Analyzing the Raman Spectra of Graphenic Carbon Materials from Kerogens to Nanotubes: What Type of Information Can Be Extracted from Defect Bands?

 , , , ,
, , , , 
Abstract
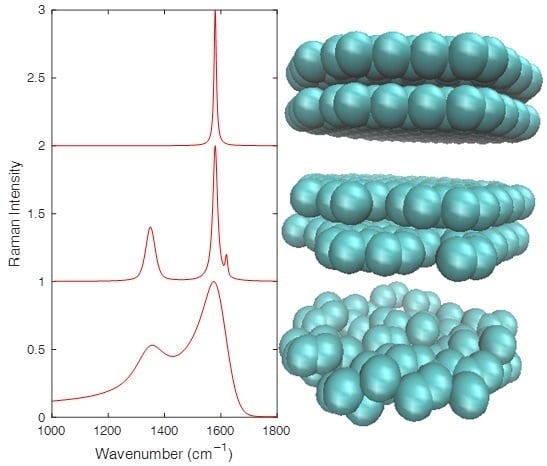
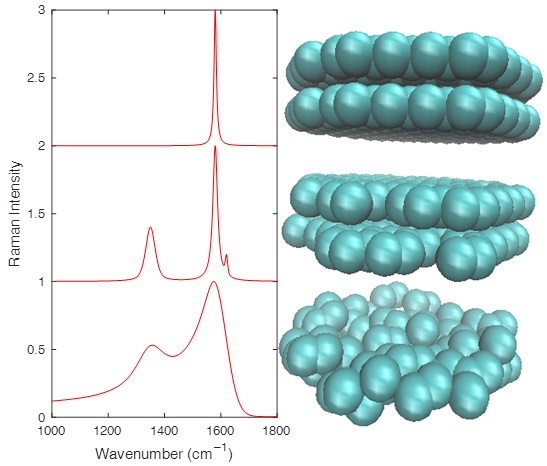
:
1. Introduction
2. Experimental Section
2.1. Samples
2.2. Experimental Conditions
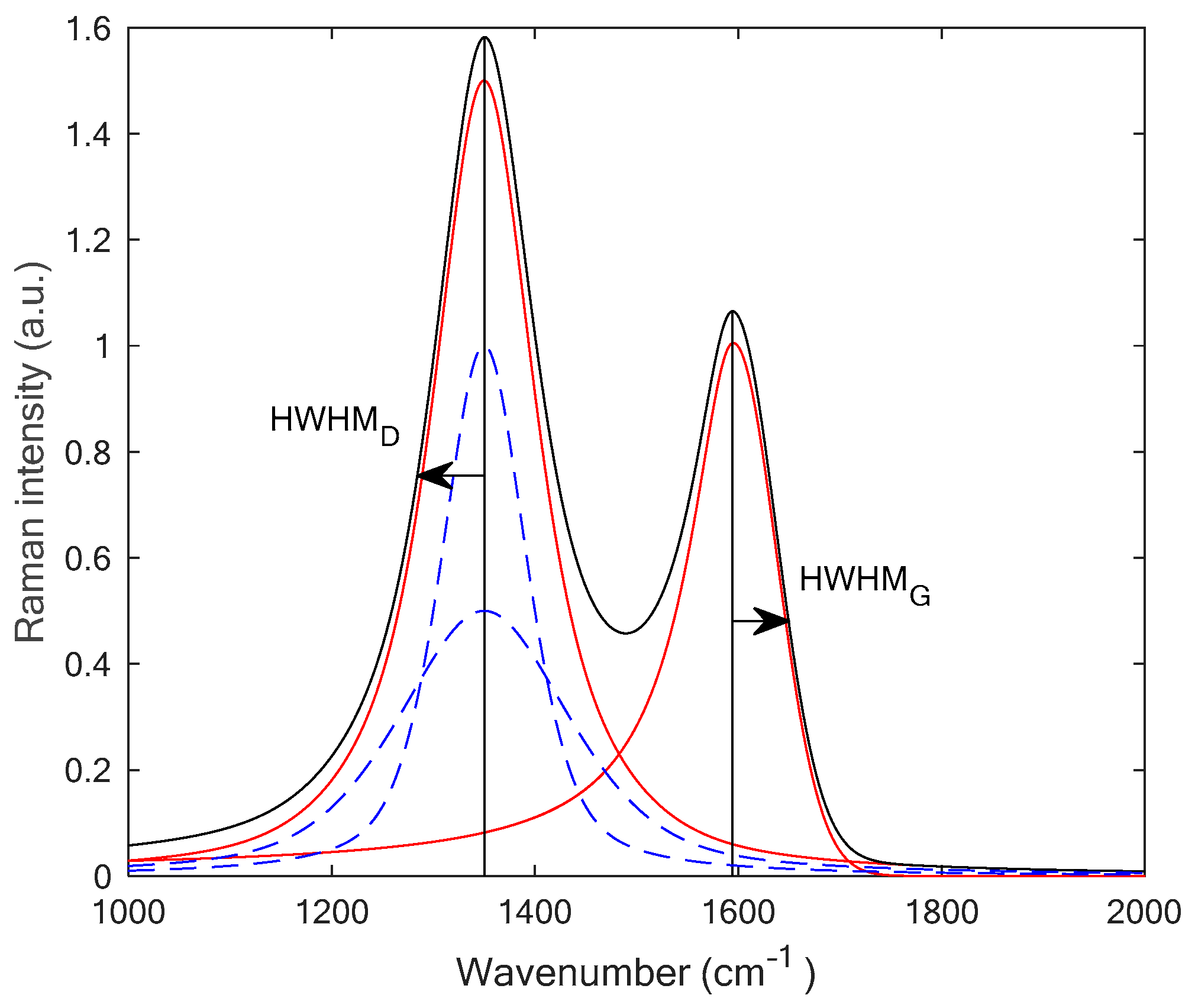
2.3. Fitting Procedure
2.4. La, Lc, and LD Parameters and Experimental Laws in the Literature
3. Experiments on Poorly Ordered Graphenic Carbon Materials
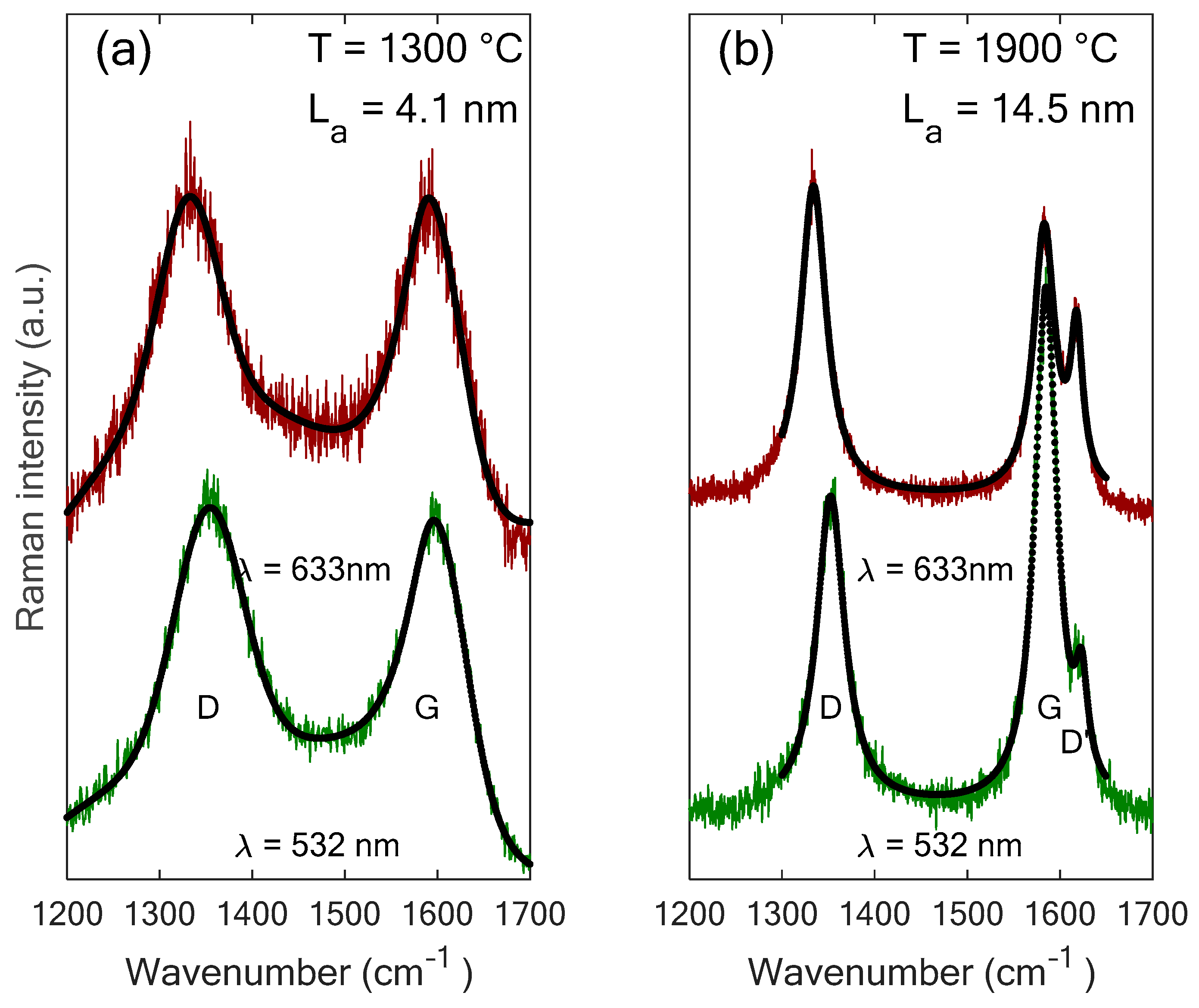
3.1. Crystallite Size Lower Than 2 nm − The Example of Gas-Shales
3.2. Crystallite Size Directly Above 2 nm: The Example of Carbon Black
3.3. La Much Larger Than 2 nm: The Example of Coal Tar Pitch Cokes
4. Experiments on Highly Ordered Graphenic Carbon Materials
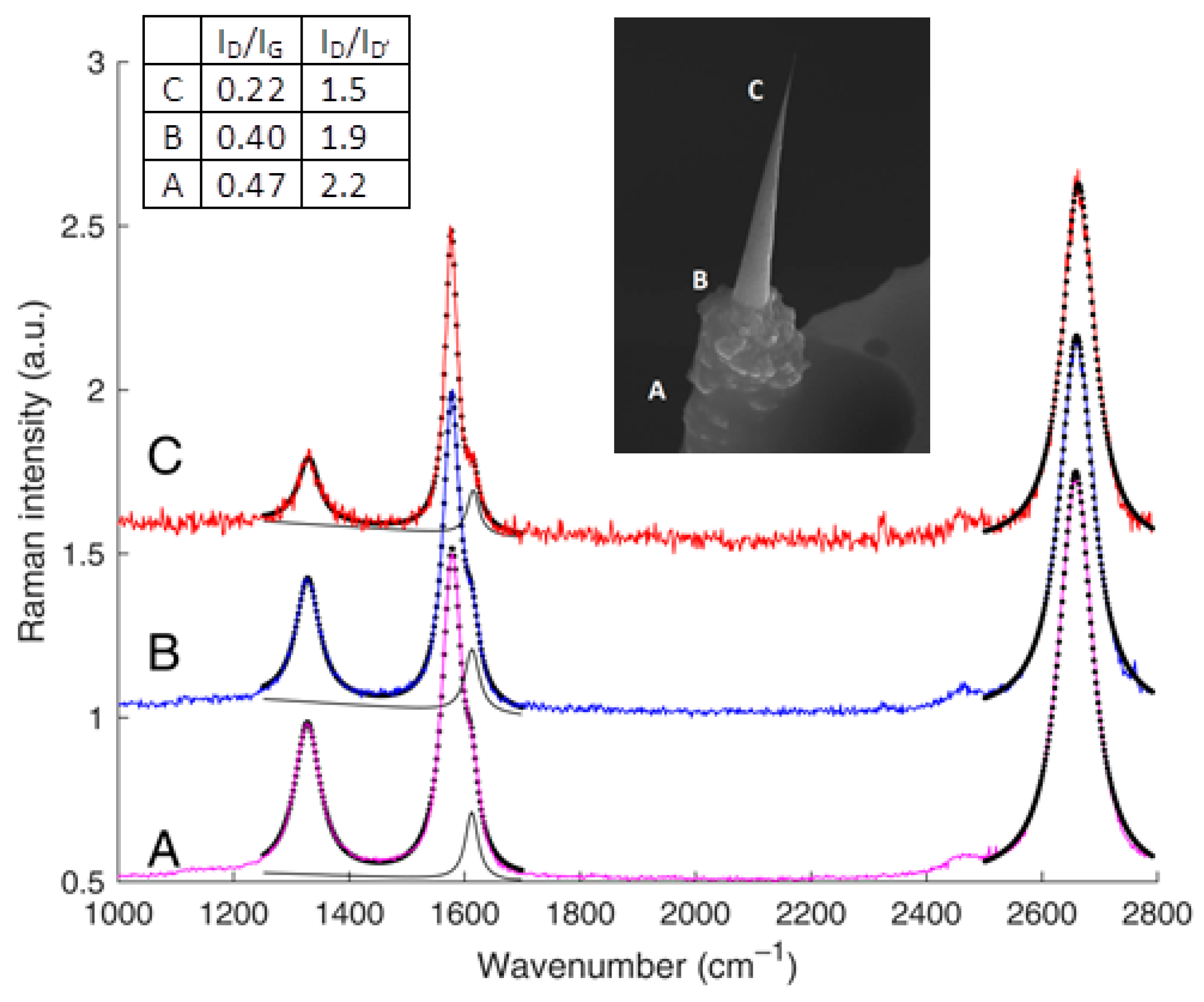
4.1. Fewly Defective, Turbostratically Stacked Graphenes – The Example of Carbon Cones
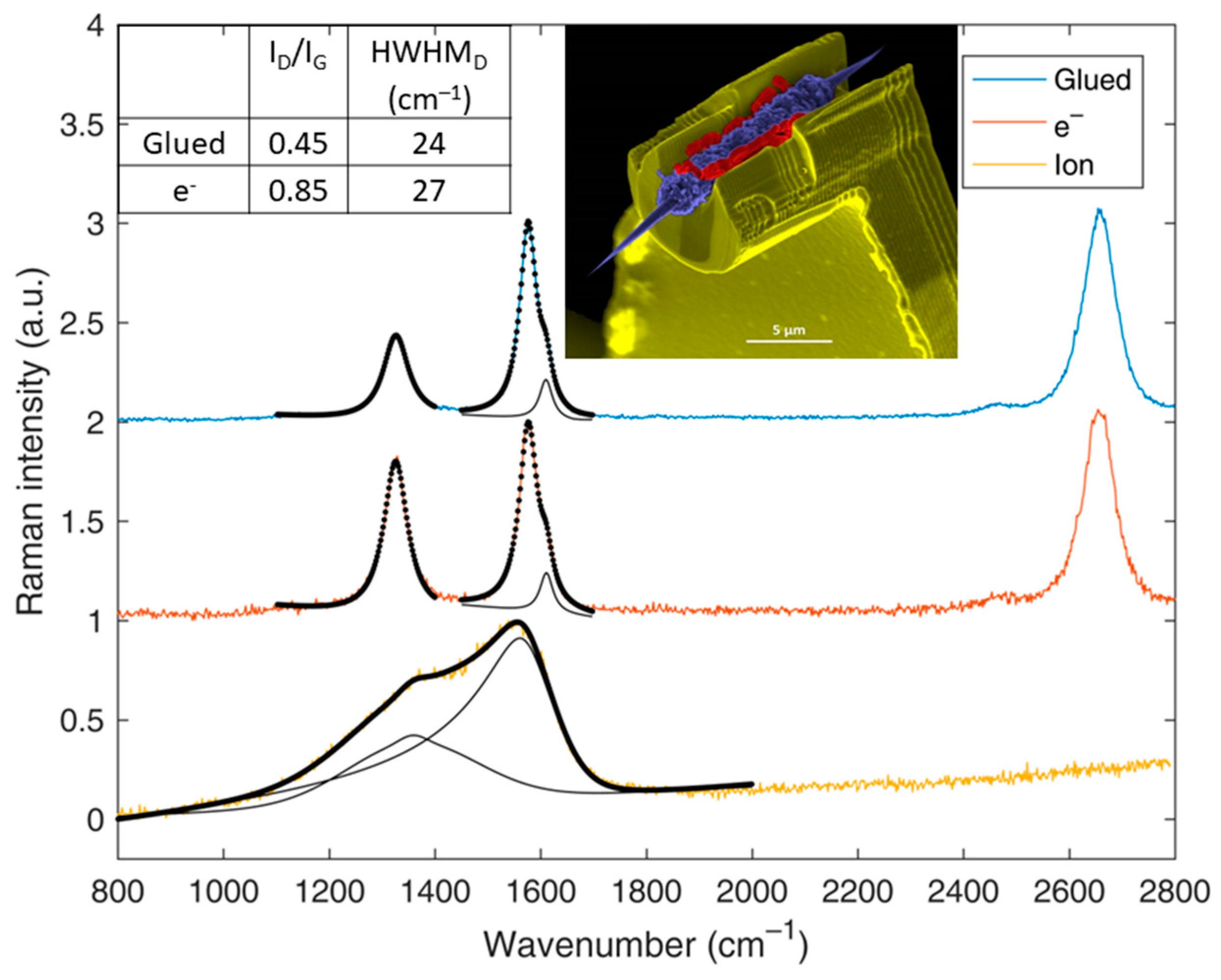
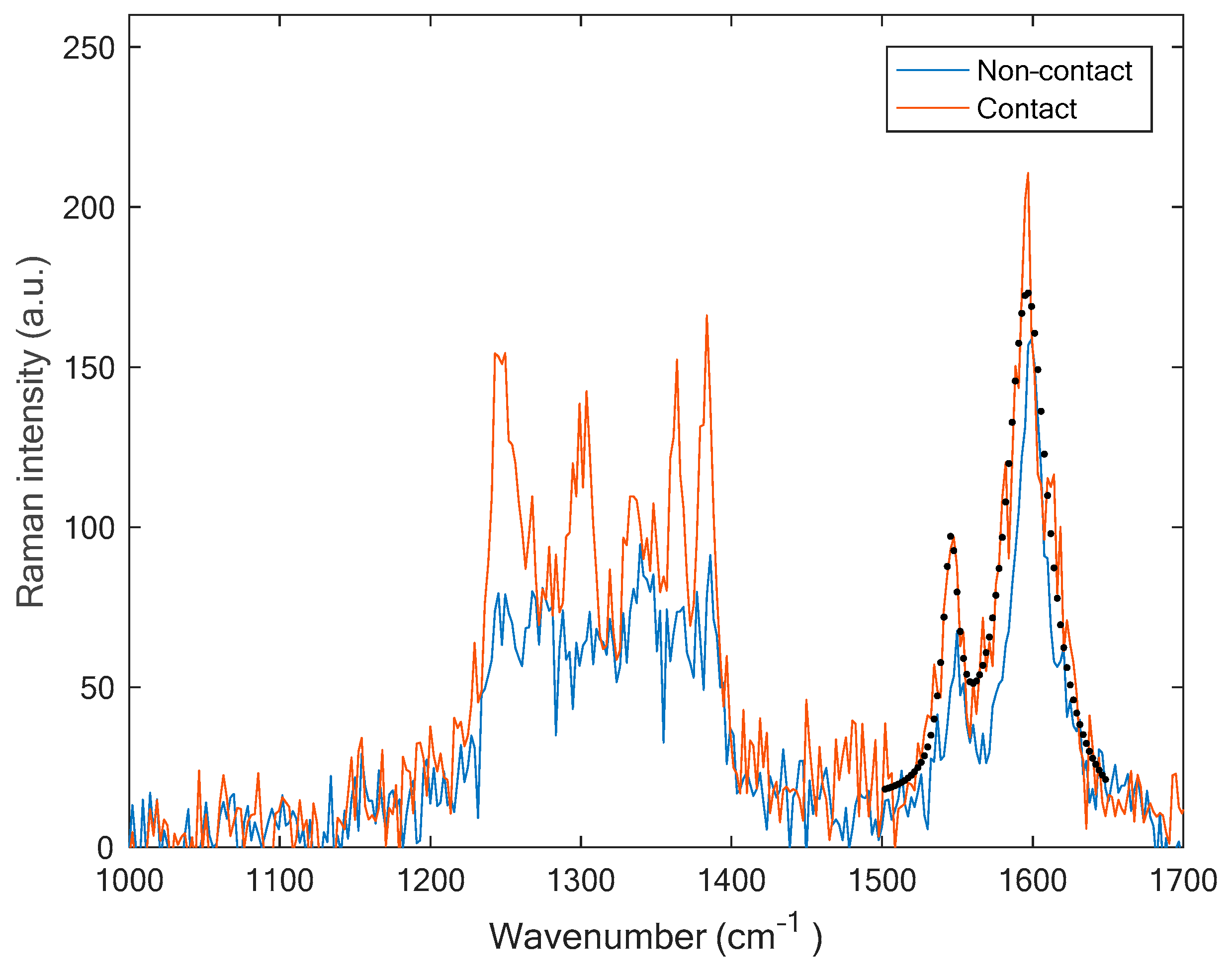
4.2. Single, Quasi-Perfect Graphene: The Case of Isolated Single-Wall Carbon Nanotubes
5. Summary
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferrari, A.C.; Robertson, J. Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 2000, 61, 14095. [Google Scholar] [CrossRef]
- Casiraghi, C.; Ferrari, A.C.; Robertson, J. Raman spectroscopy of hydrogenated amorphous carbons. Phys. Rev. B 2005, 72, 085401. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, A.C.; Robertson, J. Raman spectroscopy of amorphous, nanostructured, diamond–like carbon, and nanodiamond. Philos. Trans. R. Soc. Lond. Ser. A: Math. Phys. Eng. Sci. 2004, 362, 2477–2512. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Robertson, J. Resonant Raman spectroscopy of disordered, amorphous, and diamond-like carbon. Phys. Rev. B 2001, 64, 075414. [Google Scholar] [CrossRef]
- Johnson, C.A.; Patrick, J.W.; Thomas, K.M. Characterization of coal chars by Raman spectroscopy, X-ray diffraction and reflectance measurements. Fuel 1986, 65, 1284–1290. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Dennison, J.R.; Holtz, M.; Swain, G. Raman spectroscopy of carbon materials. Spectroscopy 1996, 11, 38–45. [Google Scholar]
- Lucchese, M.M.; Stavale, F.; Ferreira, E.M.; Vilani, C.; Moutinho, M.V.O.; Capaz, R.B.; Achete, C.A.; Jorio, A. Quantifying ion-induced defects and Raman relaxation length in graphene. Carbon 2010, 48, 1592–1597. [Google Scholar] [CrossRef]
- Cançado, L.G.; Da Silva, M.G.; Ferreira, E.H.M.; Hof, F.; Kampioti, K.; Huang, K.; Pénicaud, A.; Achete, C.A.; Capaz, R.B.; Jorio, A. Disentangling contributions of point and line defects in the Raman spectra of graphene-related materials. 2D Mater. 2017, 4, 025039. [Google Scholar] [CrossRef]
- Tuinstra, F.; Koenig, J.L. Raman spectrum of graphite. J. Chem. Phys. 1970, 53, 1126–1130. [Google Scholar] [CrossRef]
- Cançado, L.G.; Jorio, A.; Pimenta, M.A. Measuring the absolute Raman cross section of nanographites as a function of laser energy and crystallite size. Phys. Rev. B 2007, 76, 064304. [Google Scholar] [CrossRef]
- Mallet-Ladeira, P.; Puech, P.; Toulouse, C.; Cazayous, M.; Ratel-Ramond, N.; Weisbecker, P.; Vignoles, G.L.; Monthioux, M. A Raman study to obtain crystallite size of carbon materials: A better alternative to the Tuinstra–Koenig law. Carbon 2014, 80, 629–639. [Google Scholar] [CrossRef]
- Thomsen, C.; Reich, S. Double resonant Raman scattering in graphite. Phys. Rev. Lett. 2000, 85, 5214–5217. [Google Scholar] [CrossRef] [PubMed]
- Allouche, H.; Monthioux, M.; Jacobsen, R.L. Chemical vapor deposition of pyrolytic carbon on carbon nanotubes: Part 1. Synthesis and morphology. Carbon 2003, 41, 2897–2912. [Google Scholar] [CrossRef]
- Asadullah, M.; Zhang, S.; Min, Z.; Yimsiri, P.; Li, C.Z. Effects of biomass char structure on its gasification reactivity. Bioresour. Technol. 2010, 101, 7935–7943. [Google Scholar] [CrossRef] [PubMed]
- Mallet-Ladeira, P.; Puech, P.; Weisbecker, P.; Vignoles, G.L.; Monthioux, M. Behavior of Raman D band for pyrocarbons with crystallite size in the 2–5 nm range. Appl. Phys. A 2014, 114, 759–763. [Google Scholar] [CrossRef]
- Fung, A.W.P.; Rao, A.M.; Kuriyama, K.; Dresselhaus, M.S.; Dresselhaus, G.; Endo, M.; Shindo, N. Raman scattering and electrical conductivity in highly disordered activated carbon fibers. J. Mater. Res. 1993, 8, 489–500. [Google Scholar] [CrossRef] [Green Version]
- Merlen, A.; Buijnsters, J.; Pardanaud, C. A guide to and review of the use of multiwavelength Raman spectroscopy for characterizing defective aromatic carbon solids: From graphene to amorphous carbons. Coatings 2017, 7, 153. [Google Scholar] [CrossRef]
- Puech, P.; Dabrowska, A.; Ratel-Ramond, N.; Vignoles, G.L.; Monthioux, M. New insight on carbonisation and graphitisation mechanisms as obtained from a bottom-up analytical approach of X-ray diffraction patterns. Carbon 2019, 147, 602–611. [Google Scholar] [CrossRef]
- Cançado, L.G.; Jorio, A.; Ferreira, E.M.; Stavale, F.; Achete, C.A.; Capaz, R.B.; Moutinho, M.V.O.; Lombardo, A.; Kulmala, T.S.; Ferrari, A.C. Quantifying defects in graphene via Raman spectroscopy at different excitation energies. Nano Lett. 2011, 11, 3190–3196. [Google Scholar] [CrossRef] [PubMed]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Beyssac, O.; Goffé, B.; Chopin, C.; Rouzaud, J.N. Raman spectra of carbonaceous material in metasediments: A new geothermometer. J. Metamorph. Geol. 2002, 20, 859–871. [Google Scholar] [CrossRef]
- Guizani, C.; Jeguirim, M.; Gadiou, R.; Sanz, F.J.E.; Salvador, S. Biomass char gasification by H2O, CO2 and their mixture: Evolution of chemical, textural and structural properties of the chars. Energy 2016, 112, 133–145. [Google Scholar] [CrossRef]
- Schmid, J.; Grob, B.; Niessner, R.; Ivleva, N.P. Multiwavelength Raman microspectroscopy for rapid prediction of soot oxidation reactivity. Anal. Chem. 2011, 83, 1173–1179. [Google Scholar] [CrossRef]
- Puech, P.; Plewa, J.M.; Mallet-Ladeira, P.; Monthioux, M. Spatial confinement model applied to phonons in disordered graphene-based carbons. Carbon 2016, 105, 275–281. [Google Scholar] [CrossRef]
- Romero-Sarmiento, M.F.; Rouzaud, J.N.; Bernard, S.; Deldicque, D.; Thomas, M.; Littke, R. Evolution of Barnett Shale organic carbon structure and nanostructure with increasing maturation. Org. Geochem. 2014, 71, 7–16. [Google Scholar] [CrossRef]
- Dopita, M.; Rudolph, M.; Salomon, A.; Emmel, M.; Aneziris, C.G.; Rafaja, D. Simulations of X-Ray Scattering on TwoDimensional, Graphitic and Turbostratic Carbon Structures. Adv. Eng. Mater. 2013, 15, 1280–1291. [Google Scholar] [CrossRef]
- Robertson, J. Photoluminescence mechanism in amorphous hydrogenated carbon. Diam. Relat. Mater. 1996, 5, 457–460. [Google Scholar] [CrossRef]
- Pillet, G.; Sapelkin, A.; Bacsa, W.; Monthioux, M.; Puech, P. Size-controlled graphene-based materials prepared by annealing of pitch-based cokes: G band phonon line broadening effects due to high pressure, crystallite size, and merging with D′ band. J. Raman Spectrosc. 2019, in press. [Google Scholar] [CrossRef]
- Eckmann, A.; Felten, A.; Mishchenko, A.; Britnell, L.; Krupke, R.; Novoselov, K.S.; Casiraghi, C. Probing the nature of defects in graphene by Raman spectroscopy. Nano Lett. 2012, 12, 3925–3930. [Google Scholar] [CrossRef] [PubMed]
- Audier, M.; Oberlin, A.; Oberlin, M.; Coulon, M.; Bonnetain, L. Morphology and crystalline order in catalytic carbons. Carbon 1981, 19, 217–224. [Google Scholar] [CrossRef]
- Allouche, H.; Monthioux, M. Chemical vapor deposition of pyrolytic carbon on carbon nanotubes. Part. II: Texture and structure. Carbon 2005, 43, 1265–1278. [Google Scholar] [CrossRef]
- Su, W.; Kumar, N.; Krayev, A.; Chaigneau, M. In situ topographical chemical and electrical imaging of carboxyl graphene oxide at the nanoscale. Nat. Commun. 2018, 9, 2891. [Google Scholar] [CrossRef]
- Richard-Lacroix, M.; Zhang, Y.; Dong, Z.; Deckert, V. Mastering high resolution tip-enhanced Raman spectroscopy: Towards a shift of perception. Chem. Soc. Rev. 2017, 46, 3922–3944. [Google Scholar] [CrossRef]
- Rao, A.M.; Richter, E.; Bandow, S.; Chase, B.; Eklund, P.C.; Williams, K.A.; Fang, S.; Subbaswamy, K.R.; Menon, M.; Thess, A.; et al. Diameter-selective Raman scattering from vibrational modes in carbon nanotubes. Science 1997, 275, 187–191. [Google Scholar] [CrossRef]
- Jorio, A.; Saito, R.; Hafner, J.H.; Lieber, C.M.; Hunter, D.M.; McClure, T.; Dresselhaus, G.; Dresselhaus, M.S. Structural (n, m) determination of isolated single-wall carbon nanotubes by resonant Raman scattering. Phys. Rev. Lett. 2001, 86, 1118. [Google Scholar] [CrossRef]
- Araujo, P.T.; Pesce, P.B.C.; Dresselhaus, M.S.; Sato, K.; Saito, R.; Jorio, A. Resonance Raman spectroscopy of the radial breathing modes in carbon nanotubes. Phys. E Low-Dim. Syst. Nanostruct. 2010, 42, 1251–1261. [Google Scholar] [CrossRef]
- Jorio, A.; Souza Filho, A.G.; Dresselhaus, G.; Dresselhaus, M.S.; Swan, A.K.; Ünlü, M.S.; Goldberg, B.B.; Pimenta, M.A.; Hafner, J.H.; Lieber, C.M.; et al. G-band resonant Raman study of 62 isolated single-wall carbon nanotubes. Phys. Rev. B 2002, 65, 155412. [Google Scholar] [CrossRef]
- Telg, H.; Maultzsch, J.; Reich, S.; Hennrich, F.; Thomsen, C. Chirality distribution and transition energies of carbon nanotubes. Phys. Rev. Lett. 2004, 93, 177401. [Google Scholar] [CrossRef]
- Maultzsch, J.; Reich, S.; Thomsen, C. Double-resonant Raman scattering in graphite: Interference effects, selection rules, and phonon dispersion. Phys. Rev. B 2004, 70, 155403. [Google Scholar] [CrossRef] [Green Version]
- Chhowalla, M.; Ferrari, A.C.; Robertson, J.; Amaratunga, G.A.J. Evolution of sp2 bonding with deposition temperature in tetrahedral amorphous carbon studied by Raman spectroscopy. Appl. Phys. Lett. 2000, 76, 1419–1421. [Google Scholar] [CrossRef]
- Zhang, L.; Wei, X.; Lin, Y.; Wang, F. A ternary phase diagram for amorphous carbon. Carbon 2015, 94, 202–213. [Google Scholar] [CrossRef]
- Pillet, G.; Freire-Soler, V.; Eroles, M.N.; Bacsa, W.; Dujardin, E.; Puech, P. Reversibility of defect formation during oxygen-assisted electron-beam-induced etching of graphene. J. Raman Spectrosc. 2018, 49, 317–323. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Rodil, S.E.; Robertson, J. Interpretation of infrared and Raman spectra of amorphous carbon nitrides. Phys. Rev. B 2003, 67, 155306. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
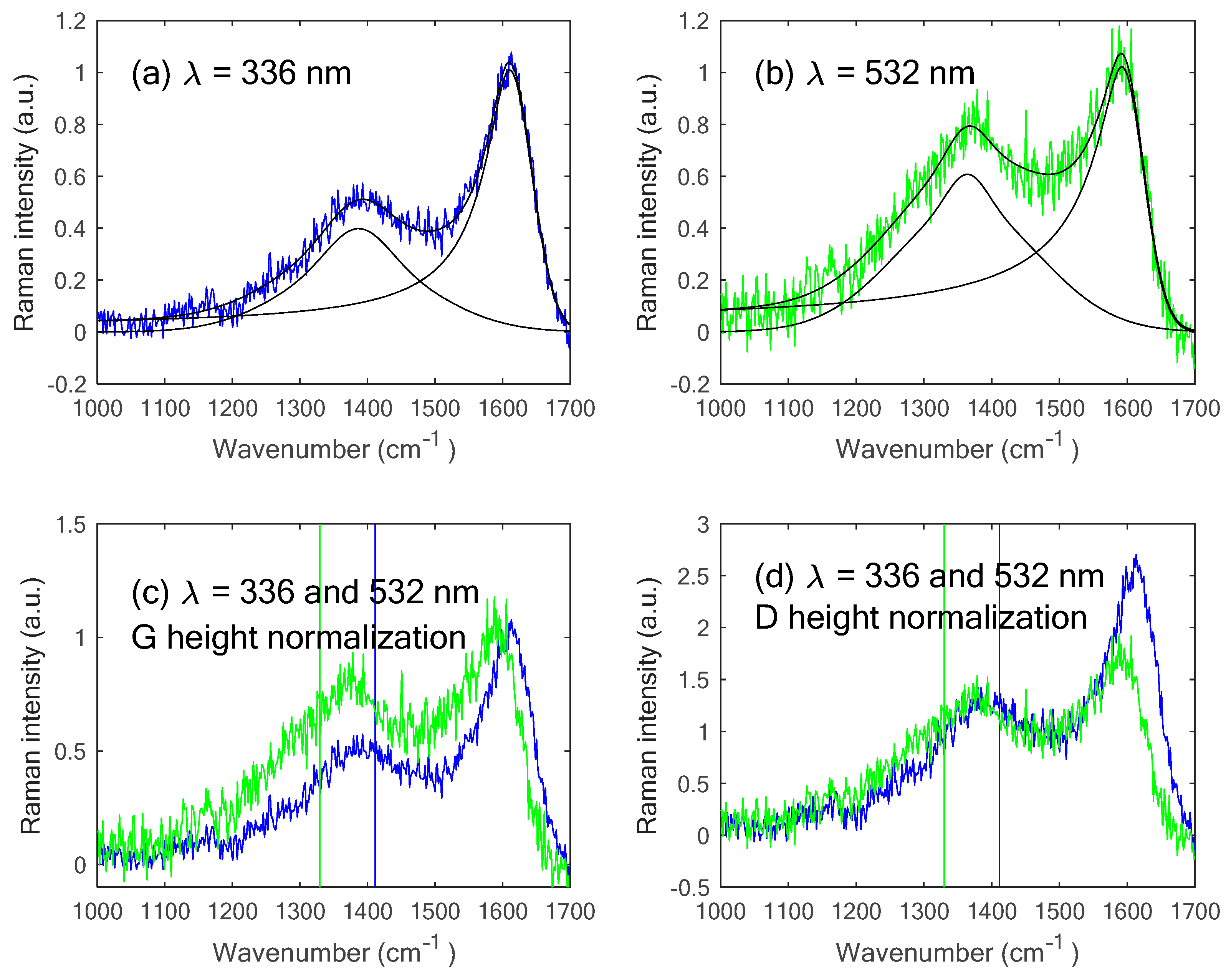
| Sample | λ (nm) | ωD (cm−1) | ωG (cm−1) | HWHMD (cm−1) | HWHMG (cm–1) | ID/IG | dωD/dEL (cm−1.eV–1) | α See Equation (1) |
|---|---|---|---|---|---|---|---|---|
| Barnett Gas and oil | 336 | 1388 | 1610 | 43 | 24 | 0.37 | 18 | 1.1 |
| 532 | 1364 | 1592 | 52 | 26 | 0.61 | |||
| Marcellus49 Gas | 336 | 1374 | 1608 | 39 | 17 | 0.44 | 26 | 0.6 |
| 532 | 1339 | 1601 | 46 | 12 | 0.59 |
| Sample | λ (nm) | ωD (cm−1) | ωG (cm−1) | HWHMD (cm−1) | HWHMG (cm−1) | ID/IG | dωD/dEL (cm−1.eV−1) | α |
|---|---|---|---|---|---|---|---|---|
| Steam | 532 | 1313 | 1601 | 144 | 42 | 1.05 | 127 | 3.7 |
| 633 | 1360 | 1594 | 125 | 46 | 1.98 | |||
| Pyro | 532 | 1357 | 1603 | 127 | 45 | 0.99 | 49 | 1.3 |
| 633 | 1339 | 1597 | 133 | 56 | 1.24 | |||
| N330 | 532 | 1344 | 1589 | 119 | 53 | 1.23 | 27 | 0.14 |
| 633 | 1334 | 1601 | 109 | 50 | 1.26 |
| Type of Defects | ID/ID’ |
|---|---|
| sp3 | 13* |
| Vacancy | 7* |
| Boundary | 3.5* |
| Loop | 1.5 (our result) |
| La | <2 nm | 2–7 nm | >7 nm |
|---|---|---|---|
| ID/IG | For amorphous carbon (a-C): proportional to La2 [1] | Varies roughly with 1/La (see Equation (2)) and is affected by the presence of heretoatoms (see Section 3) | ID/ID’ to be considered (see Section 4). Experimental laws for ID/IG (see Equations (2) and (3) for the least) |
| For disordered-C: varies with the content in chain, defects, and heteroatoms [3] | |||
| HWHMD | One component. Is dependent on both dωD/dE and 1/La. Could reach a maximum (see Section 3) | Often two different components [17]. Varies with 1/La [26] | Varies roughly with 1/La [26] |
| HWHMG | For a-C: decreases with increased La [42] | Varies with 1/La or La (because the La size range is small) [13] | Jumps due to the merging of G and D’ around 7 nm [30]. Weak dependence for larger La |
| For disordered-C: varies with the content in chain, defects, and heteroatoms [43] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puech, P.; Kandara, M.; Paredes, G.; Moulin, L.; Weiss-Hortala, E.; Kundu, A.; Ratel-Ramond, N.; Plewa, J.-M.; Pellenq, R.; Monthioux, M. Analyzing the Raman Spectra of Graphenic Carbon Materials from Kerogens to Nanotubes: What Type of Information Can Be Extracted from Defect Bands? C 2019, 5, 69. https://0-doi-org.brum.beds.ac.uk/10.3390/c5040069
Puech P, Kandara M, Paredes G, Moulin L, Weiss-Hortala E, Kundu A, Ratel-Ramond N, Plewa J-M, Pellenq R, Monthioux M. Analyzing the Raman Spectra of Graphenic Carbon Materials from Kerogens to Nanotubes: What Type of Information Can Be Extracted from Defect Bands? C. 2019; 5(4):69. https://0-doi-org.brum.beds.ac.uk/10.3390/c5040069
Chicago/Turabian StylePuech, Pascal, Mariem Kandara, Germercy Paredes, Ludovic Moulin, Elsa Weiss-Hortala, Anirban Kundu, Nicolas Ratel-Ramond, Jérémie-Marie Plewa, Roland Pellenq, and Marc Monthioux. 2019. "Analyzing the Raman Spectra of Graphenic Carbon Materials from Kerogens to Nanotubes: What Type of Information Can Be Extracted from Defect Bands?" C 5, no. 4: 69. https://0-doi-org.brum.beds.ac.uk/10.3390/c5040069