electrical properties of carbon nanofiber/polyetherimide composites. Mater. Sci. Eng. B Solid State Adv. Technol. 2007, 141, 61–70. [Google Scholar] [CrossRef]
- Wang, J.; Lin, Y. Functionalized carbon nanotubes and nanofibers for biosensing applications. TrAC Trends Anal. Chem. 2008, 27, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampalanonwat, P.; Supaphol, P. Preparation and adsorption behavior of aminated electrospun polyacrylonitrile nanofiber mats for heavy metal ion removal. ACS Appl. Mater. Interfaces 2010, 2, 3619–3627. [Google Scholar] [CrossRef] [PubMed]
- Toebes, M.L.; van Heeswijk, J.M.P.; Bitter, J.H.; Jos van Dillen, A.; de Jong, K.P. The influence of oxidation on the texture and the number of oxygen-containing surface groups of carbon nanofibers. Carbon 2004, 42, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.-H.; Sui, Z.-J.; Zhu, J.; Li, P.; Chen, D.; Dai, Y.-C.; Yuan, W.-K. Characterization of surface oxygen complexes on carbon nanofibers by TPD, XPS and FT-IR. Carbon 2007, 45, 785–796. [Google Scholar] [CrossRef]
- Pradhan, B.K.; Sandle, N.K. Effect of different oxidizing agent treatments on the surface properties of activated carbons. Carbon 1999, 37, 1323–1332. [Google Scholar] [CrossRef]
- Kyzas, G.Z.; Lazaridis, N.K.; Deliyanni, E.A. Oxidation time effect of activated carbons for drug adsorption. Chem. Eng. J. 2013, 234, 491–499. [Google Scholar] [CrossRef]
- Datsyuk, V.; Kalyva, M.; Papagelis, K.; Parthenios, J.; Tasis, D.; Siokou, A.; Kallitsis, I.; Galiotis, C. Chemical oxidation of multiwalled carbon nanotubes. Carbon 2008, 46, 833–840. [Google Scholar] [CrossRef]
- Rosca, I.D.; Watari, F.; Uo, M.; Akasaka, T. Oxidation of multiwalled carbon nanotubes by nitric acid. Carbon 2005, 43, 3124–3131. [Google Scholar] [CrossRef]
- Moreno-Castilla, C.; Ferro-Garcia, M.; Joly, J.; Bautista-Toledo, I.; Carrasco-Marin, F.; Rivera-Utrilla, J. Activated carbon surface modifications by nitric acid, hydrogen peroxide, and ammonium peroxydisulfate treatments. Langmuir 1995, 11, 4386–4392. [Google Scholar] [CrossRef]
- Hernadi, K.; Siska, A.; Thiên-Nga, L.; Forró, L.; Kiricsi, I. Reactivity of different kinds of carbon during oxidative purification of catalytically prepared carbon nanotubes. Solid State Ion. 2001, 141–142, 203–209. [Google Scholar] [CrossRef]
- Pereira, M.F.R.; Soares, S.F.; Órfão, J.J.M.; Figueiredo, J.L. Adsorption of dyes on activated carbons: Influence of surface chemical groups. Carbon 2003, 41, 811–821. [Google Scholar] [CrossRef]
- Sullivan, P.D.; Stone, B.R.; Hashisho, Z.; Rood, M.J. Water adsorption with hysteresis effect onto microporous activated carbon fabrics. Adsorption 2007, 13, 173–189. [Google Scholar] [CrossRef]
- Li, R.; Zhang, L.; Wang, P. Rational design of nanomaterials for water treatment. Nanoscale 2015, 7, 17167–17194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivera-Utrilla, J.; Sánchez-Polo, M.; Gómez-Serrano, V.; Alvarez, P.; Alvim-Ferraz, M.; Dias, J. Activated carbon modifications to enhance its water treatment applications. An overview. J. Hazard. Mater. 2011, 187, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M. Osmium tetroxide cis-hydroxylation of unsaturated substrates. Chem. Rev. 1980, 80, 187–213. [Google Scholar] [CrossRef]
- Hwang, K.C. Efficient cleavage of carbon graphene layers by oxidants. J. Chem. Soc. Chem. Commun. 1995, 2, 173–174. [Google Scholar] [CrossRef]
- Berkowitz, L.M.; Rylander, P.N. Use of ruthenium tetroxide as a multi-purpose oxidant. J. Am. Chem. Soc. 1958, 80, 6682–6684. [Google Scholar] [CrossRef]
- Manickam, S.S.; Karra, U.; Huang, L.; Bui, N.-N.; Li, B.; McCutcheon, J.R. Activated carbon nanofiber anodes for microbial fuel cells. Carbon 2013, 53, 19–28. [Google Scholar] [CrossRef]
- Bai, Y.; Huang, Z.-H.; Kang, F. Surface oxidation of activated electrospun carbon nanofibers and their adsorption performance for benzene, butanone and ethanol. Colloids Surf. A 2014, 443, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Collin, R.J.; Jones, J.; Griffith, W.P. Reaction of osmium tetroxide with alkenes, glycols, and alkynes; oxo-osmium(VI) esters and their structures. J. Chem. Soc. Dalton Trans. 1974, 10, 1094–1097. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Eder, S.J.; Scherer, W. Mehrfachbindungen zwischen Hauptgruppenelementen und Übergangsmetallen, CIX. Strukturchemische Aspekte der Fluorolefinchemie von Osmiumtetraoxid: Halogenierte Osmatester und Folgeprodukte. Chem. Ber. 1993, 126, 39–43. [Google Scholar] [CrossRef]
- Jagiello, J.; Olivier, J.P. A simple two-dimensional NLDFT model of gas adsorption in finite carbon pores. Application to pore structure analysis. J. Phys. Chem. C 2009, 113, 19382–19385. [Google Scholar] [CrossRef]
- Culmo, R.F. The Elemental Analysis of Various Classes of Chemical Compounds Using CHN; PerkinElmer, Inc.: Shelton, CT, USA, 2013. [Google Scholar]
- Balcerzak, M.; Świecicka, E.; Bystrońska, D. Simple selective spectrophotometric method for the determination of ruthenium in carbon supported Pt-Ru-Ge catalyst. Anal. Lett. 1999, 32, 1799–1805. [Google Scholar] [CrossRef]
- Chahrour, O.; Malone, J.; Collins, M.; Salmon, V.; Greenan, C.; Bombardier, A.; Ma, Z.; Dunwoody, N. Development and validation of an ICP-MS method for the determination of elemental impurities in TP-6076 active pharmaceutical ingredient (API) according to USP 〈232〉/〈233〉. J. Pharm. Biomed. Anal. 2017, 145, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Boehm, H. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 1994, 32, 759–769. [Google Scholar] [CrossRef]
- Boehm, H. Chemical identification of surface groups. Adv. Catal. 1966, 16, 179–274. [Google Scholar]
- Shafeeyan, M.S.; Daud, W.M.A.W.; Houshmand, A.; Shamiri, A. A review on surface modification of activated carbon for carbon dioxide adsorption. J. Anal. Appl. Pyrolysis 2010, 89, 143–151. [Google Scholar] [CrossRef]
- Stuart, B. Infrared Spectroscopy: Fundamentals and Applications; John Wiley & Sons: New York, NY, USA, 2004. [Google Scholar]
- McGann, J.P.; Zhong, M.; Kim, E.K.; Natesakhawat, S.; Jaroniec, M.; Whitacre, J.F.; Matyjasqewski, K.; Kowalewski, T. Block copolymer templating as a path to porous nanostructured carbons with highly accessible nitrogens for enhanced (electro)chemical performance. Macromol. Chem. Phys. 2012, 213, 1078–1090. [Google Scholar] [CrossRef]
- Dandekar, A.; Baker, R.; Vannice, M. Characterization of activated carbon, graphitized carbon fibers and synthetic diamond powder using tpd and drifts. Carbon 1998, 36, 1821–1831. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Kalijadis, A.M.; Vukčević, M.M.; Jovanović, Z.M.; Laušević, Z.V.; Laušević, M.D. Characterization of surface oxygen groups on different carbon materials by the Boehm method and temperature programmed desorption. J. Serbian Chem. Soc. 2011, 76, 757–768. [Google Scholar] [CrossRef]
- Mangun, C.L.; Benak, K.R.; Daley, M.A.; Economy, J. Oxidation of activated carbon fibers: Effect on pore size, surface chemistry, and adsorption properties. Chem. Mater. 1999, 11, 3476–3483. [Google Scholar] [CrossRef]
- Parikh, S.J.; Chorover, J. FTIR spectroscopic study of biogenic Mn-oxide formation by Pseudomonas putida GB-1. Geomicrobiol. J. 2005, 22, 207–218. [Google Scholar] [CrossRef]
- Bhadra, B.N.; Seo, P.W.; Jhung, S.H. Adsorption of Diclofenac sodium from water using oxidized activated carbon. Chem. Eng. J. 2016, 301, 27–34. [Google Scholar] [CrossRef]
- Travis, B.R.; Narayan, R.S.; Borhan, B. Osmium tetroxide-promoted catalytic oxidative cleavage of olefins: An organometallic ozonolysis. J. Am. Chem. Soc. 2002, 124, 3824–3825. [Google Scholar] [CrossRef] [PubMed]
- Schomake, J.M.; Travis, B.R.; Borhan, B. Direct lactonization of alkenols via osmium tetroxide-mediated oxidative cleavage. Org. Lett. 2003, 5, 3089–3092. [Google Scholar] [CrossRef] [PubMed]
- De la Puente, G.; Pis, J.J.; Menéndez, J.A.; Grange, P. Thermal stability of oxygenated functions in activated carbons. J. Anal. Appl. Pyrolysis 1997, 43, 125–138. [Google Scholar] [CrossRef]
- Schönherr, J.; Buchheim, J.R.; Scholz, P.; Adelhelm, P. Boehm titration revisited (part II): A comparison of Boehm titration with other analytical techniques on the quantification of oxygen-containing surface groups for a variety of carbon materials. C 2018, 4, 22. [Google Scholar] [CrossRef]
Scheme 1.
Step-wise reaction of the OsO4-mediated oxidation of a double bond.

Figure 1.
Field emission scanning electron microscopy images of (a) unmodified activated carbon nanofiber membrane (ACNF); (b) HNO3/H2SO4 oxidized ACNF; and (c) OsO4-MnO4-H oxidized ACNF.
Figure 1.
Field emission scanning electron microscopy images of (a) unmodified activated carbon nanofiber membrane (ACNF); (b) HNO3/H2SO4 oxidized ACNF; and (c) OsO4-MnO4-H oxidized ACNF.

Figure 2.
FTIR spectra of the different oxidized ACNF species indicated. Spectra are at the same absorbance scale, but are arbitrarily stacked for comparison. Sample MnO4-H was omitted as it had an identical appearance to MnO4-L.
Figure 2.
FTIR spectra of the different oxidized ACNF species indicated. Spectra are at the same absorbance scale, but are arbitrarily stacked for comparison. Sample MnO4-H was omitted as it had an identical appearance to MnO4-L.
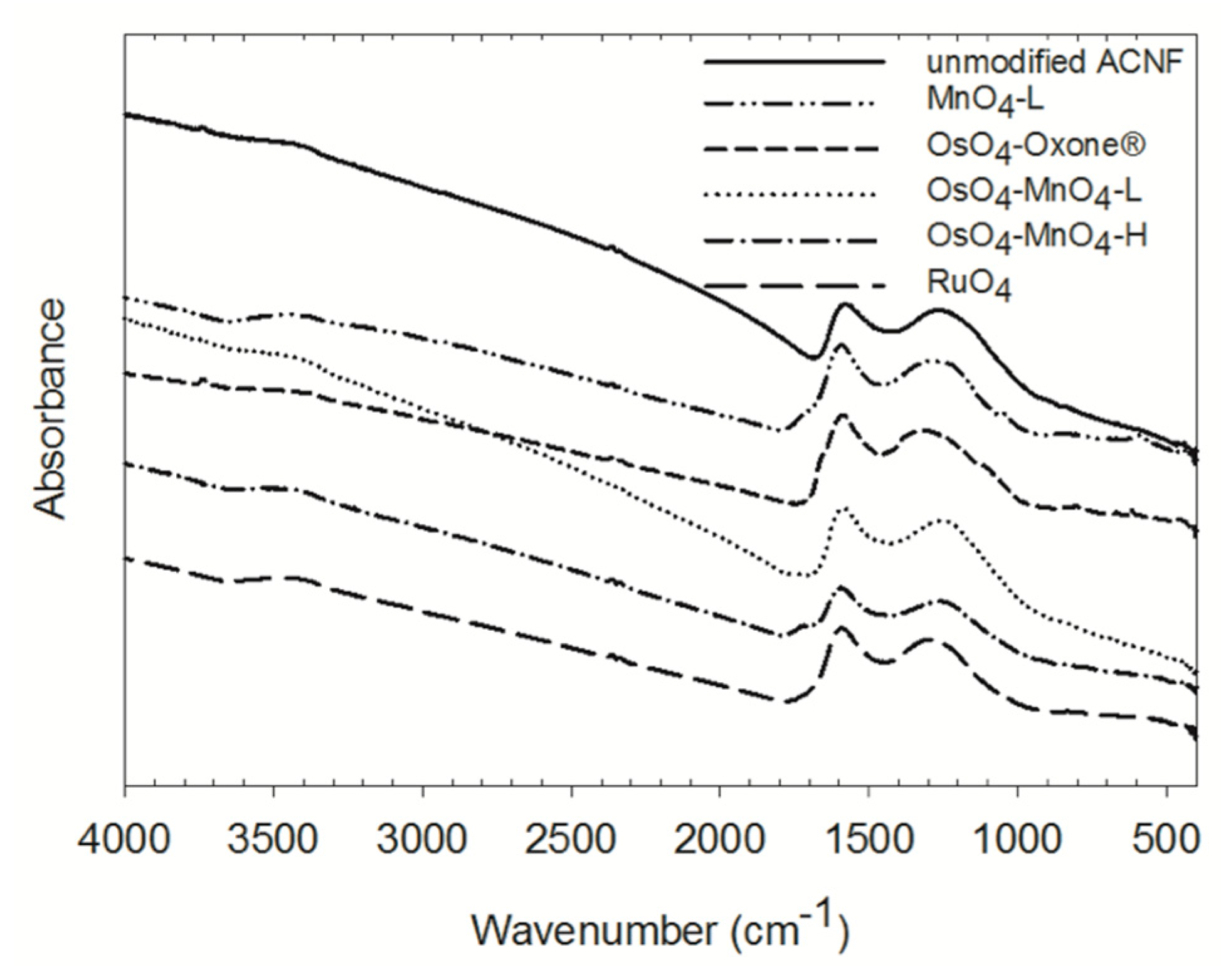
Figure 3.
Comparison of acidic functional groups composition in the ACNF samples following different oxidation pathways, as determined using the Boehm titration method.
Figure 3.
Comparison of acidic functional groups composition in the ACNF samples following different oxidation pathways, as determined using the Boehm titration method.
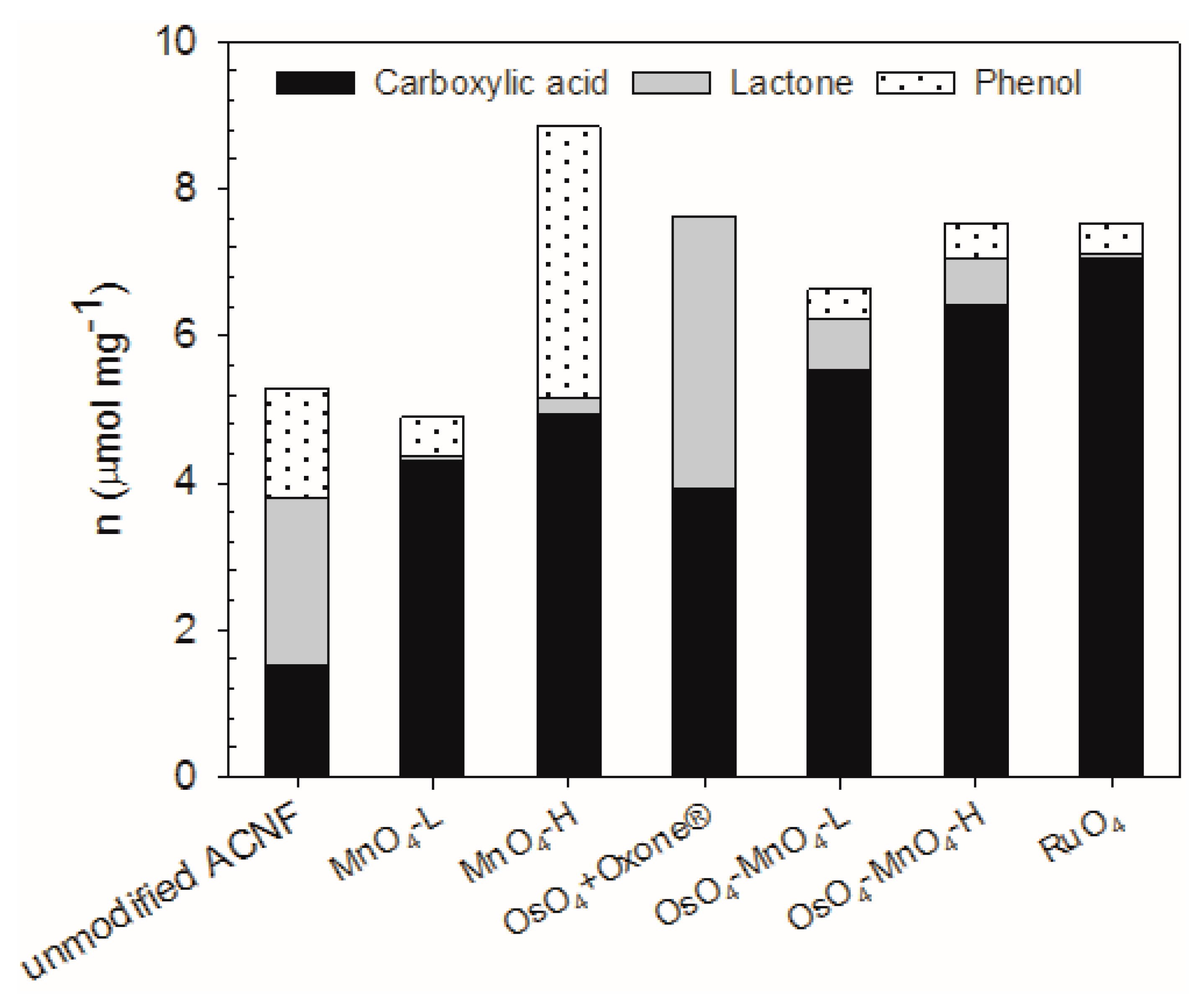
Figure 4.
Temperature programmed desorption mass spectrometry (TPD-MS) traces for the release of CO2 (a) and CO (b) of the unmodified and modified ACNF samples indicated. Note: the mass trace below 500 in the CO trace is suspected residual N2 gas flushing from the sample chamber.
Figure 4.
Temperature programmed desorption mass spectrometry (TPD-MS) traces for the release of CO2 (a) and CO (b) of the unmodified and modified ACNF samples indicated. Note: the mass trace below 500 in the CO trace is suspected residual N2 gas flushing from the sample chamber.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of physical properties after oxidation treatment.
| Treatment | Mass Yield (%) | Average Fiber Diameter (nm) | Visible Physical Change of Membrane |
|---|---|---|---|
| Unmodified | – | 430 ± 65 | – |
| HNO3 | 82.4 | 285 ± 41 | powdered material |
| HNO3/H2SO4 | 30.0 | 529 ± 73 | powdered material |
| MnO4-L | 99.1 | 534 ± 120 | none |
| MnO4-H | 105 1 | 345 ± 58 | none |
| OsO4 + Oxone | 97.2 | 300 ± 39 | none |
| OsO4 + MnO4-L | 98.8 | 384 ± 73 | none |
| OsO4 + MnO4-H | 96.5 | 359 ± 94 | none |
| RuO4 | 49.6 | 499 ± 66 | none |
1 Likely due to an incomplete washing out of the manganese oxide reaction products.
Table 2.
Brunauer-Emmett-Teller (BET) specific surface area and pore distribution change in activated carbon nanofiber membrane (ACNF) after oxidation treatments.
Table 2.
Brunauer-Emmett-Teller (BET) specific surface area and pore distribution change in activated carbon nanofiber membrane (ACNF) after oxidation treatments.
| Sample | BET Specific Surface Area (m2/g) | Average Pore Width (Å) | Micropore Volume (cm3/g) | Total Pore Volume (cm3) | Microporosity (%) |
|---|---|---|---|---|---|
| Unmodified | 604 | 20.5 | 0.24 | 0.25 | 96 |
| MnO4-L | 134 | 22.4 | 0.048 | 0.066 | 73 |
| MnO4-H | 457 | 24.9 | 0.17 | 0.17 | 100 |
| OsO4-Oxone® | 21.9 | 56.8 | 0.001 | 0.067 | 1.5 |
| OsO4-MnO4-L | 8.44 | 65.2 | 0.0003 | 0.017 | 1.8 |
| OsO4-MnO4-H | 294 | 21.7 | 0.11 | 0.13 | 85 |
| RuO4 | 7.62 | 83.9 | 0.002 | 0.015 | 13 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, Y.; Li, R.; Brückner, C.; Vadas, T.M. Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation. C 2018, 4, 40. https://0-doi-org.brum.beds.ac.uk/10.3390/c4030040
AMA Style
Han Y, Li R, Brückner C, Vadas TM. Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation. C. 2018; 4(3):40. https://0-doi-org.brum.beds.ac.uk/10.3390/c4030040
Chicago/Turabian StyleHan, Yi, Ruoshi Li, Christian Brückner, and Timothy M. Vadas. 2018. "Controlling the Surface Oxygen Groups of Polyacrylonitrile-Based Carbon Nanofiber Membranes While Limiting Fiber Degradation" C 4, no. 3: 40. https://0-doi-org.brum.beds.ac.uk/10.3390/c4030040
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.