Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution
by
 , , and
, , and
Teerachote Pakornchote
1,2 ,
,
Annop Ektarawong
1,2,
Udomsilp Pinsook
1,2 and
and
Thiti Bovornratanaraks
1,2,*
1
Extreme Condition Physics Research Laboratory, Physics of Energy Materials Research Unit, Department of Physics, Faculty of Science, Chulalongkorn University, Bangkok 10330, Thailand
2
Thailand Center of Excellence in Physics, Commission on Higher Education, 328 Si Ayutthaya Road, Bangkok 10400, Thailand
*
Author to whom correspondence should be addressed.
C 2021, 7(1), 8; https://0-doi-org.brum.beds.ac.uk/10.3390/c7010008
Submission received: 15 December 2020
/
Revised: 12 January 2021
/
Accepted: 16 January 2021
/
Published: 21 January 2021
(This article belongs to the Special Issue 2D Ultrathin Carbon Films)
Abstract
:One type of two-dimensional diamonds that are derived from [111] direction, so-called diamane, has been previously shown to be stabilized by N-substitution, where the passivation of dangling bonds is no longer needed. In the present work, we theoretically demonstrated that another type of two-dimensional diamonds derived from [110] direction exhibiting a washboard conformation can also be stabilized by N-substitution. Three structural models of washboard-like carbon nitrides with compositions of C6N2, C5N3, and C4N4 are studied together with the fully hydrogenated washboard-like diamane (C8H4). The result shows that the band gap of this type structure is only open the dangling bonds that are entirely diminished through N-substitution. By increasing the N content, the C11 and C22 are softer and the C33 is stiffer where their bulk modulus are in the same order, which is approximately 550 GPa. When comparing with the hydrogenated phase, the N-substituted phases have higher elastic constants and bulk modulus, suggesting that they are possibly harder than the fully hydrogenated diamane.
1. Introduction
Diamond, a carbon allotrope, is a material that has been reported to be the first hardest material and it also possesses the highest thermal conductivity. Its hardness originates from characteristic of sp hybridization of C atoms, which has a tetrahedral symmetry with high bonding strength. In fact, the sp hybridization of C atoms is stronger than sp hybridization, for example, graphene. Graphene can persist with a high load of an indentation without breaking bonds showing remarkably high tensile strength [1]; however, its hardness cannot be measured. Other sp carbon allotrope, i.e., lonsdaleite, is predicted to be harder than diamond, but its existence is under discussion [2,3,4].
Since the two-dimensional (2D) materials has come into the spot light, the path for creating a thin diamond-like film has been examined [5,6]. A 2D diamond that is derived from the bulk diamond thinned down in [111] direction or diamane is a promising material for adopting the property of its bulk counterpart, diamond, but its thickness is only 4 C atoms stacking [7,8,9,10]. In order to create the sp carbon film, the layers of the sp carbon films, e.g., bilayer graphene and few-layer graphene, must be closer, because its interlayer distance is much larger than a bond length of the sp carbon film [11]. The sp carbon film can be indented by a sharp indenter creating the sp hybridization and showing that the indented multi-layer graphene is harder if its number of layers is fewer [10,12,13]. Moreover, diamane can also be synthesized at high pressure, where it is expected to be stable at 5 GPa [6,14].
Even though the diamane can be created, its sp hybridization cannot persist without an external auxiliary i.e., indentation, high-pressure [6,10]. The surfaces of diamane that are full of dangling bonds need to be passivated with atoms or molecules; otherwise, diamane will transform back to a sp carbon film [11]. Several works have theoretically studied on the stability of diamane that is passivated by other atoms and molecules and also its band gap and thermal conductivity, depending on passivating molecules [15,16]. The diamane can be synthesized by exposing a bilayer graphene in order to chemically react with H or F atoms [17,18,19,20].
Lately, we have shown that the passivation of dangling bonds of the surficial C atoms is not the only method for stabilizing diamane. It can also be stabilized by substituting the N atom, which has one electron more than C atoms on its surfaces [21]. The remaining electrons that form lone electron pairs behave like the passivation that stabilizes the structure and opens a band gap. Moreover, the surface reconstruction that occurs on the outermost layer to have a particular structure can promote the stabilization of the sp structure of the 2D carbon [21,22,23]. Beneficially, the reconstructing surface also enhances a magnetism of the structure, which is typically absent in diamane [21,24].
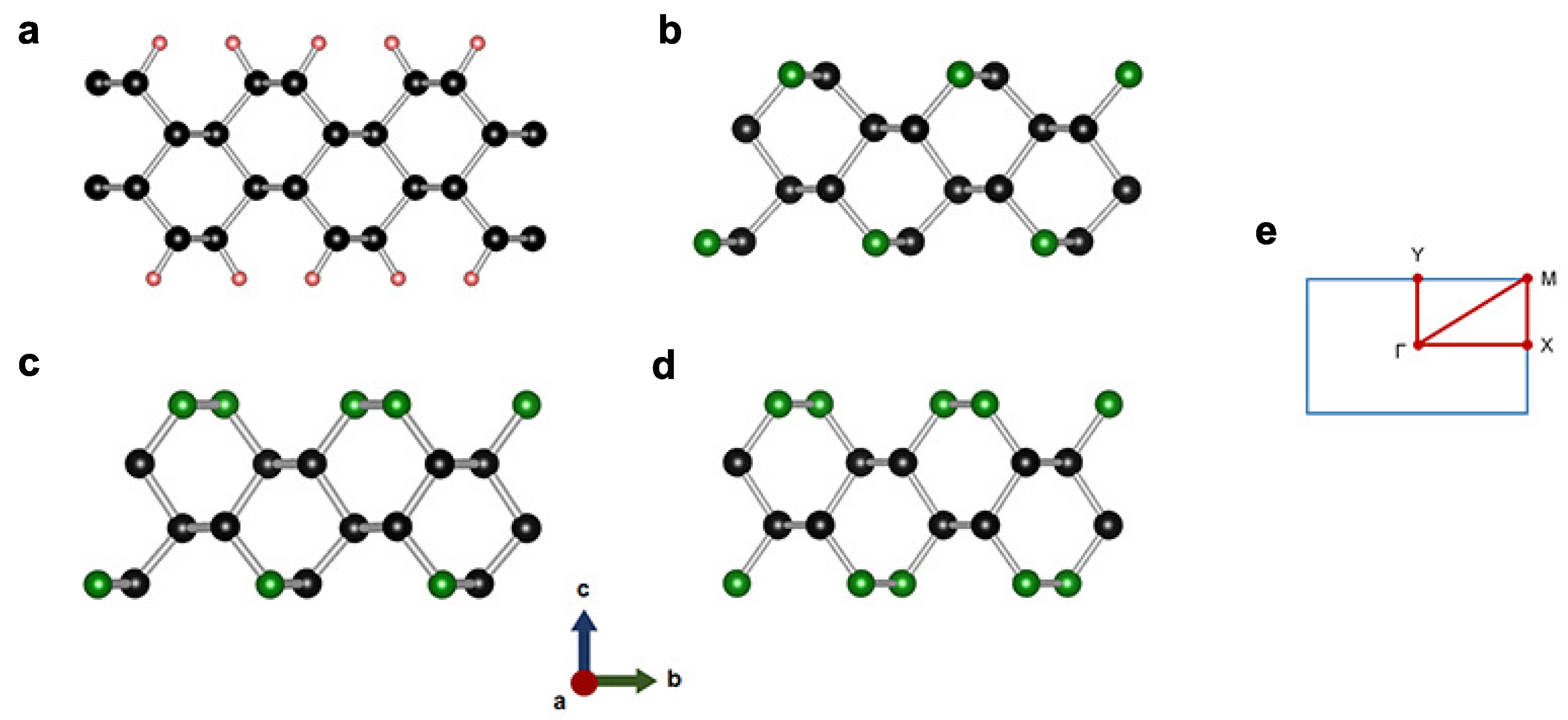
In this work, 2D diamond-like carbon nitrides (CN), where their structures are derived from the structure of diamond thinned down in [110] direction until it has eight atoms in a unit cell (see Figure 1), are studied where . Because its surfaces are washboard conformations [25], it will be represented as W-diamane for a phase with and N-subsituted W-diamanes for phases with . Their elastic and electronic properties are investigated, due to a number of N atoms in the structure. It will be shown that an accretion of N atoms on the surfaces of W-diamane improves the dynamical stability and enhances the stiffness of the structure, which are even stiffer than hydrogenated 2D diamond.
2. Computational Method
The VASP package [26,27], based on the density functional theory, was performed in order to study the properties of the 2D diamond. The projector augmented-wave method [28], which is an efficient method for reducing a cumbersome calculation of the core electrons, was used for the pseudopotential. The exchange-correlation functionals, Perdew-Burke-Ernzerhof (PBE) and Heyd–Scuseria–Ernzerhof (HSE06), were employed [29,30]. The former was used for the structural relaxation in order to find the optimum structural parameters and atomic positions at 0 GPa and 0 K, evaluating the elastic constants, and calculating phonon modes. The latter was used for the electronic density of states (DOS) and band structure calculations.
The energy cutoff was set as 600 eV in all calculations. The k-point mesh was for the relaxation and calculation of the elastic constants. In order to neglect the interaction between layers of 2D materials, the c-axis of every structure was set to be 20 Å at the start of the relaxation and asserted to be larger than 15 Å after the relaxation. The phonon modes were calculated while using Phonopy package [31] with supercell and k-point mesh. Every DOS was calculated by using the tetrahedron method to integrate over the Brillouin zone [32].
In calculations of the elastic constants, lattice parameters of the studied structures were applied by 1% and 2% compressive and tensile strains, while the atomic positions were relaxed. For the elastic constants corresponding to a direction that is perpendicular to the layer, z-positions of the top and bottom atoms were fixed during the atomic relaxation to constrain a c-parameter of the strained lattice, which is a distance between top and bottom atoms. Otherwise, the top and bottom atoms would be relaxed to new z-positions, yielding unintended strained lattice, which should be fixed to be 1% or 2% strains [21].
3. Results
3.1. 2-Dimensional Structures
The structure of W-diamane is relaxed without any passivations or N substitutions. As expected, W-diamane with entire dangling bonds on its surfaces cannot be stabilized after relaxation, but it can be stabilized if the dangling bonds are passivated through hydrogenation (CH, see Figure 1a). We find that, instead of hydrogenation, the substitution of N atoms for surficial C atoms can also result in the structural stabilization of W-diamane. Nevertheless, the substitution is needed on both surfaces of W-diamane in order to keep the washboard-like shape stable on two sides after relaxation. For CN, an amount of N atoms is not enough to substitute on both of its surfaces, hence it is not considered. Noting that the studied phases have N atoms arranging in highly order by considering the substitution in the unit cell of W-diamane with eight atoms, while the configurational disorder of C and N atoms in W-diamane is beyond the scope and, thus, not considered in this work.
Table 1 reports the relaxed structures of CH, CN, CN, and CN. Howeve, noting that c-axes were not fixed during the relaxation, the relaxed c-axes are long enough to keep all of structures as 2D materials. The lattice parameters, a and b, are smaller as the number of N atoms increasing, while their thicknesses from top to bottom layers () are thicker, as shown in Table 1. Two parameters presenting the characteristics of the structures are bond length and bond angle. Typically, the C-N and N-N bond lengths are shorter than C-C bond length, so average bond lengths of N-substituted W-diamanes are shorter if the number of N atoms is higher. For an illustration, the average C-C bond length of CH is 1.558 Å, while the average bond lengths of CN, CN, and CN are 1.508, 1.495, and 1.506 Å, respectively. On the contrary, bond angles of atoms between layers of CN, CN, and CN (see Figure 1) are 101.85, 110.08, and 112.37, respectively, showing an upward trend that corresponds to the number of N atoms.
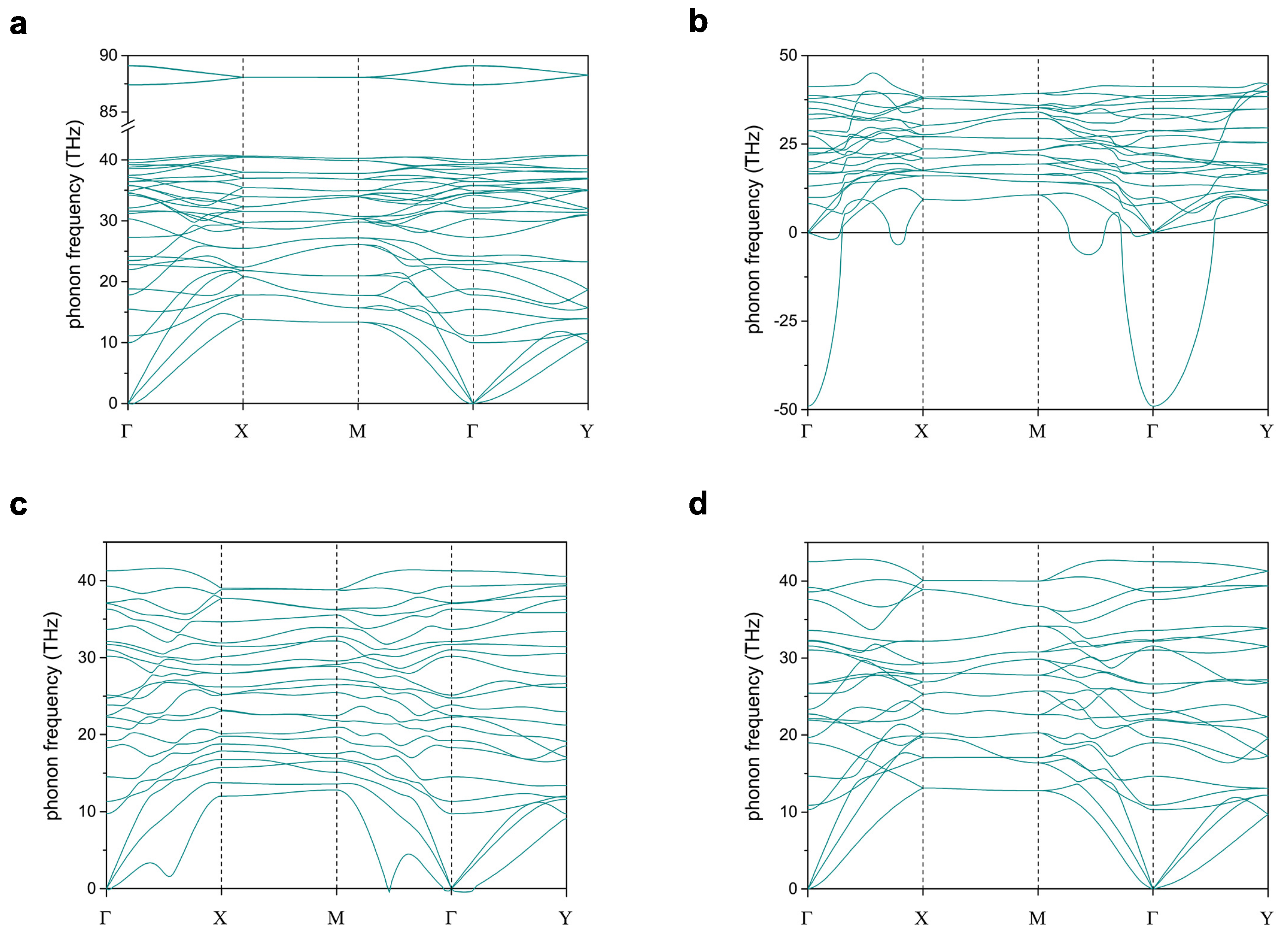
Figure 2 shows phonon dispersions of hydrogenated and N-substituted W-diamanes, where the negative value of phonon frequency represents an imaginary mode of the phonon. CH and CN, which are fully passivated and N substituted, respectively, have no imaginary modes, so they are dynamically stable. On the other hand, CN and CN, which are partially N-substituted, have some amounts of the imaginary phonon, so they are not dynamically stable. Figure 2b–d show the evolution of the imaginary phonon, which is fewer with respect to the number of N substitution in W-diamanes. This is clearly indicating that the stabilization of such a structure can be promoted by suppressing a number of the dangling bonds on the surfaces.
3.2. Electronic Property and Bonding
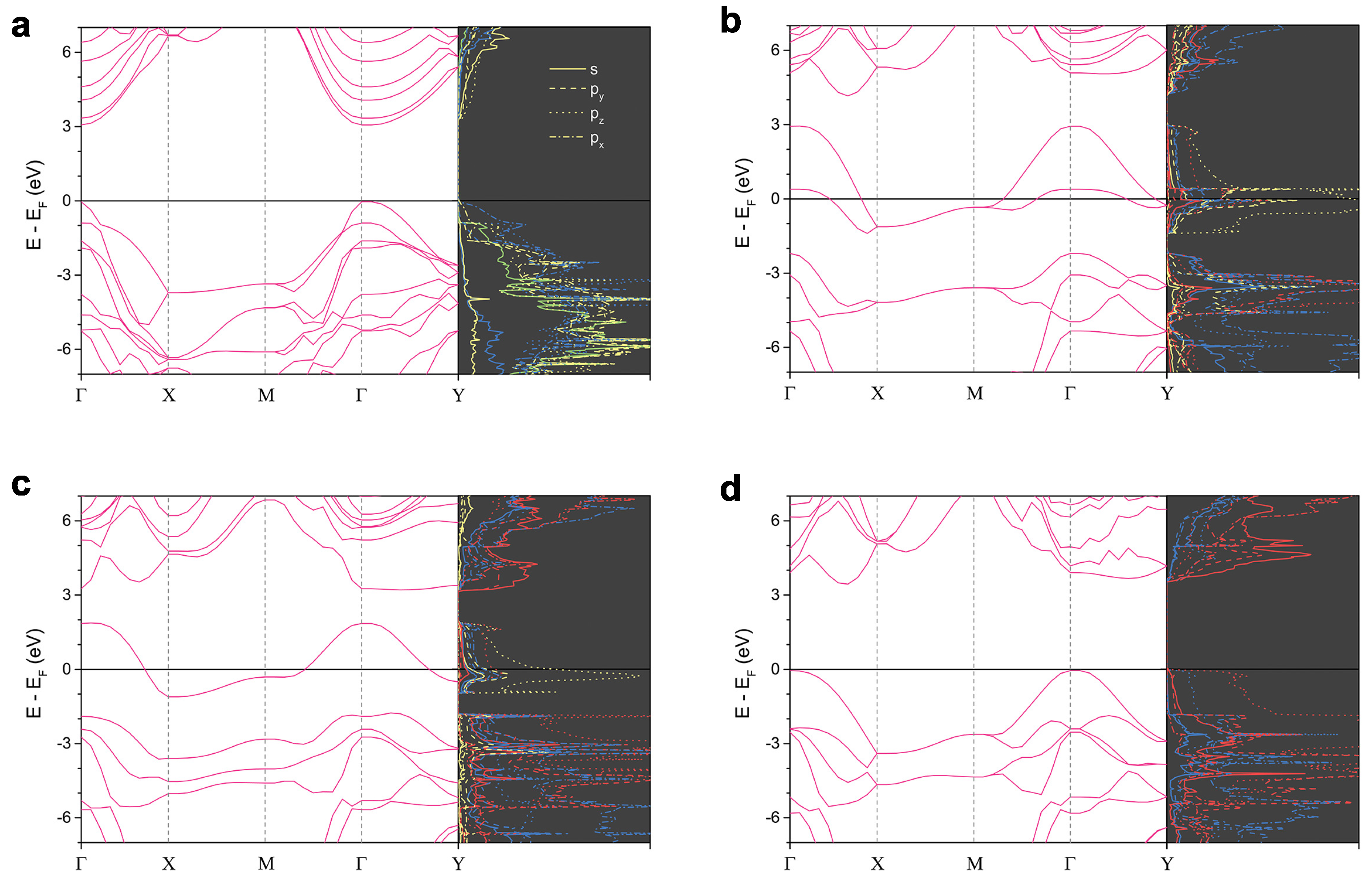
In the sp 2D carbons, the dangling bonds on their surfaces form energy bands across the Fermi level (E) closing the band gap, but the band gap is open if the dangling bonds are all passivated [11]. For example, the dangling bonds of NCCN and CNCN phases on the surfaces that are substituted by N atoms are replaced by lone pair electrons that open the band gap wildly. CH and CN are semiconducting with HSE06 band gap, 3.2 and 3.5 eV, respectively, while CN and CN are metallic. Thus, the band gap of W-diamane is open if no dangling bond is left to be passivated.
The right side of each figure shown in Figure 3 shows the electronic DOS corresponding to the electronic band dispersion on the left side of each structure. CH, where its dangling bonds of C atoms are fully passivated by H atoms, has the p-orbital of C atoms hybridizing with the s-orbital of H atoms at energy state −1 eV below the E. The electronic density of states (DOS) being occupied near the E are of the p-orbitals of C atoms in majority and of the p-orbitals of C next below. CN, instead fully substituted by N atoms on the surfaces, has the electronic DOS, which is C-N hybridization near the E in contrast to CH, where its C-H hybridization is at a lower energy level. The p-orbital of N atoms obviously dominates the valence states near the E.
The electronic band structures of CN, CN, and CN have similar dispersing feature across the reciprocal space (see Figure 3b–d). One that makes the electronic property of these three structures different is top two valence bands. CN, CN, and CN have two, one, and none valence bands, respectively, crossing the E. Despite that, their valence states are dominated by p-orbital of C atoms for CN and CN, and p-orbital of N atoms for CN. This is opposite to the diamane that its partial N-substituted phase, CNCN, has an opening band gap, while the p-orbital of C also dominates the valence states [21]. Moreover, the lowest conduction state of CN is on a -X path, and its highest valence state state is at point. Thus, CN has an indirect band gap, while CH has a direct band gap.
It is worth noting that N-substituted W-diamanes have no magnetism, while the CNCN phase has a tiny magnetization [21]. The magnetism occurs in 2D carbons when the 2D carbons are structurally distorted or defected, for example, hydrogenated graphene, 2D carbon nitrides that are porous structure, diamond surface with Pandey’s reconstruction [24,33,34,35,36,37]. The latter needs HSE06 for calculation in order to obtain magnetism [24], while magnetism can be acquired using PBE for the former two [33,34]. However, the N-substituted W-diamanes investigated while using PBE and HSE06 are non-magnetism.
3.3. Elastic Constants
The hardness is one of the distinguishing properties of diamond and carbon nitrides, where their 2D counterparts are expected to be adopted. However, the hardness of 2D materials, to the best of our knowledge, has not been well-defined. Therefore, the elastic constants that implicitly reflect the hardness are herein considered. Despite the fact that the elastic constants can be calculated by a second derivative of energy with respect to applied strain and devided by a volume of non-strained structure, the 2D materials, such as graphene, are lacking a third dimension. The elastic constants defined for three-dimensional (3D) materials are thus reduced to 2D elastic constants [38,39],
where is an unstrained area of CN, and is an applied strain up to in order to fit the . The 2D elastic constants of W-diamanes are reported in Table 2, by comparing with other 2D sp carbons and carbon nitrides. However, Pakornchote et al. [21] discussed that the 2D elastic constants cannot be used in order to compare across the 2D materials that have different thickness. Therefore, the 2D elastic constants have to be divided by of the 2D materials in order to make the values become intrinsic [21,40],
where is reported in Table 1. The and are reported in Table 2 and Table 3, respectively.
As the number of N atoms in the unit cell of W-dimane increasing, the of N-substituted W-diamanes are lower, but their and are higher. Figure 1 shows that, in the x direction, the surficial atoms are bonding with the surficial atoms, which are at the same level, but, in the y direction, the surficial atoms are bonding with the inner atoms, which are at the lower level. Thus, the N substitution in the surficial layers can enhance (reduce) the stiffness in the direction that the atoms are bonding in the different (same) level. Although, the Voigt bulk modulus () of CN, CN, and CN, which are 569, 553, and 533 GPa, respectively, are approximately the same if they are compared with the of NCCN and CNCN phases that are above 600 GPa. The result is in accordance with the simulation in Ref. [10], showing that diamane has the tensile strength higher than other conformations. Noting that the is typically valid for polycrystals and might not be valid for 2D materials, so the values of reported in this work are used for the purpose of comparison.
For CH, its , except C, are smaller than those of N-substituted W-diamanes. Therefore, of CH is 310 GPa, which is much smaller that of CN. One might argue that of CH is much larger than of CN, so is unsurprisingly small. However, even if we consider , CH has , , , , and smaller than CN. The result is in accordance with the result in the H-diamane, which is a fully hydrogenated diamane that its and are smaller than that of NCCN and CNCN phases. Thus, it can be concluded that the hydrogenation seem to soften the 2D diamond, while the N substitution makes the 2D diamond stiffer.
Moreover, the elastic constants can be calculated from group velocities of phonons for cross-validation [41]. In the orthorhombic system, the sound waves can be expressed as [42]
where is a wave vector, is a phonon eigenvector, is a phonon eigenvalue that is limited to acoustic modes, and is a 3D density of W-diamanes. By solving Equations (3)–(5), each yields three values of sound velocities, where and are herein considered,
The above two equations lead to a condition that
Superscripts of the velocities are just numbering, but have not yet been assigned to any phonon modes. The sound velocities can be computed from three acoustic phonons while using Phonopy package that takes the derivative on a dynamical matrix with respect to q divided by at . Noting that only CN and CH are discussed, since the dispersion of acoustic phonons of CN and CN are dropping, causing the instability of the structures (see Figure 2).
In Table 4, the were calculated from the group velocities of three acoustic phonons, ZA, TA, and LA modes while using Equations (6) and (7). If we assigned to be , this satisfies an unrestricted condition of Equation (8) that . Therefore, from Equations (6) and (7), is either 725 or 716 GPa, which is similar to the 719 GPa of that was reported in Table 3. Thus, if we assigned to be , GPa and GPa. These two values are similar to and reported in Table 3. By these assignments, calculated using Equations (2), (6) and (7) are in correspondence. Noting that, because of a convex dispersion of ZA mode around -point, abruptly changes along q, yielding inconsistent values of and , so it needs to be further investigated in the future work.
3.4. Formation Energy
An equation that is used to calculate a formation energy is
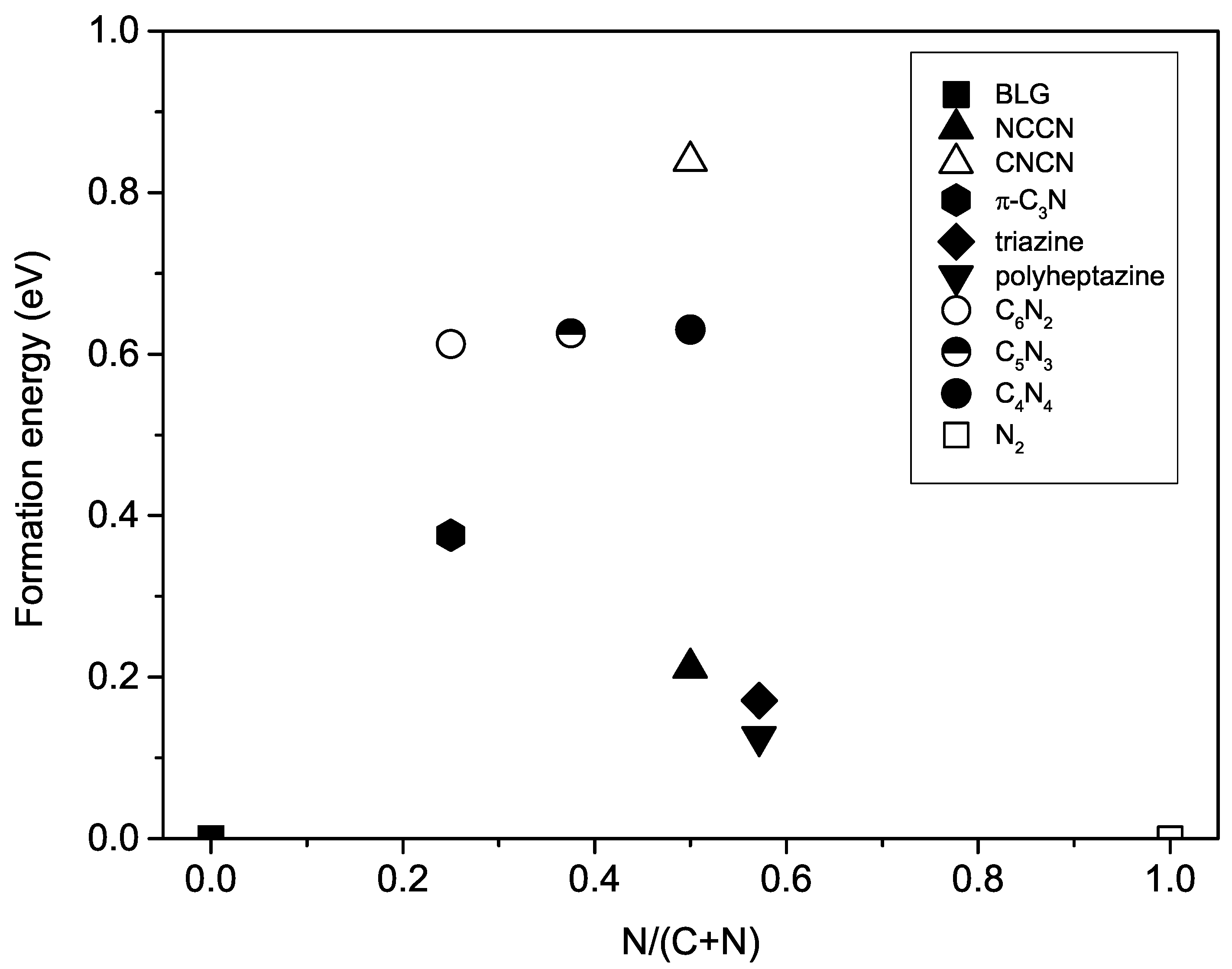
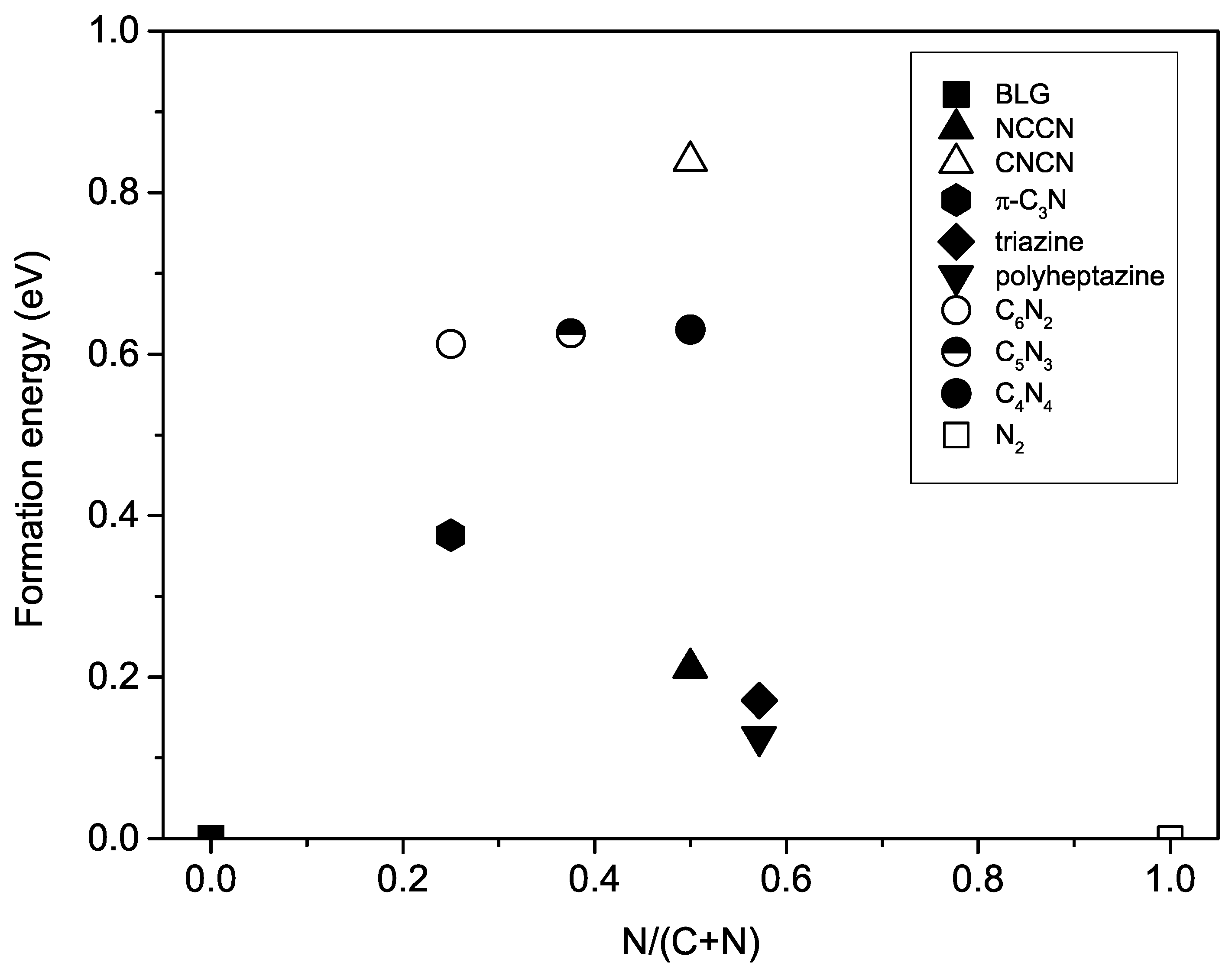
where is the energy of 2D carbon nitrides, is an energy of graphene, and is an energy of N molecule. The formation energies of N-substituted W-diamanes plotted in circle symbols in Figure 4 are 612, 626, and 630 meV for CN, CN, and CN, respectively. They are relatively high by comparing with the formation energy of synthesizable phases, triazine and polyheptazine [43], which are 171 and 126 meV, respectively.
On the one hand, the NCCN phase has the lowest formation energy among 2D diamond-like carbon nitrides. Its formation energy is positive at 0 GPa, but the formation energy is on a convex hull at 10 GPa if only layered phases of carbon nitrides are considered. Hence, it is possible to be synthesized if the precursor is limited to be 2D materials [21]. On the other hand, the N-subsituted W-diamanes have the formation energy as triple the NCCN phase (see Figure 4), which is too high for synthesizing such materials. Nw starting materials other than graphene and N molecule must be examined in order to find the possible pathway to synthesize W-diamane, but this is beyond the scope of the present work.
4. Conclusions
We have shown that the stiffness and of N-substituted W-diamane increase with the N content. The band gap of W-diamane is only open if the N atoms are entirely substituted on two surfaces. Even though their formation energies is positive when evaluated with respect to graphene and N2 molecule, which are both assumed to be precursors for W-diamane in the present work, the N-containing diamane is likely to possess higher stiffness than that of (hydrogenated) diamane. However, further investigation regarding a possible pathway to fabricate N-containing diamane, such as W-diamane, must be deserved in order to serve as guidelines for furture experimental synthesis of the material.
Author Contributions
T.P. and T.B. designed the research; T.P., A.E. and U.P. performed the research; All authors analysed the data; T.P. and T.B. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project is funded by National Research Council of Thailand (NRCT): (NRCT5-RSA63001-04). This research is partially funded by Chulalongkorn University; Grant for Research.
Data Availability Statement
Not applicable.
Acknowledgments
This research project was supported by the Second Century Fund (C2F), Chulalongkorn University. This project is funded by National Research Council of Thailand (NRCT): (NRCT5-RSA63001-04). This research is partially funded by Chulalongkorn University; Grant for Research.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| 2D | 2-dimensional |
| PBE | Perdew-Burke-Ernzerhof |
| HSE06 | Heyd-Scuseria-Ernzehof (2006) |
| DOS | density of states |
References
- Lee, C.; Wei, X.; Kysar, J.W.; Hone, J. Measurement of the Elastic Properties and Intrinsic Strength of Monolayer Graphene. Science 2008, 321, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Ye, H.Q. Ab initio elastic constants for the lonsdaleite phases of C, Si and Ge. J. Phys. Condens. Matter 2003, 15, 5307. [Google Scholar] [CrossRef]
- Németh, P.; Garvie, L.A.J.; Aoki, T.; Dubrovinskaia, N.; Dubrovinsky, L.; Buseck, P.R. Lonsdaleite is faulted and twinned cubic diamond and does not exist as a discrete material. Nat. Commun. 2014, 5, 5447. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, D.G.; Wong, S.; Shiell, T.B.; Haberl, B.; Cook, B.A.; Huang, X.; Boehler, R.; McKenzie, D.R.; Bradby, J.E. Investigation of Room Temperature Formation of the Ultra-Hard Nanocarbons Diamond and Lonsdaleite. Small 2020, 16, 2004695. [Google Scholar] [CrossRef]
- Barboza, A.P.M.; Guimaraes, M.H.D.; Massote, D.V.P.; Campos, L.C.; Neto, N.M.B.; Cancado, L.G.; Lacerda, R.G.; Chacham, H.; Mazzoni, M.S.C.; Neves, B.R.A. Room-Temperature Compression-Induced Diamondization of Few-Layer Graphene. Adv. Mater. 2011, 23, 3014–3017. [Google Scholar] [CrossRef]
- Martins, L.G.P.; Matos, M.J.S.; Paschoal, A.R.; Freire, P.T.C.; Andrade, N.F.; Aguiar, A.L.; Neves, B.R.A.; de Oliveira, A.B.; Mazzoni, M.S.C.; Filho, A.G.S.; et al. Raman evidence for pressure-induced formation of diamondene. Nat. Commun. 2017, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Leenaerts, O.; Partoens, B.; Peeters, F.M. Hydrogenation of bilayer graphene and the formation of bilayer graphane from first principles. Phys. Rev. B 2009, 80, 245422. [Google Scholar] [CrossRef]
- Samarakoon, D.K.; Wang, X.Q. Tunable Band Gap in Hydrogenated Bilayer Graphene. ACS Nano 2010, 4, 4126–4130. [Google Scholar] [CrossRef]
- Chernozatonskii, L.A.; Sorokin, P.B.; Kvashnin, A.G.; Kvashnin, D.G. Diamond-Like C2H Nanolayer, Diamane: Simulation of the Structure and Properties. JETP Lett. 2009, 90, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Cao, T.; Cellini, F.; Berger, C.; de Heer, W.A.; Tosatti, E.; Riedo, E.; Bongiorno, A. Ultrahard carbon film from epitaxial two-layer graphene. Nat. Nanotechnol. 2018, 13, 133–138. [Google Scholar] [CrossRef]
- Pakornchote, T.; Ektarawong, A.; Alling, B.; Pinsook, U.; Tancharakorn, S.; Busayaporn, W.; Bovornratanaraks, T. Phase stabilities and vibrational analysis of hydrogenated diamondized bilayer graphenes: A first principles investigation. Carbon 2019, 146, 468–475. [Google Scholar] [CrossRef]
- Kvashnin, A.G.; Sorokin, P.B. Lonsdaleite Films with Nanometer Thickness. J. Phys. Chem. Lett. 2014, 5, 541–548. [Google Scholar] [CrossRef]
- Kvashnin, A.G.; Avramov, P.P.V.; Kvashnin, D.G.K.; Chernozatonskii, L.A.; Sorokin, P.B. Features of Electronic, Mechanical, and Electromechanical Properties of Fluorinated Diamond Films of Nanometer Thickness. J. Phys. Chem. C 2017, 121, 28484–28489. [Google Scholar] [CrossRef]
- Pimenta Martins, L.G.; Silva, D.L.; Smith, J.S.; Lu, A.Y.; Su, C.; Hempel, M.; Occhialini, C.; Ji, X.; Pablo, R.; Alencar, R.S.; et al. Hard, transparent, sp3-containing 2D phase formed from few-layer graphene under compression. Carbon 2021, 173, 744–757. [Google Scholar] [CrossRef]
- Zhu, L.; Li, W.; Ding, F. Giant thermal conductivity in diamane and the influence of horizontal reflection symmetry on phonon scattering. Nanoscale 2019, 11, 4248–4257. [Google Scholar] [CrossRef]
- Raeisi, M.; Mortazavi, B.; Podryabinkin, E.V.; Shojaei, F.; Zhuang, X.; Shapeev, A.V. High thermal conductivity in semiconducting Janus and non-Janus diamanes. Carbon 2020, 167, 51–61. [Google Scholar] [CrossRef]
- Piazza, F.; Gough, K.; Monthioux, M.; Puech, P.; Gerber, I.; Wiens, R.; Paredes, G.; Ozoria, C. Low temperature, pressureless sp2 to sp3 transformation of ultrathin, crystalline carbon films. Carbon 2019, 145, 10–22. [Google Scholar] [CrossRef]
- Piazza, F.; Monthioux, M.; Puech, P.; Gerber, I. Towards a better understanding of the structure of diamanoïds and diamanoïd/graphene hybrids. Carbon 2020, 156, 234–241. [Google Scholar] [CrossRef]
- Piazza, F.; Cruz, K.; Monthioux, M.; Puech, P.; Gerber, I. Raman evidence for the successful synthesis of diamane. Carbon 2020, 169, 129–133. [Google Scholar] [CrossRef]
- Bakharev, P.V.; Huang, M.; Saxena, M.; Lee, S.W.; Joo, S.H.; Park, S.O.; Dong, J.; Camacho-Mojica, D.C.; Jin, S.; Kwon, Y.; et al. Chemically induced transformation of chemical vapour deposition grown bilayer graphene into fluorinated single-layer diamond. Nat. Nanotechnol. 2020, 15, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Pakornchote, T.; Ektarawong, A.; Busayaporn, W.; Pinsook, U.; Bovornratanaraks, T. Roles of nitrogen substitution and surface reconstruction in stabilizing nonpassivated single-layer diamond. Phys. Rev. B 2020, 102, 075418. [Google Scholar] [CrossRef]
- Pandey, K.C. New dimerized-chain model for the reconstruction of the diamond (111)-(2 × 1) surface. Phys. Rev. B 1982, 25, 4338. [Google Scholar] [CrossRef]
- Iarlori, S.; Galli, G.; Gygi, F.M.C.; Parrinello, M.; Tosatti, E. Reconstruction of the diamond (111) surface. Phys. Rev. Lett. 1992, 69, 2947–2950. [Google Scholar] [CrossRef] [Green Version]
- Pamuk, B.; Calandra, M. Exchange-driven dimerization, magnetism, and insulating state in diamond (111). Phys. Rev. B 2019, 99, 155303. [Google Scholar] [CrossRef] [Green Version]
- Artyukhov, V.I.; Chernozatonskii, L.A. Structure and Layer Interaction in Carbon Monofluoride and Graphane: A Comparative Computational Study. J. Phys. Chem. A 2010, 114, 5389–5396. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106. [Google Scholar] [CrossRef]
- Togo, A.; Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 2015, 108, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E.; Jepsen, O.; Andersen, O.K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 1994, 49, 16223. [Google Scholar] [CrossRef] [PubMed]
- Sofo, J.O.; Usaj, G.; Cornaglia, P.S.; Suarez, A.M.; Hernández-Nieves, A.D.; Balseiro, C.A. Magnetic structure of hydrogen-induced defects on graphene. Phys. Rev. B 2012, 85, 115405. [Google Scholar] [CrossRef] [Green Version]
- Rudenko, A.N.; Keil, F.J.; Katsnelson, M.I.; Lichtenstein, A.I. Exchange interactions and frustrated magnetism in single-side hydrogenated and fluorinated graphene. Phys. Rev. B 2013, 88, 081405. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, S.Y.; Huang, H.; Li, W.T.; Qiao, J.B.; Wang, W.X.; Yin, L.J.; Bai, K.K.; Duan, W.; He, L. Scanning Tunneling Microscopy of the π Magnetism of a Single Carbon Vacancy in Graphene. Phys. Rev. Lett. 2016, 117, 166801. [Google Scholar] [CrossRef] [Green Version]
- Bu, S.; Yao, N.; Hunter, M.A.; Searles, D.J.; Yuan, Q. Design of two-dimensional carbon-nitride structures by tuning the nitrogen concentration. Npj Comput. Mater. 2020, 6, 128. [Google Scholar] [CrossRef]
- Bafekry, A.; Neek-Amal, M.; Peeters, F.M. Two-dimensional graphitic carbon nitrides: Strain-tunable ferromagnetic ordering. Phys. Rev. B 2020, 101, 165407. [Google Scholar] [CrossRef]
- Wei, X.; Fragneaud, B.; Marianetti, C.A.; Kysar, J.W. Nonlinear elastic behavior of graphene: Ab initio calculations to continuum description. Phys. Rev. B 2009, 80, 205407. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wang, Y. Density Functional Theory Study of the Silicene-like SiX and XSi3 (X = B, C, N, Al, P) Honeycomb Lattices: The Various Buckled Structures and Versatile Electronic Properties. J. Phys. Chem. C 2013, 117, 18266–18278. [Google Scholar] [CrossRef]
- Wei, Q.; Peng, X. Superior mechanical flexibility of phosphorene and few-layer black phosphorus. Appl. Phys. Lett. 2014, 104, 251915. [Google Scholar] [CrossRef]
- Chernozatonskii, L.A.; Sorokin, P.B.; Kuzubov, A.A.; Sorokin, B.P.; Kvashnin, A.G.; Kvashnin, D.G.; Avramov, P.V.; Yakobson, B.I. Influence of Size Effect on the Electronic and Elastic Properties of Diamond Films with Nanometer Thickness. J. Phys. Chem. C 2011, 115, 132–136. [Google Scholar] [CrossRef] [Green Version]
- Mahan, G.D. Condensed Matter in a Nutshell; Princeton University Press: Princeton, NJ, USA, 2011. [Google Scholar]
- Miller, T.S.; Jorge, A.B.; Suter, T.M.; Sella, A.; Corà, F.; McMillan, P.F. Carbon nitrides: Synthesis and characterization of a new class of functional materials. Phys. Chem. Chem. Phys. 2017, 19, 15613–15638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Structures of (a) CH, (b) CN, (c) CN, and (d) CN where the black, red and green balls are C, H, and N atoms, respectively. The indicates to the angle measured between surficial and inner atoms. (e) Paths between high symmetry points in the Brillouin zone.
Figure 1.
Structures of (a) CH, (b) CN, (c) CN, and (d) CN where the black, red and green balls are C, H, and N atoms, respectively. The indicates to the angle measured between surficial and inner atoms. (e) Paths between high symmetry points in the Brillouin zone.

Figure 2.
Phonon dispersions of (a) CH, (b) CN, (c) CN, and (d) CN.
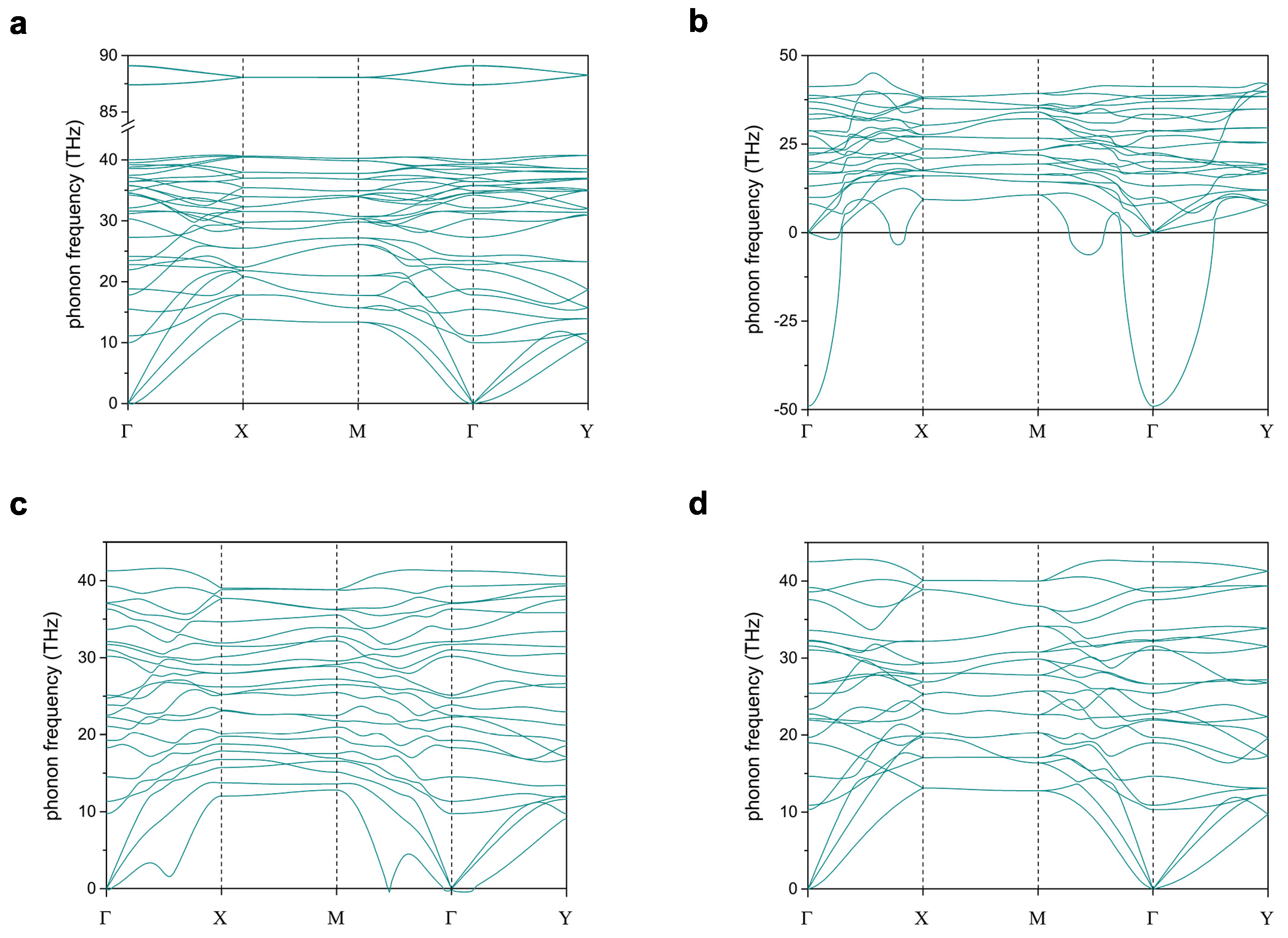
Figure 3.
The electronic band dispersions (left) and DOS (right) of (a) CH, (b) CN, (c) CN and (d) CN. The DOS of each atom are presented by blue, yellow, green and red lines for C, C, H and N atoms, respectively. The DOS of each orbital are presented by solid, dashed, dotted, and dash-dotted lines for s, p, p, and p orbitals, respectively.
Figure 3.
The electronic band dispersions (left) and DOS (right) of (a) CH, (b) CN, (c) CN and (d) CN. The DOS of each atom are presented by blue, yellow, green and red lines for C, C, H and N atoms, respectively. The DOS of each orbital are presented by solid, dashed, dotted, and dash-dotted lines for s, p, p, and p orbitals, respectively.

Figure 4.
Formation energy of two-dimensional (2D) carbon nitrides is plotted with respect to the N concentration in the structures, where pure C phase is graphene and pure N phase is N molecule. The formation energies of NCCN, CNCN, -CN, triazine, and polyheptazine phases are from Ref. [21].
Figure 4.
Formation energy of two-dimensional (2D) carbon nitrides is plotted with respect to the N concentration in the structures, where pure C phase is graphene and pure N phase is N molecule. The formation energies of NCCN, CNCN, -CN, triazine, and polyheptazine phases are from Ref. [21].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The relaxed structures of CH, CN and CN are reported. The atomic species with subscript sf and in indicate an atom on the surface and atom in the inner layer, respectively.
Table 1.
The relaxed structures of CH, CN and CN are reported. The atomic species with subscript sf and in indicate an atom on the surface and atom in the inner layer, respectively.
| Phase | Space Group | Lattice Parameters | Atomic Species | Wyckoff Sites | Positions |
|---|---|---|---|---|---|
| CH | Å | C | 4 h | (0.000, 0.626, 0.027) | |
| Å | C | 4 h | (0.000, 0.367, 0.079) | ||
| Å | H | 4 h | (0.000, 0.519, 0.120) | ||
| Å | |||||
| CN | Å | C | 2 a | (0.000, 0.616, 0.538) | |
| Å | C | 2 a | (0.000, 0.359, 0.463) | ||
| Å | C | 2 a | (0.000, 0.333, 0.601) | ||
| Å | N | 2 a | (0.000, 0.628, 0.397) | ||
| CN | Å | C | (0.000, 0.126, 0.154) | ||
| Å | C | (0.500, 0.377, 0.155) | |||
| Å | C | (0.500, 0.624, 0.226) | |||
| C | (0.000, 0.881, 0.226) | ||||
| Å | C | (0.000, 0.851, 0.092) | |||
| N | (0.500, 0.636, 0.090) | ||||
| N | (0.500, 0.374, 0.293) | ||||
| N | (0.000, 0.190, 0.292) | ||||
| CN | Å | C | 4 h | (0.000, 0.378, 0.031) | |
| Å | N | 4 h | (0.000, 0.625, 0.089) | ||
| Å | |||||
| Å |
Table 2.
The two-dimensional (2D) elastic constants of four phases of W-diamanes are reported in N/m unit by comparing with those of other 2D sp carbons and carbon nitrides.
Table 2.
The two-dimensional (2D) elastic constants of four phases of W-diamanes are reported in N/m unit by comparing with those of other 2D sp carbons and carbon nitrides.
| Phases | |||||||
|---|---|---|---|---|---|---|---|
| CH | 577 | 41 | 22 | 384 | 8 | 451 | 210 |
| CN | 709 | 64 | 27 | 362 | 148 | 300 | 245 |
| CN | 666 | 73 | 26 | 392 | 120 | 335 | 256 |
| CN | 645 | 71 | 17 | 429 | 87 | 375 | 270 |
| NCCN [21] | 568 | 66 | 51 | 217 | 243 | ||
| CNCN [21] | 526 | 61 | 38 | 170 | 220 | ||
| -CN [21] | 595 | 106 | 27 | 510 | 16 | 159 | 244 |
| H-diamane [11] | 487 | 38 |
Table 3.
The elastic constants and Voigt bulk modulus () of four phases of W-diamanes are reported in GPa unit by comparing with those of other 2D sp carbons and carbon nitrides.
Table 3.
The elastic constants and Voigt bulk modulus () of four phases of W-diamanes are reported in GPa unit by comparing with those of other 2D sp carbons and carbon nitrides.
| Phases | ||||||||
|---|---|---|---|---|---|---|---|---|
| CH | 1036 | 73 | 40 | 688 | 15 | 810 | 378 | 310 |
| CN | 1963 | 177 | 76 | 1002 | 410 | 830 | 678 | 569 |
| CN | 1808 | 199 | 71 | 1062 | 3217 | 909 | 694 | 553 |
| CN | 1715 | 190 | 56 | 1140 | 226 | 996 | 719 | 533 |
| NCCN [21] | 2191 | 253 | 198 | 836 | 939 | 718 | ||
| CNCN [21] | 1968 | 227 | 142 | 635 | 825 | 618 | ||
| -CN [21] | 2030 | 361 | 91 | 1739 | 199 | 541 | 831 | 624 |
| H-diamane [21] | 1026 | 81 |
Table 4.
The elastic constants in GPa unit calculated from the group velocities of acoustic phonons at .
Table 4.
The elastic constants in GPa unit calculated from the group velocities of acoustic phonons at .
| Phases | ||||||
|---|---|---|---|---|---|---|
| CH | 102 | 370 | 1003 | 39 | 394 | 692 |
| CN | 147 | 725 | 1666 | 60 | 716 | 1097 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pakornchote, T.; Ektarawong, A.; Pinsook, U.; Bovornratanaraks, T. Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution. C 2021, 7, 8. https://0-doi-org.brum.beds.ac.uk/10.3390/c7010008
AMA Style
Pakornchote T, Ektarawong A, Pinsook U, Bovornratanaraks T. Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution. C. 2021; 7(1):8. https://0-doi-org.brum.beds.ac.uk/10.3390/c7010008
Chicago/Turabian StylePakornchote, Teerachote, Annop Ektarawong, Udomsilp Pinsook, and Thiti Bovornratanaraks. 2021. "Modifying Electronic and Elastic Properties of 2-Dimensional [110] Diamond by Nitrogen Substitution" C 7, no. 1: 8. https://0-doi-org.brum.beds.ac.uk/10.3390/c7010008
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.