
Towards Controlled Degradation of Poly(lactic) Acid in Technical Applications
by
 , and
, and
Stefanie Teixeira
, 
Katarzyna Morawa Eblagon
*,
Filipa Miranda
,
M. Fernando R. Pereira
and
José Luis Figueiredo
Laboratory of Separation and Reaction Engineering-Laboratory of Catalysis and Materials (LSRE-LCM), Faculdade de Engenharia, Universidade do Porto, Rua Dr. Roberto Frias, 4200-465 Porto, Portugal
*
Author to whom correspondence should be addressed.
C 2021, 7(2), 42; https://0-doi-org.brum.beds.ac.uk/10.3390/c7020042
Submission received: 24 February 2021
/
Revised: 15 April 2021
/
Accepted: 19 April 2021
/
Published: 30 April 2021
(This article belongs to the Collection Carbon in the Circular Economy)
Abstract
:Environmental issues urge for the substitution of petrochemical-based raw materials with more environmentally friendly sources. The biggest advantages of PLA over non-biodegradable plastics are that it can be produced from natural sources (e.g., corn or sugarcane), and at the end of its lifetime it can be returned to the soil by being composted with microorganisms. PLA can easily substitute petroleum-based plastics in a wide range of applications in many commodity products, such as disposable tableware, packaging, films, and agricultural twines, partially contributing to limiting plastic waste accumulation. Unfortunately, the complete replacement of fossil fuel-based plastics such as polyethylene (PE) or poly(ethylene terephthalate) (PET) by PLA is hindered by its higher cost, and, more importantly, slower degradation as compared to other degradable polymers. Thus, to make PLA more commercially attractive, ways to accelerate its degradation are actively sought. Many good reviews deal with PLA production, applications, and degradation but only in the medical or pharmaceutical field. In this respect, the present review will focus on controlled PLA degradation and biodegradation in technical applications. The work will include the main degradation mechanisms of PLA, such as its biodegradation in water, soil, and compost, in addition to thermal- and photo-degradation. The topic is of particular interest to academia and industry, mainly because the wider application of PLA is mostly dependent on discovering effective ways of accelerating its biodegradation rate at the end of its service life without compromising its properties.
1. Introduction
Many reviews deal with the technology and degradation of PLA, mostly in a plethora of medical applications, including tissue engineering, implants, skin and tendon healing, and medical tools and equipment [1,2,3,4,5]. However, reviews dealing with the technical applications of PLA are very scarce. The present review’s objective is to show the current progress in the controlled degradation of PLA in commodity applications, where it can substitute in the future the non-degradable poly(ethylene terephthalate) (PET), polystyrene (PS), polypropylene (PP) and others.
With an approximate production of 140 million tons per year, synthetic polymers have become an indispensable part of our life [6]. Intense use of petrochemical-based polymers for more than 70 years has resulted in many environmental issues concerning non-degradable waste accumulated in the landfills and, sadly, in the oceans. The well-known “great Pacific garbage patch,” which is mainly composed of plastics (an estimated 80,000 tones) covering over 1.6 million square kilometers, is shocking evidence of the size of the global problem with plastic waste management. The improper disposal of plastic materials can endanger living organisms’ health and life. Thus, with the ever-increasing use of plastics, significant funding worldwide is directed towards either improving the disposal of petrochemical-based polymers or substituting them with more environmentally benign materials, e.g., produced from biomass. Additionally, alternative bio-resources for plastic production can help decrease the depletion rate of fossil fuels, minimize the political conflicts around the world, and lessen climate changes connected with emissions of greenhouse gasses [7].
Notably, bio-based polymers can replace petrochemical plastics in various sectors and provide new and sometimes improved combinations of properties. Unfortunately, biodegradable plastic represents only a small-scale market compared with conventional petrochemical materials [6]. Globally, bioplastics account for less than 1% of the 181 million metric tons of synthetic plastics the world produces each year [8]. Lately, research and development on biodegradable plastics have been mainly driven by the restrictions in the total “carbon footprint,” particularly in applications such as packaging, automotive, electrical, and electronics industries [9].
PLA is a biodegradable plastic produced from natural resources, such as starch (mainly harvested from corn), combining ecological advantages with reasonable mechanical properties. One of the crucial advantages of PLA is that it requires up to 55% less energy to be produced than petroleum-based polymers, and estimations have shown that this could be further reduced to less than 10% in the near future [10]. Further, at the end of its service life, it can be degraded to CO2 and biomass in the environment [11], allowing a reduction in landfill volumes. The lifecycle of PLA demonstrates that PLA is a sustainable alternative to olefins, because it is produced from natural resources, such as starch (mainly harvested from corn), and at the end of its service life it can be degraded to CO2 and biomass in the environment [11], thus serving as so-called carbon sink. Biodegradability in conjunction with selected disposal systems of PLA such as composting and anaerobic digestion offers an end-of-life solution to completely remove the plastic substrate from the environment and to close the carbon cycle.
Disposable products that would benefit from the advantage of PLA biodegradability should be disposed of alongside organic waste (e.g., PLA twines together with the green residues) in industrial composting facilities, without the need for separation. In some European countries, e.g., the Netherlands, lower surcharges are placed on biodegradable and compostable waste disposal compared to mixed waste [12]. However, PLA is mostly resistant to attack by microorganisms in soil or sewage under ambient conditions, and in fact, it is less susceptible to degradation than other aliphatic biodegradable polymers such as poly(ε-caprolactone) (PCL) in the natural environment [8]. The slow degradation of PLA in the environment limits its application. Thus, both industry and academy have a tremendous interest in searching for new ways of increasing the degradation rate of PLA and its derivatives. However, improving the PLA’s biodegradability is a big challenge to overcome, mainly because it should not compromise the material’s mechanical properties during their service lifetime [13].
Thus, the review will focus on PLA applications in food packaging, textile, and agricultural industries, excluding drug delivery and medical uses of PLA and its decomposition in vivo. Firstly, PLA’s general characteristics that directly influence its degradation will be briefly discussed in Section 4, Properties of PLA, followed by its main degradation paths, along with a description of the factors that influence these processes (Section 5, Degradation of PLA). The mechanism of PLA hydrolytic degradation and biodegradation will be described in more detail in Section 5.1, Hydrolytic Degradation, including enzymatic and microbial degradation in compost (Section 5.4, Microbial Degradation, and Section 6.1, Composting Conditions). Finally, an overview of some selected methods for controlling the PLA’s biodegradation in technical applications will be presented (Section 7, Control and Improvement of PLA Degradation).
2. Properties of Lactic Acid and Lactides
The monomeric unit of PLA is lactic acid with the formula CH3–CH(OH)–COOH, chemically known as 2-hydroxy-propionic acid. Lactic acid is chiral and has two stereoisomers (i.e., L-(+)-lactic acid or S-lactic acid and D-(−)-lactic acid or (R)-lactic acid), as shown in Figure 1 [14]. As the result of these stereoisomers, three different PLA materials can be expected, depending on the substrate material, such as poly(L-lactide) (PLLA), poly(D-lactide) (PDLA), or a racemic mixture called poly(D,L-lactide) or PDLLA. Most commercial PLA consists of copolymers of PLLA and PDLLA [15].
All the stereoisomers above occur naturally as the products of microorganism activity (bacterial systems). However, the most commonly observed forms are L-type lactic acid or a racemic mixture (mainly 50% L and 50% D) [16]. Additionally, the L-form constitutes the main fraction of PLA derived from natural resources [15] (such as biomass), and it is also the only form of lactic acid produced by humans and other mammals, which has led to the wide application of PLA in the biomedical industry [17].
The optical properties of PLA are essential for packaging applications, where clarity is desirable [18].
In general, the polymer’s physical and mechanical properties, which may be tailored to fit the specific requirements of the application, are mainly determined by its stereochemistry [19]. The degree of crystallinity of PLA depends on the type and composition of the stereoisomers of lactic acid monomers and the polymer’s thermal history. Control of the physical properties and biodegradability of PLA can be achieved by the racemization of D- and L-isomers [20]. Thus, when the amount of D-(−)-lactic acid is higher than 6%, the resulting PLA can be amorphous; when it is lower than 6%, it is considered semicrystalline.
The degree of crystallization of PLA affects its melting temperature (Tm), glass transition temperature (Tg), and its mechanical properties, including elastic modulus and mechanical strength. Moreover, as the percentage of the D-isomer increases, both the Tm and Tg decrease. High crystallinity PLA might be desirable for durable products with long-term performance in the automotive and electronic industries due to improved thermal and chemical resistance. On the other hand, the presence of many amorphous domains, which increase the degradation rate of PLA, is highly beneficial for disposable products [4,20,21,22].
3. Methods of PLA Production
Currently, NatureWorks LLC Northford, USA, is the world leader in lactic polymer technology and markets. The company off-set their non-renewable carbon emissions by buying wind energy certificates and thus claim carbon neutrality for their PLA (marketed under the trade names Natureworks™ or Ingeo™).
Corn-derived PLA presents a competitive advantage versus petroleum-based plastics due to significantly diminished greenhouse gas emissions (approximately 30–55%) [23] and price independence from oil fluctuations. Over the past ten years, NatureWorks LLC has developed patent-protected technologies for lactic acid-based products for a wide range of specific applications such as extrusion and thermoforming, injection molding, films, foams and 3 D printing. Recently, one of their patented lactic acid products was used to produce reusable N95 protection masks against Covid 19 [24]. Nevertheless, from an economic point of view, bioplastics continue to be more expensive than traditional petroleum-derived plastics due to their less efficient production [25,26]
Nowadays, PLA production accounts for over 39% of the total lactic acid demand worldwide. Moreover, 70% of the total worldwide production of lactic acid is carried out by microbial fermentation [27], using enzymatic hydrolysates from starch [28]. The high cost of pure carbon and nitrogen sources commonly employed for lactic acid production with Lactobacillus or Lactococcus has motivated several studies using food wastes. Numerous candidates have been explored, including sugarcane molasses [29], paper sludge [30], waste paper [31], apple pomace [32], among others.
Fermentation techniques produce the desired stereoisomer, optically pure D (−) or L (+) lactic acid, or a racemic mixture. The majority of the commercially produced lactic acid uses modified strains of genus Lactobacilli [33]. The type of bacteria used governs the type of stereoisomers obtained and the yield of lactic acid produced in the process. The most technically and economically feasible alternative method of lactic acid production is chemical synthesis using the lactonitrile route and applying acetaldehyde derived from petroleum resources [28]. However, this method is expensive, and it can only produce a racemic mixture of the lactic acid isomers [34]. As a result, approximately 90% of the total lactic acid produced worldwide is made by bacterial fermentation, and only the small remaining portion is produced synthetically by the hydrolysis of lactonitrile [28].
PLA of variable molecular weights can be produced from lactic acid. However, mostly the high Mw PLA has commercial value in the fiber, textile, plasticulture, and packaging industries [4].
The synthesis of PLA can be divided into chemical and biological polymerization. Within chemical polymerization, there are three methods, (i) direct polycondensation (DP) of lactic acid (ii) azeotropic condensation polymerization (iii) ring-opening polymerization (ROP) of the cyclic dimer of lactic acid called l lactide [4,21,35].
The main steps of the direct condensation polymerization process involve: (i) removal of free water, (ii) oligomer polycondensation, and (iii) melt polycondensation of high Mw PLA [4,18,36]. A detailed description of the condensation polymerization method can be found in [33].
On the other hand, azeotropic condensation polymerization (ACP) and ROP are two methods for directly synthesizing PLA with high Mw. The former method (i.e., ACP) is similar to DP per se; the main difference lies in the use of the solvent in ACP to reduce the systems’ viscosity at high degrees of conversion [37]. Moreover, ACP produces high molecular weight polymer without chain extenders or adjuvants (unlike DP). The general steps of DP and ROP are shown in Figure 2. The main difference between these methods is that ROP involves an intermediate called lactide in the first step, which will be discussed later. Due to the existence of different preparation methods, two names can be interchangeably used to describe PLA, namely: poly(L-lactide) denotes the origin of the polymer from the cyclic monomer, or poly(L-lactic acid) to indicate the repeating acid unit in the polymer backbone, respectively. Thus, polymers obtained by polycondensation are usually referred to as poly(lactic acid) and those prepared from lactide as polylactides. In the present work, both types are abbreviated as PLA for simplification.
ACP was first reported by Mitsui Toatsu Chemicals, Inc. Singapore [38]. It involves continuous removal of condensation water by azeotropic distillation and consists of the following steps: (i) distillation of lactic acid under vacuum; (ii) addition of catalyst and diphenyl ether and oligomer polycondensation; (iii) polycondensation of melt to obtain high Mw PLA while refluxing the solvent with additional drying using 3-A molecular sieves (iv) isolation or precipitation of PLA [4,21,33]. It should be noted that the PLA produced by ACP is very likely to contain traces of the catalysts used during its synthesis, which may, later on, lead to unwanted side reactions or even PLA degradation. Thus, additional steps of PLA purification procedures have to be carried out after the synthesis, such as precipitation of the catalyst followed by filtering in the presence of, e.g., sulfuric acid. Tin catalysts can be deactivated by the addition of phosphoric acid [38].
ROP is the industrially most preferred method of obtaining high-Mw polylactic acid and copolymers of lactide and glycolide. The method is based on the original Cargill-Dow patented process [8,18,33,37,39] and allows obtaining high Mw PLA with narrow molecular weight distribution. ROP can be carried out in the melt, in solution, or suspension, and a catalyst is usually required to start the polymerization process [40]. The primary intermediate of ROP, lactide, is a cyclic lactic acid dimer formed in the first step of the reaction (condensation polymerization), while the condensation water is removed via evaporation during oligomerization. All enantiomers of lactic acid (i.e., L-lactic acid, D-lactic acid, or mixtures thereof) can be polymerized to low Mw PLA (prepolymer PLA), which is subsequently depolymerized by thermal unzipping reaction to form lactide [40]. Three stereoisomers of lactide can be produced, namely: L-lactide, D-lactide, and Meso-Lactide [9,34], as shown in Figure 3, which can result in three stereochemical forms of PLA, namely: PLLA, PDLLA and PDLA [41].
4. Properties of PLA
4.1. Thermophysical Properties and Crystallinity
The thermophysical properties and crystallinity of PLA have a direct influence on its degradation. The thermophysical properties of PLA depend on molar mass, thermal history, and purity of the polymer sample The influence of these features on the thermal behavior of PLA has been a subject of many published works; thus, here, only general trends that are strongly connected with the degradation of PLA will be pointed out, which involve heat capacity, thermal transition, and crystallization of PLA. Interested readers can find more detailed information about these properties elsewhere [16,43,44,45,46,47,48,49].
Polymers prepared from meso- or racemic-lactide are amorphous, as mentioned before, but by applying stereoselective catalysts, PLA having tacticity high enough for crystallization can be obtained [20]. The crystallization rate of PLLA is unusually high at temperatures between 100 and 118 °C [50]. PLA can crystalize in three structural forms: α, β, and γ, which differ in the helix conformation and symmetries [50,51]. The PLA’s crystallinity can be increased with nucleating agents in injection-molding and extrusion [52]. In this way, the α form mentioned above is produced. Alternatively, the β form of PLA develops upon mechanical stretching of the more stable α form from solution-spinning processes conducted at high temperatures or high hot-drawing ratios (i.e., stress-induced crystallization) [50,52,53].
Moreover, crystallization of PLA for applications requiring high thermal stability can be initiated by annealing PLA at temperatures higher than Tg but below its melting point [15]. It should be pointed out that the rate of crystallization of PLA is much slower than other thermoplastics, limiting its use in high-performance applications. The rate of crystallization influences the PLA degradation rate, thermal resistance, optical, mechanical, and barrier properties.
Typically, Tm, Tg, and crystallinity of PLA decrease with decreasing content of L-isomer. However, it also depends on the Mw of the polymer. Oligomers with a minor number of lactyl units show a Tg directly dependent on Mw [20,54]. The Tg of PLA directly impacts the material’s processability and service temperature.
In comparison to other thermoplastics, PLA has a relatively high Tg and low Tm. Metastable states of high molecular weight amorphous polylactides, based on the reports of Auras et al. [55], are shown in Figure 4. As follows, below the so-called transition temperature of −45 °C, PLA is entirely brittle. Between temperatures of −45 and 58 °C, amorphous polylactide undergoes physical ageing and behaves similarly to glass, capable of creep until it is cooled to its transition temperature [15]. In this range of temperatures, PLA can show ductile or even brittle fracture [55]. As the temperature increases above Tg (~58 °C), PLA becomes rubbery. At temperatures below Tg, no large-scale molecular motion is feasible inside the polymer chains, and the material is hard, which explains its behavior. On the other hand, if the temperature increases above Tg, some motion can occur, and the material becomes softer [16].
Thus, PLA with a low Tg is not suitable for storing hot liquid, as with the increased temperature, the material would soften and would easily deform [9]. With further increase of the temperature, in the range of 110–150 °C, PLA changes its phase from rubbery solid to viscous liquid, and this transformation depends mainly on the Mw of the specimen and the applied shear stress [55]. As shown in Figure 4, at temperatures above 215 °C, PLA undergoes decomposition. Overall, the properties of amorphous PLA depend on the difference between its Tg and the temperature at which it is stored and used.
For semicrystalline PLA, both Tg and Tm are critical for predicting the polymer´s behavior. For amorphous PLA, the Tg is one of its most essential parameters since dramatic polymer chain mobility changes occur at and above this temperature. As a result, amorphous plastics, such as PLA containing a mixture of D and L-lactides, perform best at T < Tg, but elastomers must be used above the brittle point [18]. Moreover, it should also be mentioned that the Tg of PLA depends on its optical purity and, to a large extent, on the particular sample’s thermal history.
Concerning the Tm, it is usually influenced by the level of crystallinity of the polymer [52]. Pure poly(D-Iactide) or poly(L-lactide) equilibrium crystalline melting temperature can be as high as 207 °C. However, the lower temperature of around 180 °C is typically measured in practice due to small and imperfect crystallites, slight racemization, and impurities usually present in the materials. The Tm = 180 °C can be decreased down to 130 °C by adding meso lactide, depending on the amount incorporated into the polymer [15]. As obtained, lower crystalline melting points allow lower melt-processing temperatures of the final polymer, expand the process window, decrease lactide formation and reduce degradation [52].
Blending of PLLA and PDLA can form a stereocomplex with an equilibrium crystalline melting point as high as 230 °C, which is considerably higher than the Tm of the pure polymers. This increase in Tm is due to the side-by-side packing of stereo complexes, forming a compact, and ordered structure [56,57]. The mentioned Tm of enantiomerically pure semicrystalline PLA is higher than 130 °C, which is typical for amorphous meso-lactide [20,21,54,58]. Precisely, for engineering applications, which demand high thermal stability, the general tendency is to use a PLA matrix with extremely low D-isomer content, which can exhibit higher Tm and degree of crystallinity. Moreover, Tm of PLA depends strongly on its molar mass. For example, PLA with Mn of 2000 displays a 60 °C lower Tm than that of PLA with Mw close to 20,000 [58].
4.2. Miscibility with Other Polymers
New PLA-based products (composites, nanocomposites, tailored formulations, etc.) with improved characteristics and performance are always under research and development. Reducing the cost of the material by blending PLA with other polymers is also crucial since the cost of PLA is roughly twice to three times the cost of oil-based plastics. On the other hand, employing copolymerization, blending, or additives to PLA is one way to control its degradation by influencing its mechanical and thermal properties. Thus, we will briefly discuss the most commonly found blends of PLA.
Lactic acid can be polymerized with other monomers, hydrophilic macromonomers (e.g., PEG), or other monomers with functional groups (such as amino and carboxylic groups) [18]. For example, the copolymerization of lactide with another lactone-type monomer such as polyglycolide results in poly-lactic-glycolic acid (PLGA), which has lower crystallinity and melting point than PLA [59].
Polyethylene glycol (PEG) is an excellent plasticizer for PLA and can significantly enhance its elongation at break [60]. Moreover, PLA is very often mixed with starch in the presence of glycerin due to economic reasons and to improve the mechanical properties of PLA (e.g., increase toughness) by changing the concentration of starch in the composite [61]. Another study reported the improvement of tensile strength and elongation after blending PLA with starch particles. The enhancement of the properties mentioned above depends on the starch granules’ average particle size—the improvement declined when the granules were greater than 45 µm [15].
Several PLA blends with other polymers have been studied over the past years, including: poly(4-vinyl phenol) [20,62,63,64], poly(vinyl acetate) [20,62,65,66], poly(vinyl chloride) [20,67], poly(ethylene) [20,68], poly(ethylene oxide) and copolymers [20,62,69,70], poly(ethylene glycol) [20,62,71], poly(ethylene-co-vinyl acetate) [20,62,72], poly(cis-1,4-isoprene)s [20,73], poly(ε-CBZ-L-lysine) [20,74], polyacrylates [20,75,76,77], poly(butylene succinate) [20,48,62,78], hydrolytically degradable rubbers [20,79], silicates [20,80], glycolide [20,62,81,82], and polyhydroxyalkanoates [20,62,83,84].
In general, to improve the targeted property, phase separation during blending should be avoided. On the other hand, immiscible blends produce a rubber toughening effect on PLA that could benefit some applications [20,62]. Under normal conditions, a compatibilizer (i.e., catalyst or a coupling agent) must make the polymers miscible with PLA [20,62,85].
5. Degradation of PLA
According to the European Union standard, a biodegradable material should be converted in more than 90% into CO2, water, and minerals by biological processes within six months. More generally, the European norm for biodegradable plastics (CEN/TR 159325) states that the characteristics of biopolymers and bioplastics can be used to describe:
- Biogenic or biobased plastic, originating from renewable sources;
- Biodegradable plastics, in terms of their functionality;
- Biocompatible plastics, in medical applications only.
The term biodegradation is often used to refer to the degradation of PLA occurring in biological environments. The process is based on microorganisms (bacteria, fungi, and algae) using a polymer material as a source of energy for their life [86].
PLA is a biodegradable commodity plastic that needs to be stable during the service life but quickly degrade when it enters the waste stream, should the recycling be not feasible. Initially, PLA biodegradation occurs on the PLA surface and ultimately spreads inside the PLA matrix to facilitate the material’s total degradation.
PLA can undergo hydrolytic, thermal, oxidative, bacterial, enzymatic degradation and photodegradation [87]. However, most of the studies published were focused on enzymatic degradation [88,89,90] or hydrolytic stability of PLA [41,91,92]. These mechanisms are essential for reducing PLA to CO2, water and biomass material in the context of increasing environmental issues with plastic waste disposal.
Degradation of any polymer produces changes in its mechanical, optical, or electrical characteristics through crazing, cracking, erosion, discoloration, or phase separation [93]. It can take place via (a) scission of the leading chains, (b) scission of the side chains, or (c) scission of the intersectional chains [41]. It should be noted that all of the PLA degradation mechanisms usually involve the ester bonds’ scission. Depending on the type of factor inducing the degradation process, we can divide the degradation of PLA into non- biotic processes (i.e., hydrolysis, thermal degradation, oxidation, photolysis) and biotic processes (i.e., digestion by microorganisms) [4,21]. Out of the processes mentioned earlier, abiotic hydrolysis isa rate-limiting step in the biodegradation of PLA [94].
Regarding the hydrolytic stability of PLA, one of the significant challenges that the PLA users have to face is its tendency to undergo degradation during processing from the molten state. The degradation is accelerated if the material is not correctly dried. Thus the PLA substrate has to be carefully dried beforehand, accordingly to the indications from the supplier.
The rates of thermal (isothermal at 220 °C), biological, and photo-degradation (under ultraviolet (UV) light) of PLA with 4 mol% D were compared by monitoring the mass decrease of the polymer samples. The study concluded that the primary step under all of the conditions studied was a random chain excision, and the degradation rate of PLA specimens followed the progression: thermal > photo > biological. Overall, the degradation in the soil was found to be the slowest of the three processes considered. After the same degradation time, photodegraded specimens showed about half the molar mass of soil-degraded samples [95].
The degradation of PLA depends not only on the characteristics of the specimen (i.e., degree of crystallinity, molecular weight, sample morphology, its molecular and supramolecular structures, etc.) but also on the conditions of the surrounding environment, including the presence of water and moisture, temperature, acidity conditions, presence of oxygen, or type and activity of microorganisms [96]. For example, water or moisture are essential in the degradation environment for the microorganisms to grow and reproduce. Additionally, the increase in water amount maximizes the rate of hydrolytic degradation of PLA. Thus, it can be expected that the overall biodegradation of PLA will be faster in moisture-rich environments than in dry conditions [96].
In general, the rate of PLA degradation is mainly determined by polymer reactivity with water and catalysts (if present) [55]. Temperature is another critical factor affecting the degradation of PLA. In general, the hydrolysis rate and the microbial activity increase monotonically with temperature. However, at too high temperatures, microorganisms’ activity can drastically decrease or stop altogether.
Another environmental factor influencing the degradation of PLA is exposure to UV light. It was found that UV light decreases the physical integrity of PLA, increases its brittleness, stress at break, and average Mw [21]. The photodegradation and other modes of PLA degradation will be described in more detail in the following paragraphs.
Last but not least, the degradability of PLA depends on macromolecular architecture. Thus, branched structures display faster degradation rates than the corresponding linear ones [97,98].
5.1. Hydrolytic Degradation
An in-depth understanding of the hydrolysis process of PLA is imperative to control the degradation properties of most of the PLA products at the end of their lifetime. During hydrolysis, the main chain’s ester groups are cleaved until the polymer is completely converted into soluble oligomers and monomers [4,20,21,99]. The ester bonds of PLA fragment into carboxylic acid and alcohol by chemical hydrolysis due to hydrion (see Equation (1)). However, in the absence of any catalyst, the hydrolysis of PLA consumes plenty of energy and time [41,87].
The degradation of PLA in water can be described as a non-enzymatic chain scission followed by a chain-end, which causes a decrease in the Mw of PLA [4,20,21].
−COO + H2O → −COOH + OH
Hydrolysis can take place not only in water but also in alcohol [100], alcoholic solutions, such as water-ethanol mixtures [101], or acetonitrile solutions [99]. Conversely, it has been found that PLA will plasticize and crystallize in the presence of other organic solvents, which swell the polymer matrix, increasing chain mobility and rapid solvent-induced crystallization [8,102].
The hydrolysis of PLA is a self-catalyzed reaction since the carboxylic acid end groups of PLA and their oligomers can catalyze the breakage of ester linkages. The chain’s scission during hydrolysis releases H+ acid groups. The acid is retained in the material or the reaction medium, resulting in a decrease in the pH and acceleration of the hydrolysis rate [87]. The pKa of PLA’s carboxylic acid end group and its oligomers is lower (~3) than most carboxylic acid groups (4.5–5), leading to a faster rate of degradation (autocatalysis).
As it can be expected, the hydrolysis of the amorphous PLA chains is always easier than that of crystalline ones because water diffuses readily into the less organized amorphous regions. Thus, hydrolysis tends to start in these regions and later propagates with a decreased rate into the sample’s crystalline parts [8,21,103]. As a result, the hydrolytic degradation of PLA causes a substantial increase in the specimen’s crystallinity and reduces its physical and mechanical properties [104].
Chen et al. studied molecular ordering of amorphous PLLA during hydrolysis in alkaline media at temperatures in the range of 40–60 °C [105]. The authors found that at temperatures of 40–50 °C, a locally ordered structure was created, and many α-form crystallites were induced when the temperature reached 60 °C. Subsequently, the amount of ordered structure increased gradually with the time of hydrolytic degradation.
It should be mentioned that the hydrolytic degradation of PLA in medical applications is not the focus of the present review. Interested readers can find information on this subject in various excellent articles and reviews [106,107,108].
5.1.1. Hydrolysis Mechanism
In general, the hydrolytic degradation of a polymer results from the interplay between chemical hydrolysis and diffusion of water and oligomers [2]. From the point of view of organic chemistry, the reaction is a bimolecular nucleophilic substitution (shown in Figure 5), and it can be catalyzed by either acids or bases [109]. Interestingly, the mechanism and kinetics of the hydrolytic degradation of PLA are governed by the pH of the sample’s environment. In particular, at neutral or basic pH, the hydrolytic degradation occurs preferentially through backbiting reactions, although a minor contribution of random scission hydrolysis was observed [91]. On the other hand, hydrolytic degradation of D, L-lactic acid oligomers in basic media showed random ester cleavage to be the primary mechanism [110], which is controlled by the rate constant, the amount of absorbed water, the diffusion coefficient of the chain fragments and finally by the solubility of the degradation products: lactic acid and lactide [7,99,110].
Tsuji and Ikaida reported that alkaline conditions are different from the others. The formed oligomeric acids are dissociated into RCOO− and hence must have high hydrophilicity to diffuse into the outer solution in contrast to the non-dissociated oligomers RCOOH formed in the neutral environment [104]. On the other hand, at acidic pH, it was shown that the hydrolysis of PLA proceeds through a preferential scission of the polymer end-groups, or so-called “unzipping”.
The kinetic constant of the scission of the terminal groups of PLA was reported to be 10 times higher than that of the internal esters [111].
The mechanism shown in Figure 5 seems to be relatively straightforward. However, due to the long-chain structures and condensed states of polymers such as PLA, the detailed steps of the PLA hydrolysis are far more challenging. For example, polymer bonds at the end of the chain can have different reactivity from those situated in other sites, as well as the reactivity of the crystal domain being different from its amorphous counterpart.
During hydrolysis, different diffusion-transport phenomena of water, ions, and PLA degradation products occur. These phenomena can lead to several changes in the reaction mechanism. For example, in larger PLA specimens, the carboxylic acid groups concentrate inside the polymer due to the restricted diffusion of the chain ends. Thus, a pH gradient develops because the polymer’s surface is kept at a neutral pH, which slows down the degradation of the polymer matrix surface compared to the centre. The surface layer only gives up when a critical osmotic pressure builds up inside the matrix due to the accumulation of degradation products [109] .
The hydrolysis of PLA is a third-order kinetic reaction, as initially proposed by Pitt et al. [112] in their study of the degradation of similar polyester material, poly(ϵ-caprolactone). They reported that the hydrolysis rate of PLA depends on the concentration of the polymer bonds, the quantity of adsorbed water, and the concentration of acidic hydrolysis products, as well as on diffusion rate and coefficient of chain fragments within the polymer, but interestingly, not on the amount of H+ itself [87,109]. This theory predicted a linear relationship between the logarithm of the polymer molecular weight with degradation time, but it did not consider the transport phenomena during polymer degradation.
Schliecker et al. reported that the degradation constant decreases with increasing Mw of PLA sample [110]. Lyu et al. [109] reported that the hydrolysis rate displayed a slow-to-fast transition at a particular Mw. This transition was not affected by the mass loss and water uptake of samples or the changes in the media’s pH values. Thus, it was speculated that this transition was due to the slow diffusion of the polymer chain ends. The authors found that the concentration of the chain ends has to reach a specific critical value, at which the hydrolysis starts to be promoted by the carboxylic acid chain ends. Other works point out that the kinetics of chain scission of PLA indicates an autocatalytic process. The carboxylic acid end groups generated by ester hydrolysis contribute to the process acidity and participate in the transition state [99] .
The corresponding rate equation for hydrolytic degradation is given in Equation (2), where [H2O], [E], and [COOH] are concentrations of water, ester (reactant), and carboxyl end groups [99,112,113,114,115].
According to this equation, the term “self-catalysis” can be defined as taking place when the acid group (catalyst) concentration increases and, simultaneously, the ester (reactant) concentration decreases in 1:1 mole proportion.
Pseudo-first-order kinetics is a good approximation for hydrolysis catalyzed by a strong external acid but is not entirely suitable for self-catalyzed hydrolysis. Thus, a model was proposed (see Equation (3)) considering partial dissociation effects and half-order dependence on a carboxylic acid group. This model fitted the experimental data well except for the data set extrema [92,99,114].
In general, the hydrolysis of PLA can be described with four subsequent steps: (i) water sorption, (ii) reduction of mechanical properties, (iii) reduction of Mw and (iv) complete loss of weight of the material [115,116]. During the process, a loss in strength and modulus is observed due to the plasticizing effect of water and the drop in Mw of the polymer.
5.1.2. Erosion
It should be mentioned that hydrolysis is only an initial step of a multistep process in which the polymer eventually degrades. In this context, the whole process from the polymer hydrolysis to its complete “disappearance” into the environment is called erosion [117,118]. Two types of erosion have been classified according to the way of degradation. These processes are: surface erosion (heterogeneous) or bulk erosion (homogenous) [87,96,117]. For example, hydrolysis occurs heterogeneously because it proceeds faster in bulk than on the surface [92,117]. Furthermore, it was reported that heterogeneous erosion is faster than its homogenous counterpart [7].
Surface erosion happens when the hydrolysis rate is higher than the water diffusion into the material’s bulk [87]. The surface eroding polymers lose mass only in the water/polymer interface. In this case, the sample’s shape remains the same, but its volume decreases with time [117,118]. On the other hand, bulk erosion is when the hydrolysis occurs throughout the entire sample, and during the process, water diffuses rapidly into the polymer structure, and as a consequence, mass loss within the bulk of the sample is observed [118]. Since the bulk erosion is not confined to the surface, the polymer sample size usually remains constant for a substantial amount of time. It should be also mentioned that it is generally assumed that the degradation of PLA becomes a bulk process above Tg. At temperatures below Tg, degradation of the polymer matrix is restricted to its surface [3,119].
According to the literature, polymers that contain less reactive functional groups, such as PLA, were reported to be predominantly bulk eroding materials [120]. Tsuji and Ikada studied the degradation of PLLA films in alkaline solutions and reported that the degradation proceeded through surface erosion mechanism [104]. On the other hand, some authors believe that the type of erosion taking place mainly depends on the formation velocity of diffusing oligomers [121]. It is generally accepted that PLA degradation is heterogeneous (bulk erosion), which is assigned to an internal autocatalytic effect of the carboxyl end group. During the infiltration of the material with water, the hydrolysis develops from the inside of the polymer towards its surface, causing sudden and rapid loss of strength and structural integrity [115,117].
A significant advance in this field was achieved by Burkersroda et al. [122], who have developed a theoretical model that allows predicting the erosion mechanism of water-insoluble biodegradable polymers. Following this model, all of the polymers can undergo bulk or surface erosion, and the type of the mechanism depends on the diffusivity of water inside the matrix, the degradation rate of the polymer’s functional groups, and the matrix dimensions. Accordingly, the erosion mechanism is defined by the critical dimension of the device to be degraded. It is defined by the thickness normal to the reaction surface (abbreviated Lcritical = 7.4 cm in PLA). Thus, surface erosion happens when the thickness of the matrix is larger than Lcritical; if the dimensions are smaller than Lcritical, bulk erosion prevails [115,122].
On the other hand, Lyu and co-workers found that the erosion rate and the erosion front width (defined as a product of induction time and the erosion rate) can be expressed as simple functions of the rate of polymer bond hydrolysis, water diffusivity, and solubility, and other experimentally defined parameters [118]. Considering this theory, the type of erosion that the sample undergoes can be predicted considering the ratio of the front width to the polymer specimen’s thickness. Moreover, the authors’ modelling work allows predicting the type and rate of erosion for a given sample. These results allow better understanding and control of the degradation of PLA.
5.1.3. Factors Affecting Hydrolytic Degradation of PLA
The most important factors that influence the hydrolysis rate of PLA are temperature, pH, relative humidity (RH), Mw of the sample and its crystallinity, chain stereo-configuration, and the chemistry of the solvent. The rate of hydrolysis increases with temperature as the chain scission is accelerated. For example, the abiotic hydrolysis rates differ considerably in the range of temperatures from 20 to 60 °C [109]. Furthermore, it should be noted that the dielectric constant of liquid water decreases with increasing temperature leading to the improved affinity of water for the polymer and acceleration of the hydrolysis [123]. Most of the studies on the influence of the temperature on the PLA’s hydrolysis were conducted at temperatures that were either higher or lower than Tg or Tm of PLA, and they all suggested Arrhenius-dependent kinetics with activation energies in the order of 40–100 kJ/mol [55].
In contrast, Lyu et al. studied hydrolysis of PLA in the range of temperatures from 37–90 °C, which included the Tg of PLA (55 °C), and they showed an essential complication in the general understanding of biodegradation of PLA. The authors found that the molecular weight data at various temperatures could not fit the existing equations describing polymer degradation. Moreover, the data points at 37 °C did not fit the Arrhenius equation. It appeared that the lack of mobility of the –COOH chain ends in the early stages of biodegradation was not adequately accounted for in the existing theories. However, the authors found that the degradation kinetic constant followed a Vogel–Tamman–Fulcher (VTF) temperature dependence [109].
On the other hand, Han and Pan [124] developed a polymer degradation model which accounted for the interplay between the autocatalytic hydrolysis reaction, oligomer diffusion, and degradation-induced crystallization. Later, this model was further improved and simplified by Han and co-workers [125]. Another study reported that the rate of PLA degradation is higher above Tg because polymer chains become more flexible at this temperature, which increases the adsorption of water, accelerating both hydrolysis and microbial attachment [21].
Concerning the influence of the solvent chemistry on the hydrolytic degradation of PLA films, the rate of the process was found to be higher in 50% solutions of ethanol in water than in 95% ethanol or pure water [101]. It was explained by the fact that ethanol molecules diffused more efficiently inside the polymer matrix.
Furthermore, Coszach et al., in their patent [126], reported that when alkyl lactates are used as a solvent for hydrolysis of PLA to lactic acid, they significantly aided PLA dissolution and added supplementary benefit of removing other polymers. Hydrolysis of PLA in alkyl lactates was achieved with or without NaOH at 80−180 °C and pressures of up to 10 bar.
The effect of pH on the hydrolytic degradation of PLA has been studied in a few works. It is known that the hydrolysis of polyesters is slowest in pH 4 solutions. The study of lactic acid degradation showed that on increasing pH values from 1 to 10, the observed rate constant values first decreased and reached a minimum at pH of about 4, and then increased for higher pH values [127]. Notably, the increase mentioned above is about 4 orders of magnitude. It can be explained taking into consideration that the pKa of lactic acid is 3.84, which means that in solutions with pH > 4, lactic acid will be present mainly in a dissociated form, thereby accelerating hydrolysis. Alternatively, in solutions with pH < 4, lactic acid at the chain ends exists in an associated acid form, which increases the rate of hydrolysis via auto-acceleration. In agreement with these results, Göpferich reported that a fast rate of hydrolytic degradation of PLA could be realized at low and high pH conditions [117], which confirmed that both acids and bases could catalyze hydrolysis.
Conversely, the degradation rates of solid polylactide samples were reported to be almost the same for pH values of 0, 4, and 7. It was explained by the fact that the ions (H+ and OH−) from the solutions have very low solubility in polymers, so they cannot effectively catalyze the degradation of solid polymer samples [128].
Another study reported higher hydrolytic degradability of poly(D, L-lactic acid) in strongly basic conditions than in strongly acidic conditions, but very low degradability in moderate acidic, basic and neutral conditions [129]. Interestingly, in a buffered pH media, the auto-acceleration of PLA’s hydrolysis was found to be suppressed at neutral and low pH [109,117].
Concerning the influence of the RH, it was reported that the rate of hydrolysis of PLA increases with a high RH (>60%) due to faster absorption of water molecules [4,21,99]. Another study focusing on hydrolytic degradation of PLA films revealed that the state of water (i.e., liquid vs. vapor) has an interesting influence on the hydrolysis rate. It was found that prolonged storage of PLA specimens (2 months) in water or 100% RH conditions was slightly faster than in the liquid water conditions [130].
It was reported that the higher degree of crystallinity and higher dispersity of PLA decrease the degradation rate. As mentioned before, the incorporation of D-units in the PLA material perturbs the crystallization process and thereby reduces the degree of crystallinity of the sample. Thus, increasing the amount of D-units in the PLA specimen lowers the regularity of the material and increases the water diffusion rate, leading to the acceleration of the hydrolysis. Moreover, hydrolytic degradation was also enhanced by a higher molar mass distribution of PLA [131].
The hydrolytic degradation of PLA products can be tailored to broaden its applications by adopting the following methods: (i) incorporation of hydrophilicity into the surface of the polymer, (ii) addition of hydrophilic fillers or polymers, (iii) incorporation of proteinase, which facilitates hydrolytic attack, (iv) introduction of chain extenders [132], (v) decrease of activation energy of hydrolysis [127]. Some of these approaches will be discussed in more detail in the following paragraphs.
5.2. Thermal Degradation
One of the main disadvantages of PLA is its limited thermal stability, especially at temperatures above 190 °C [58]. It is mainly because the ester groups of PLA responsible for polymer degradation are also thermally unstable [133]. Thus, the thermal degradation of PLA usually is enhanced by moisture, oxygen, metal catalysts, or mechanical forces [134]. Depending on the combination of external forces, the degradation of PLA under elevated temperatures can be purely thermal, thermo-oxidative (degradation due to temperature and the presence of oxygen in the atmosphere), or thermo-mechanical (degradation due to temperature and mechanical stress) [135].
In general, structural changes in the PLA start to be visible at Tg when the mobility and the volume of the polymeric chains are modified [136]. Thermal degradation of PLA is associated with the hydrolysis initiated by moisture during processing and is influenced by several factors such as initial Mw, the amount of residual water, and traces of polymerization catalysts (if present).
Under an inert atmosphere, thermoplastic polymers’ thermal degradation occurs at Tm when the polymer is transformed from a solid into a viscous liquid. In the case of PLA, it depends on the Mw of the sample and occurs at temperatures from 159–178 °C [136].
Several studies have addressed the complex mechanism of the thermal degradation of PLA. Zou et al. interpreted the thermal degradation kinetics of PLA in multi-step degradation mechanisms [137]. Some of the most commonly-reported PLA degradation mechanisms can be observed in Figure 6.
McNeal and Leiper [103] reported that thermal degradation occurs predominantly by random chain scissions through non-radical reactions along the PLA backbone. Dynamic oscillatory measurements showed that the extent of these phenomena could be increased by raising the temperature, extending time, or applying mechanical stress [135]. Additionally, some radical reactions can be present at temperatures above 270 °C.
Kopinke and Mackenzie [138,139] reported transesterification and non-radical reactions to be the major mechanisms of PLA’s pyrolysis, apart from non-selective radical reactions observed at temperatures above 300 °C. The authors found that intramolecular transesterification gives rise to cyclic oligomers.
Due to acrylic acid found in the reaction products, cis-elimination (shown in Figure 6) was also suggested as another reaction pathway [103]. The ester interchange has lower energy of activation than the cis-elimination. Therefore, the ratio between these reactions shifts towards the cis-elimination at more elevated temperatures. Apart from acrylic acid, other products of cis-elimination include oligomers with open-chain structure and carboxyl groups. O-phthalic acid or 1,4-butanediol can also be produced if the reaction starts in the nonactivated C-H bonds [139,140].
Apart from the mechanisms discussed above, Jamshidi and co-workers [58] found others such as: thermohydrolysis, depolymerization, and cyclic oligomerization. These mechanisms can lead to the appearance of a wide range of products, such as lactide and cyclic oligomeric units, CO, CO2, acetaldehyde, and methyl ketone, which might further degrade into methane and butanedione [103,137,139].
According to Kopinke [139], acetaldehyde formation above 270 °C increased with temperature, whereas McNeal and Leiper [103] observed that the maximum concentration of acetaldehyde was around 230 °C and decreased at higher temperatures. It was due to acetaldehyde degradation involving chain reactions to CH4 and CO by-products.

At elevated temperatures (230–440 °C) and in a closed system, the dominant reaction pathway of the isothermal degradation of PLA was found to be a non-radical, backbiting ester interchange reaction involving the OH chain ends. It should be pointed out that the type of products from the backbiting reaction depends on the point in the backbone of the PLA where the reaction occurred. The products include lactide, oligomers, acetaldehyde, and CO [103,139]. Moreover, when the temperature was raised above 270 °C, the homolysis of the polymer backbone was additionally observed to give other minor side products [103].
Zou studied thermal degradation of PLA in nitrogen and reported that the reaction initiated at ≈275 °C and complete decomposition of the sample was obtained at ≈420 °C with the evolution of the following products: cyclic oligomers, lactide, acetaldehyde, carbon monoxide, and carbon dioxide. The products were attributed to the hydroxyl end-initiated ester interchange process and chain homolysis [137].
Carrasco and co-workers [142] studied the thermal decomposition of processed and raw PLA specimens and determined the activation energy (Ea) to be 280 ± 5 kJ/mol for the former and 318 kJ/mol for the latter sample. They demonstrated that there was only one first-order reaction for the entire conversion range.
In comparison, a detailed study of the isothermal degradation of PLA in the temperature range of 240–270 °C was accomplished by McNeill and Leiper. They also showed a first-order kinetic evaluation of isothermal measurements of PLA volatilization. Moreover, a considerably low value of Ea = 119 kJ/mol was obtained in their study. It was attributed to the feasibility of transition state formation under the reaction conditions [103]. Other studies reported Ea of thermal degradation of PLA changing irregularly in a range of 70–270 kJ/mol as the degradation progresses [143,144]. It was also found that the value of Ea tends to increase at higher conversions of PLA, due to more complex kinetics [139].
The onset of thermal degradation of PLA is strongly affected by several factors, including residual polymerizing catalyst (residual metals), Mw of PLA, and moisture [145,146]. For example, the effect of these residual catalysts on the thermal decomposition of PLA was studied by Cam and Mauruci [147], and their experimental results enable the comparison of the influence of different metal impurities on PLLA thermal stability, which follows a decreasing order: Fe > Al > Zn > Sn. Among these metals, the Sn catalyst is of particular importance because only Sn 2-ethylhexanoate (Sn(Oct)2) is approved by the Food and Drug Administration (FDA) as a catalyst for food contact applications [144]. The authors found that the presence of Sn, Zn, Al, and Fe in trace amounts (i.e., 0–15,000 ppm) in purified PLLA accelerated the inter-and intra-molecular transesterification reactions as well as the backbiting reaction (as shown in Figure 6 at higher temperatures (i.e., T > 240 °C).
Figure 7 shows the changes in the decomposition temperature vs impurity concentration measured by Cam and Marucci [147]. The authors showed that the decomposition of PLLA was accelerated even with a very low level of impurities below 2000 ppm. It was also concluded that transition metals (e.g., Fe) have a high capacity to coordinate ester groups and, as a result, accelerate depolymerization and transesterification reactions [147].
Several techniques showed that the thermal decomposition of PLA samples containing a higher amount of residual Zn compounds in the matrix can occur via intermolecular transesterification and selective unzipping depolymerization [4,144,148].
Cam and Maurucci [147] concluded that Sn- and Zn-initiators could give rise to interchange and depolymerization at high temperatures, but more modestly than Al and Fe since they are known as the most selective catalysts for the polymerization of lactide (as it was mentioned before); thus they are less efficient in thermodegradation of PLA [147,149,150].
On the other hand, Jamshidi et al. [58] described an accelerating effect of Sn(Oct)2 (5 wt %) on the PLLA pyrolysis. They presented thermogravimetric analysis (TG) under isothermal conditions, which showed that the Sn-containing sample exhibited a linear weight loss curve with time, in contrast to the pure PLLA, which showed a sigmoid curve. These observations suggested a change in the weight loss behavior from a random process to a zero-order reaction induced by the addition of an Sn atom.
Regarding the influence of Mw of PLA on its thermal degradation, Cam and Marucci [147] reported that the lower the Mw of the PLA oligomer, the lower the decomposition temperature. It was attributed to the fact that the specimens with lower Mw have a higher concentration of hydroxylic groups, which trigger the degradation of PLA. Once a specific Mw is reached (around 140,000 g/mol), the amount of terminal hydroxyl groups becomes negligible compared to the ester repeating units within the polymer chain, and the influence of Mw on the thermal degradation temperature becomes smaller [147].
The influence of moisture or hydrolyzed monomers or oligomers on the thermal stability of PLA is far lesser than that of residual metals. For example, Cam and Maurucci [147] reported that a sample containing 1 wt % of monomers decomposed at a temperature slightly lower than that of the PLA (1–2 °C), whereas the PLA samples containing the same amount of metals (10,000 ppm of Sn, Zn, Al, Fe) decomposed respectively at 50, 60, 70 and 110 °C lower than that of the pure polymer [147].
There are many ways to control the thermal stability of PLA. For example, end protection of the hydroxyl group has been considered a possible method, and some reports have been published dealing with the application of an acetyl group [4,148]. Acetylation of PLA end groups by 2 h refluxing with acetic anhydride was found to increase the thermal stability of PLA by 26 °C [139].
Besides, some chain extenders can be used, such as, e.g., polycarbodiimide (PCDI) and tris (nonyl phenyl) phosphite (TNPP) or Joncryl®, to increase the initial thermal decomposition temperature of PLA. These extenders increase the PLA’s stability by reducing active sites on the chain end per mass by producing longer polymer chains [4].
Stabilizers can also be added to diminish the impact of shear and temperature during the processing of PLA. Furthermore, the molecular mass of these stabilizers is an important parameter affecting the thermostabilization of PLA [151].
Oliveira et al. reported that the B900 stabilizer (i.e., a mixture of Irganox 1076, a primary stabilizer, and Irgafos 168, a secondary antioxidant) acts directly on the deactivation of free radicals and hydroperoxide decomposition [134].
On the other hand, in heterogeneous systems, such as PLLA/alkali earth metal oxides, such as CaO or MgO, the oxides lower the degradation temperature range of PLLA and completely suppress the production of oligomers except for lactides [144]. Moreover, Kopinke et al. reported that blends of PLA with poly(methylmethacrylate) (PMMA) as a source of radicals accelerate the decomposition of PLA, whereas the PMMA is stabilized [139].
Concerning the thermo-oxidative degradation of PLA (i.e., in the presence of oxygen), the process induces a chain scission mechanism of alkoxyl radicals (alkyl- and acyl-oxygen), thus leading to the formation of new free radicals that can cause chain scission and formation of oxidation product degradations (mainly ester and carbonyl groups) [135].
According to Cameron and Kerr [152], during thermal oxidation in air, the initial change in Mw of a polymer is due to the scission of bonds at various weak links that might be present/or created during heating. Another work reported that the thermo-oxidative degradation of PLA at the average processing temperature (around 200 °C) follows a random chain scission mechanism causing a significant molecular degradation and leading to the formation of various degradation products (e.g., linear hydroxyl, ester, and carbonyl groups, etc.) [139]. It is all reflected in a dramatic change of the molecular structure, resulting in a decrease in the material’s melt viscosity and elasticity. In addition, the Mw changes of PLA during thermo-oxidative degradation cause a decrease in Tg according to the Fox–Flory theory and an increase in the degree of crystallinity due to a chemicrystallization process [153]. Importantly, the processing equipment can be damaged by the formation of the volatile lactide [134,135].
Concerning kinetics of thermal degradation of PLA in air, only a few assessments of Ea, kinetic parameters, and reaction order can be found in the literature [133,154,155]. TG studies of PLA degradation in the air suggested that the degradation kinetics follows the Avrami–Erofeev equation for solid-gas equilibria, which indicates that the decomposition of PLA is due to the growth and nucleation of decomposition sites in the solid [156].
Gupta and Deshmukh studied PLA degradation in the air using TG and reported a first-order reaction with Ea = 92–126 kJ/mol [133,155]. The authors also discussed the difference between degradation of PLA in air and nitrogen. The derivative thermogravimetric (DTA) curve in air exhibited two exotherm peaks and two endotherm peaks compared to two endotherm peaks in the nitrogen atmosphere. The peaks obtained in air were assigned to thermal decomposition (310 °C) and oxidative degradation (321 °C and 351 °C). On the other hand, the peaks obtained in nitrogen were identified as a phase change (140 °C), volatilization of low boiling point products (240 °C), and thermal cracking (310 °C) [133]. Liu et al. [154] compared the kinetics of PDLA thermal decomposition in air and nitrogen and concluded that the process in both atmospheres had two or three stages, each with different Ea. The authors applied the Arrhenius equation and calculated an Ea of 130 and 87.9 kJ/mol for the first and second stages of PDLLA in air, and 142 and 138 kJ/mol for the first and second stages of PDLLA in nitrogen, respectively. They concluded that the atmosphere had little effect on the degradation in stage one. However, in stage two, oxygen had a clear promoting effect on the thermodegradation, which resulted in a substantial drop in the value of Ea. Babanalbandi et al. [143] also compared the degradation of PLA under nitrogen and air over the temperature range 180–280 °C and obtained Ea values for isothermal weight loss rate between 71.1 and 105 kJ/mol depending on the conversion of PLA.
Finally, the degradation of PLA under vacuum was reported to take place at appreciable rates at T ≥ 230 °C, and the products observed included oligomers, lactide, acetaldehyde, and carbon monoxide. At T = 277 °C, CO2 appeared as an additional product, whereas at T ≥ 320 °C, methylketene became one of the major products. Small amounts of methane were additionally recorded at higher temperatures [103].
Concerning the thermo-mechanical degradation, Amorin et al. [151] studied the extrusion of PLA. They reported that the two types of degradation reactions could occur during the process, radical and nonradical. The authors also reported that the presence of the additives could prevent the decrease in Mw of PLA when exposed to melting temperature and shearing. However, nonradical reactions suppressed this stabilizing effect during the exposition of PLA to higher temperatures and shear.
Other authors reported that a high level of stress applied to PLA at elevated temperatures produces a decrease in the carbon-oxygen bonding energy in PLA and, consequently, favors the chain scission. Moreover, radical reactions typically observed during oxidative degradation of PLA are more likely to occur in the presence of a high level of stress apart from oxygen [135].
5.3. Photodegradation
In several applications, such as, e.g., PLA twines in greenhouses, the polymer is exposed to ultraviolet (UV) radiation, leading to discoloration or even brittle fracture caused by photodegradation [4,41]. The photodegradation of polymer materials is particularly accelerated outdoors due to the strong intensity of sunlight (around 245 nm [156]), which includes invisible lower wavelength and higher energy UV radiation [157].
The main mechanism of photodegradation of polymers is the so-called “Norrish type photocleavage”, typical for aliphatic polyesters having carbonyl groups, like PLA or PCL [158]. Primarily, photodegradation mechanisms are initiated by the transformation of the polymer via photoionization (Norrish Type I) and followed by polymer chain scission (Norrish Type II) [87]. It is known that the carbonyl group (i.e., C=O) absorbs UV radiation around 220–280 nm due to n-π* electron transition and that this energy can cause chemical reactions [4,157]. Even though the extinction coefficient for PLA at 280 nm is very low (less than 100 L/(mol cm)), photodegradation of PLA products certainly takes place during exposure. Since the excited state of the C=O group of PLA via n-π* presents antibonding and biradical characteristics, the photoexcitation of C=O can result in α and β cleavage, atom abstraction, radical addition or electron abstraction or transfer. These excited states of PLA are shown in Figure 8.
Jeon and Kim reported that UV irradiation enhances PLA degradation due to making PLA sheets more hydrophilic after exposure, which results in easier absorption of water by the polymer structures [156]. In turn, the chemical hydrolysis causes chain cleavage within the PLA polymer, forming oligomers and monomers, which can be later biodegraded by microorganisms [21].
UV irradiation also affects the tensile strength of PLA, reduces polymer integrity, and finally turns it into a white brittle solid that is difficult to be digested by microorganisms [156]. Some authors claimed that faster degradation of PLA takes place with more prolonged exposure to UV lights [95]. Other effects of UV irradiation on PLA include changes in Mw of the polymer, decreased stress, and strain at break [4,160]. McNeil and Leiper [103] studied the photodegradation of PLA under vacuum in a photolysis cell at 30 °C for 72 h. Based on the volatile reaction products analysis, they suggested that PLA decomposition by UV light happens at the O-C bond in the ester linkage. The proposed mechanism of the reaction is shown in Figure 9 [103,157]. The proposed mechanism is very similar to that of thermal degradation.
Ikada studied photodegradation of PLA and reported a rapid (1 h) decrease of Mw of PLA, attributed to a random main chain scission in the chemical bonds by absorption of a photon[158]. The main chain scission occurred via the Norrish II type photo-cleavage (shown in Figure 10). In addition, the studies comparing the photodegradation of PLA with that of PCL showed that the chemical structure of the two sequential groups adjacent to the ester oxygen has a decisive impact on the photodegradation mechanism [157,160]
Tsuji et al. [160] studied PCL and PLA’s photodegradation behavior and suggested bulk erosion as the main mechanism. Bulk erosion observed in all samples studied indicated that UV penetrated the specimens with no significant reduction in their intensity, irrespective of the chemical structure and the crystallinity of biodegradable polyesters. The authors also concluded that although PLLA chains are photodegradable, even in the crystalline regions, however, their photodegradability is lower than in the amorphous regions. As a result, the anhydride groups are formed, and the crystallization rate is decreased [4]. This is in contrast to hydrolytic degradation, where the chain cleavage occurs only in the amorphous regions [160].
Janokar et al. [161] studied the effect of UV radiation wavelength within a range of 232–500 nm by irradiating the PLA films directly in atmospheric conditions or through a Pyrex container (not optically transparent at wavelengths below 300 nm). They concluded that the adsorption of photons by C=O and other relevant groups occurred at wavelengths between 200 and 300 nm, resulting in the samples’ photodegradation. Furthermore, the PLA Mw was less affected when the irradiated films were enclosed in a Pyrex container. Based on their results, two different mechanisms of PLA photodegradation were proposed. As schematically shown in Figure 11, mechanism A involves a photolysis reaction leading to the breakage of the backbone C-O bond. On the other hand, mechanism B is based on photo-oxidation of PLA, leading to the formation of a hydroperoxide derivative and its subsequent degradation to compounds containing a carboxylic acid and di-ketone end groups.
A similar mechanism of photodegradation was proposed by Bocchini et al. [162]. They suggested that the photodegradation of PLA proceeds with the usual radical mechanism beginning with the abstraction of a tertiary hydrogen atom from a PLA chain and the formation of a radical. Then this radical reacts with oxygen to form peroxide and subsequently hydroperoxide. The authors proposed that the most probable β-scission leads to the formation of anhydride groups.
Concerning factors affecting the photodegradation of PLA, Copinet et al. simultaneously studied the effect of UV, temperature, and humidity. They concluded that with increased RH and temperature, there is a faster decrease of the degree of crystallinity, Tg, and Mw apart from increased hydrolysis rate at the ester linkage. It was suggested that the increased rate of hydrolytic degradation of PLA could be the result of an accelerated autocatalytic process upon UV treatment (as described in the hydrolytic degradation paragraph). Moreover, the UV irradiation, together with elevated temperature and higher RH, caused a significant reduction in the polyester’s mechanical properties and accelerated its degradation. In general, increased exposure time is expected to induce faster polyester degradation [4,95]. However, Jeon and Kim [156] proved that the biodegradability of PLA was enhanced as UV irradiation was increased to 8 h and then decreased with a further increase in UV irradiation.
There is not much information about the chirality effect on photodegradation of PLA. However, as photodegradation starts at the monomer’s functional group, it would be expected that D and L isomers should have the same mechanism of decomposition. The chirality effects should be related to crystalline effects because the isomer ratio influences the polymer crystallinity, as mentioned earlier in this review.
In addition to UV radiation, PLA can be exposed to X-ray or gamma-rays, which are more intense than UV. The degradation mechanism of PLA is different under these two types of radiation, which may lead to unselective ionization of molecules by interactions with cloud electrons. The produced electrons are kinetically active to trigger subsequent ionization. Intermediate products are formed due to side reactions as homolytic cleavage, ionic scission, electron transition, and energy transition [141].
The effect of gamma and e-beam radiation on PLA has been recently studied by Benyathiar et al. [163]. The authors reported a decline in the average Mw number of PLA after exposure to both types of radiation. Moreover, the authors reported a decrease in Tg and Tm upon ionizing irradiation. Mechanical properties such as tensile strength, elongation at break, and elastic modulus also deteriorated [4,160,161].
Another study analyzed the effect of gamma-irradiation on PLA crystallinity in two different atmospheres (air and vacuum) under constant radiation dose and rate. Tg and Tm were found to be independent of the atmosphere and dose parameters, but the crystallinity and the enthalpy decreased considerably in the air. On the other hand, the reaction yield of damaged units was higher in vacuum [141,164].
Birkinshaw et al. [165] studied the influence of gamma-irradiation in molded poly-(D, L-lactide) and observed changes in mechanical properties. The reduction in Mw made the sample brittle due to the random chain scission of the polymer. Increasing the doses of the radiation resulted in more substantial embrittlement.
The PLA integrity was also affected by electron beam irradiation. It was reported that pre-treating of PLA by electron beam irradiation increased PLA brittleness and decreased molecular weight during compost degradation compared to non-irradiated samples [166].
Interestingly, PLA properties such as low wettability and poor thermal resistance above Tg can be upgraded using irradiation. It was reported that wettability and biocompatibility could be improved by ionized beam irradiation [141,167], UV/ozone irradiation [141,168], or by oxygen radio frequency glow discharge [141,169]. Since intermolecular interactions among polymer chains make the polymer more thermally resistant, a standard method is to induce cross-links in the polymer matrix by photoirradiation [141,170]. Moreover, reconstructing the PLA surface with hydrophilic molecules is also possible by UV-induced graft polymerization [141,171].
5.4. Microbial Degradation
As mentioned before, composting involving microbial activity (e.g., actinomycetes, bacteria or fungus) is a desirable method for environmentally friendly plastic waste management if recycling of the polymer is not feasible. The digestion of PLA by microorganisms can only occur after successfully breaking the ester bonds in chemical hydrolysis to obtain low Mw oligomers, CO2, CH4, and carboxylic acid [4,21,87]. Thus, random non-enzymatic chain scission of the ester groups occurs, leading to a reduction in molecular weight. Embrittlement of the polymers generally happens in this step, reducing their molecular weight [55]. Importantly, PLA is more resistant to microbial attacks in the environment than other synthetic polymers [172]. When mineralization is not complete, biotransformation occurs, creating organic and inorganic metabolites or transformation products. Mechanical degradation can also occur through meso- and micro-faunal activities, such as earthworms, that fragment polymer waste and incorporate it in the mineral soil [98].
Enzymatic mechanisms can be divided into two categories: enzymatic oxidation (by aerobic microorganisms only) and enzymatic hydrolysis (by either aerobic or anaerobic microbes) [96]. Microorganisms present in the soil begin to digest the lower molecular weight lactic acid oligomers, producing CO2 and water, only when the average Mw of the PLA specimen reaches approximately 10,000 g/mol [52]. The mechanism of enzymatic degradation is shown in Figure 12.
Different microorganisms can digest the products of hydrolytic degradation of PLA, which can be classified as actinomycetes, bacteria, and fungus [174]. Environmental factors such as humidity, temperature, pH, salinity, the presence or absence of oxygen, and the supply of different nutrients have important effects on the microbial activity in PLA degradation [8]. Different authors considered the influence of some of the parameters mentioned above. The optimum temperature for digestion of PLA oligomers for the same microorganism in a specific strain can vary depending on the sample’s environment.
Following Bikiaris [89], the majority of polyesters are degraded by enzymes called lipases. These enzymes usually are too large to penetrate inside the polymeric material, and as such, they are only active after conformational changes have taken place. In addition, due to the low solubility of PLA in water and the large size of its molecule, the microorganisms are unable to transport the polymeric matter directly into the cells, where the biochemical reactions take place. As a result, the microbes first release the enzymes (called depolymerase) that work outside the cells until generating water-soluble compounds, which can be transported inside the cells and digested in the appropriate metabolic pathway [173]. Typically, inducers such as gelatin, elastin, silk fibroin, and some amino acids and peptides are needed to stimulate the process [41,87]. This mechanism the supply of different nutrients have important effects on the microbial activity in PLA degradation [8]. Different authors considered the influence of some of the parameters mentioned above. The optimum temperature for digestion of PLA oligomers for the same microorganism in a specific strain can vary depending on the sample’s environment.
Microbes that can degrade PLA belong mostly to actinomycetes, a family of antibiotic-producing filamentous bacteria commonly found in soil. Most of the PLA degrading actinobacteria are Pseudonocardiaceae, and other families include Micromonosporaceae, Streptomycetaceae, Streptosporangiaceae, and Thermomonosporaceae, etc. [175]. Apart from actinomycetes, the PLA-degrading bacteria include Bacillus brevis, Pseudomonas, Stenotrophomonas, Laceyella sacchari, Nonomuraea, Thermoactinomyces vulgalis, and Bordetella petrii [175]. Additionally, thermophiles from the thermophilic genus Bacillus were reported to play a leading role in fermentation [174]. Further, there were reports of PLA oligomers (molecular weight ~1000) degradation by Fusarium moniliforme and Penicillium roqueforti and the degradation of PLA by Amycolatopsis sp. [6].
Concerning the fungal degradation of PLLA, the process occurs at the chain-ends of PLA. The microorganisms consume the monomer in the chain’s extremity, which leads to depolymerization of PLLA [172]. The most commonly reported fungi that can assimilate DL-lactic acid belong to the following types: Fusarium Moniliforme, Penicillium roqueforti, Tritirachium album, Aspergillus fumigatus, and Thermomyces laniginosa, and partially soluble racemic oligomers [172,176]. These microorganisms can colonize even extreme environments, where their ability to produce enzymes (e.g., lactases, hydrolases, esterases, and dehydrogenases) allows them to degrade many chemical compounds with different functional groups, including polyesters [177]. Moreover, it was found that the temperature is the predominant parameter governing the fungal degradation of PLLA in soil and compost [172].
The selection of the most commonly studied microorganisms for biodegradation of PLA, together with the secreted enzymes and the optimum temperatures for PLA degradation, are gathered in Table 1. As shown in Table 1, the key enzymes that play an important role in the depolymerization of PLA are carboxylesterase, cutinases, lipases, and serine proteases. Proteinase K was also reported to degrade neat PLA films and PLA composites, and the rate of the degradation of the former being faster [178]. Besides, enzymatic degradation of low molecular weight PLA (molecular weight ~2000) has been shown using esterase-type enzymes such as Rhizopus delemer lipase [6].
Serine protease usually follows a two-step reaction of hydrolysis. In the first step, PLA substrate binds to the surface of serine protease at the active site. The following second step involves the cleavage of peptide-like bonds in PLA through catalytic amino acids with water [179]. Enzymatic involvement can produce pores and fragmentation, making more polymer regions accessible to the enzymes [55] The activity of the enzymes depolymerizing the PLA depends on the following factors: pH values, temperatures, chain stereochemistry, and material crystallinity [87].
As mentioned earlier, PLA is resistant to microbial attack compared to other biodegradable polymers [41,180]. Thus, pretreatments of PLA waste, such as gamma-irradiation (described before) or mechanical grinding, help disorganize the macromolecular aggregate structures of the material and increase enzymatic rate degradation.
Single culture, co-culture, and the microbial consortium have been studied to optimize the best biodegradable conditions. It was reported that PLA degradation efficiency could be enhanced using the simultaneous PLA degradation and dialysis method. The maximum conversion efficiency of 89% was achieved after incubation for 72 h under optimized conditions [175]. Moreover, modern molecular biological techniques provide a powerful tool to explore the diversity of PLA degrading microorganisms in the environment. These techniques involve analyzing nucleic acids extracted directly from the environments, thus avoiding the limitations of the culture-dependent approach [94].
6. Degradation of PLA in Different Environments
6.1. Composting Conditions
In horticulture, PLA has already substituted partially conventional PP as material for twines supporting greenhouse crops such as tomatoes and peppers, mainly due to the increasing need for sustainable cultivation and environmentally friendly and cheaper waste processing. The cost of removing and disposing of PP twines into landfills is high, and disposal facilities are not always available. Conventional plastics such as PP are not biodegradable and thus add a sorting step into the composting process. On the contrary, compostable polymers, such as PLA, can be decomposed by biological activity with no visible and toxic residues [21,181]. For example, The Elite Bio Twine from Lankhorst Euronete Portugal, produced using PLA, can degrade in industrial compost in 5 to 8 weeks into an unrecognizable, hummus-like substance. If there are remains of string left in the soil after the composting phase, they break down without leaving behind toxins in contrast to the PP twines that have to be separated from the green waste before composting.
The composting process is used as a versatile approach to treat biodegradable solid waste [119]. A mixed microbial population carries out composting in a moist, warm, aerobic environment under controlled conditions [86]. As compared to sorting and reprocessing for recycling, composting is a less energy-demanding process, although it requires more energy than landfilling [119].
Plastic material can be considered compostable if it is biodegradable and can undergo degradation by biological processes during composting at a rate similar to other known compostable materials. It should be underlined that not all biodegradable materials are compostable [96]. In general, PLA can be expected to decompose entirely into CO2 and water in an adequate and controlled composting environment in less than 90 days [8].
The PLA degradation under composting conditions involves two-steps. The first step is hydrolysis (abiotic process), followed by micro-organisms digestion to end-products (biotic process). In this sense, the first step’s mechanism is very similar to that previously described for hydrolytic degradation of PLA; however, composting also involves digestion of soluble oligomers by bacteria and evolution of CO2 along with water as final products. The last step might be under aerobic or anaerobic conditions. In particular, abiotic hydrolysis was suggested as the primary depolymerization mechanism and the rate-controlling step of PLA biodegradation in compost [94,95]. The described two-step mechanism is a unique characteristic of biodegradable polymers [21].
When it comes to composting conditions, they are organically rich with a large diversity of microbial populations, as listed in Table 1. During composting, it is necessary to consider that both; biotic and abiotic factors coexist in the same space. The complex process of degradation in compost involves the following intermediate phenomena: (i) water adsorption, (ii) ester bond cleavage and formation of oligomer fragments, (iii) solubilization of oligomer fragments, and finally (iv) diffusion of soluble oligomers by bacteria in terms of CO2 evolution [173,182]
The composting starts with the abiotic process, chemical hydrolysis, and occurs in the presence of water at temperatures higher than 50 °C. The biotic process follows hydrolysis and happens in the presence of microorganisms (listed in Table 1) responsible for decomposing the polymer to the final products. As mentioned before, these products include CO2, water, and biomass under aerobic process conditions, while methane, hydrocarbons, and biomass are generated under anaerobic conditions [21].
The elevated temperature, high moisture content, and abundance of microorganisms in compost are the most suitable environment for the biodegradation of PLA. However, the disintegration of PLA under composting conditions made by microorganisms starts when the polymer’s molecular weight reaches about 10.000–20.000 g/mol [96]. Importantly, this natural process of degradation requires the availability of carbon, nitrogen, water, and oxygen. Nitrogen is necessary for microorganisms to build their cell structures, and they use carbon as a source of energy. The ideal composting conditions are with a carbon-to-nitrogen ratio (C:N) of 30:1. These conditions are ideal for the reproduction of thermophilic microorganisms [96].
Moreover, composting biodegradable plastics with other organic compostable materials, namely yard, food, and agricultural waste, can create much-needed carbon-rich compost for soil enrichment [182]. In turn, the temperature and the type of compost used affect chain mobility, which plays an essential role in PLA biodegradation [183].
In general, a maximum of 10% PLA should be used in compost piles [55]. PLA levels higher than 30% (w/w) in pre-composted yard waste were reported to decrease pH due to disintegration of the polymer to lactic acid, which suppresses the microbial growth and slows down the overall decomposition [182].
The aerobic reaction observed in the composting process with the aid of microbial metabolism can be described as follows [184].
FRESH ORGANIC WASTE + O2 = STABILIZED ORGANIC MATERIAL + CO2 + H2O + HEAT
The composting process starts with a mesophilic phase (temperatures between 10 and 45 °C), where microorganisms break-down PLA into simple molecules. A thermophilic phase follows this step, which takes place at temperatures optimum for microbial activity (between 45 and 70 °C) [185]. Subsequently, the cooling phase starts, which continues until the compost achieves the ambient temperature and hummus-like end-products are observed. Degradation occurs at a slower rate and continues until all the utilizable carbon is converted to carbon dioxide, and the humification coincides with the degradation of the organic matter [119,184]. The general scheme of PLA bottle degradation under composting conditions is represented in Figure 13.
Gattin [186] classified the PLA degradation in composting media to be more efficient than in inert solid media but slower than in liquid media. Other researchers reported that complete PLA degradation takes about one year in the soil, whereas it takes 60 to 100 days in a compost environment [176].
A different study carried out by Saadi and collaborators [172] compared the purely fungal degradation of PLA to the natural biodegradation (bacteria and fungi together). The obtained results indicated that the kinetics of PLLA mineralization were faster under natural biodegradation, attributed to a synergy between bacteria and fungi and the diversity of enzymes released in the medium. The authors reported a decrease of Tg from 60.8 to 43.8 °C and Tm from 165 to 142 °C during the 70 days of the degradation test. The decrease in Tm can indicate that degradation occurred not only in the amorphous phase of PLLA samples but also in the crystalline regions.
Comparisons between laboratory set compost and field exposure (soil in the banana-growing farm) degradation of PLA films were made by Ho and collaborators [187]. The results indicated that PLA plastic films lost their mechanical properties faster when exposed to natural conditions than during simulated compost exposure in the laboratory. They also confirmed that the degradation of PLA was enhanced at higher temperatures and relative humidity.
Husárová et al. reported the influence of Mw on the biodegradation of PLLA samples under composting conditions [94]. They found that with the increasing Mw of the polymer, the biodegradation rate decreased, and initial retardation was discernible in parallel. Further, the addition of a limited amount of low Mw PLA did not accelerate the biodegradation of high Mw PLA, suggesting that the process is not limited to the number of specific degraders and the induction of specific enzymes.
Kale et al. [119] compared the degradation of three types of commercial packaging made of poly (LD-lactide) (PLA) under compost and ambient conditions. The samples were subjected to composting for 30 days, and the degradation of the physical properties was monitored. PLA packages made of 96% L-lactide exhibited lower degradation than PLA packages made of 94% L-lactide, mainly due to the former’s higher crystallinity. Further, the authors reported that the degradation rate changed with the initial crystallinity. Other factors that significantly influenced the PLA degradation in compost were: L-lactide content of the samples, temperature, relative humidity, and pH of the compost pile. They also reported that PLA trays and deli containers’ degradation time in a commercial facility was not more than 30 days.
As a result, any factors described before, which allow controlling the PLA matrix’s hydrolytic degradation, will ultimately allow tuning the decomposition of PLA in compost. Thus, similarly to the hydrolytic degradation, the rate of degradation of PLA in compost can be affected by different properties of the polymer material, including first and higher-order structures (e.g., Mw, optical purity, crystallinity, Tg, and Tm) as well as the environment conditions (humidity, temperature, pH and the enzymes or microorganisms present) [21,181]. A study by Karamanlioglu and Robson [188] suggested that sterilization of soil or compost decreased the PLA decomposition rate by tested fungal strains, which has proven that soil micro-biota directly or indirectly stimulates the biodegradation process.
6.2. Soils
PLA degradation in soils is complex and much slower than in compost due to the latter’s higher moisture content and temperature range, which increases the PLA hydrolysis and the assimilation of PLA by thermophilic microorganisms [181,182]. The appropriate PLA-degrading microorganisms and the optimal environmental conditions, e.g., pH, humidity, temperature, oxygen, are compulsory for the successful biodegradation of PLA in the environment [41]. The soil’s geographic location could also impact the rate of PLA degradation [21].
For example, a 20 month PLA soil burial trial caused 20% and 75% degradation of PLA100 (crystalline PLA) and PLA75 (amorphous PLA), respectively [21]. Calmon et al. [189] compared the degradation in soil burial of PLA, PCL, cellophane, polyethylene (PE), and polyhydrobutyrate by burying the samples in four different areas of France for two years. Surprisingly at the end of the experiments, the authors found a PLA sample with significant degradation signs, whereas the PCL specimen was entirely degraded. As was expected, no degradation was observed in PE. Similar studies were done by Karamanlioglu and collaborators [188,190], who also demonstrated minimal PLA degradation at ambient temperatures. In their study, PLA coupons were buried in soil or compost at 25 °C and showed minimal changes after one year. A further study done by these authors, let to the isolation of several fungi from the PLA’s surface; however, the sample showed no signs of degradation.
Saadi et al. [172] studied the degradation of PLLA in sterilized and inoculated soil at 30 °C and reported a low rate of fungal degradation of only 8% after 220 days incubation. In another study, the soil burial for a year (T = 0–22 °C) was reported not to affect the physical properties of PLA test bars [191]. Therefore, while compostable, PLA cannot be regarded as hydrolyzable or entirely biodegradable under normal environmental conditions. Similar results were obtained by Kolstad and collaborators [192] who studied the behavior of Ingeo PLA in landfills and performed two tests: (i) under accelerated landfill conditions, at 21 °C, up to 390 days; and (ii) anaerobic digestion test under optimal and significantly accelerated conditions at 35 °C for 170 days. The tests had accelerated the biological degradation sufficiently to be equivalent to approximately a century of a “typical” landfill. The tests were used to evaluate the methane generation potential of PLA under these conditions. None of the PLA samples exhibited a statistically significant generation of biogas in the first test, and no signs of PLA degradation were observed. In the second test, at 35 °C, the amorphous PLA sample degraded, with a biogas contribution suggesting approximately 36% degradation. The authors concluded that semicrystalline PLA (typical of >96 wt % of resin used to manufacture products), under anaerobic conditions of a landfill under ambient conditions, does not release a significant amount of methane, and there is no available population of organisms to degrade high molecular weight PLA directly under anaerobic conditions. The obtained results confirmed that semicrystalline PLA samples at ambient temperature would require many decades to hydrolyze to the point where microbes begin to consume the oligomers.
Rudnik and Briassoulis [193] carried out experimental studies on the biodegradation of fibres and films of PLA and poly(hydroxyalkanoates) (PHA) under natural conditions (in soil) and in the laboratory -simulated soil burial for seven months. The results showed that under both testing conditions, PLA needed much more time and higher temperatures (at least 58 °C) to show any signs of degradation compared to PHA. In fact, the PLA degradation at temperatures below the Tg occurs slowly because of polymer molecules’ inactivity to change from a glass-like state to a rubber-like state [192,194]. Furthermore, Rudnik and Briassoulis reported that PLA fibers degraded more slowly than PLA films, which was related to the higher crystallinity of the former. After seven months of burial in the simulated laboratory soil, the PLA films became brittle and creased but were not disintegrated in contrast to PHA films, which took only one month to biodegrade.
When PLA degrades in soil, its physical, mechanical, and chemical properties change. Commonly, transparent PLA samples become opaque after soil burial degradation [193,194]. Santonja-Blasco and co-workers [195] studied commercial PLA degradation in soil burial for 15 days, monitoring the samples with thermal techniques. The results showed that the cold crystallization temperature decreased with degradation time, while the related enthalpy of crystallization increased. The degradation of PLA in soil affected the PLA chain reorganization, leading to an increase in the polylactide chains’ free volume in the amorphous phase that favorably affected the bulk properties.
Weng and collaborators [196] studied the degradation of PLA samples buried 40 cm deep under the soil surface for four months and reported a decrease in organic carbon contents in the degraded samples while the oxygen atom contents increased. The authors also reported a slight decrease in Tm of the degraded samples.
The degradation of elongation at break, tensile strength at break, and Young modulus of PLA after 50 days of soil burial degradation was reported by Vasile et al. [194]. These authors studied the influence of chitosan (CS) on soil burial degradation of plasticized PLA bio-composites sheets obtained by melt processing. Samples were buried in natural active soils (with known water retention) and sterile soils under aseptic conditions. The rate of degradation of the bio-composites was higher compared to the neat PLA sample. The chitosan was found to favor the degradation process, which was demonstrated by increasing weight loss with increasing CS content. Moreover, the mass losses were higher in active soil than those found in sterile soil. The authors concluded that the yeasts, molds, and total aerobic mesophiles present in the active soil promoted the bio-composite biodegradation process.
Another group studied methods for accelerating the degradation of commercial beverage cups produced using PLA granules 2003D (Ingeo™, Natureworks LLC, Northford, CT, USA) in soil burial. The PLA cups were cut into sheets and buried in soil containing various sources of bacteria and fungi, including cow-manure, green yard-waste, wastewater sludges from dairy, etc. UV light was used to reduce the Mw of the samples. Interestingly, the PLA cups degraded entirely after 15 days of burial in the soil with dairy wastewater sludge, having a high total nitrogen content under thermophilic conditions. In comparison, the neat PLA cups (not UV radiated) were found to be more challenging to degrade [180].
6.3. Aquatic Environment
As described before, degradation of PLA in aquatic environment occurs through hydrolysis, which increases with the presence of high moisture in the environment. The aquatic environment conditions are very stringent for the microorganisms because the polymer becomes the sole carbon and energy source, and there are no co-substrates and vitamins, apart from inorganic nitrogen source and the elements essential for microbial growth. Thus, only some microbes capable of growing under harsh conditions can survive.
Similar to the degradation of PLA in soil, PLA degradation in the aquatic environment takes a long time at ambient conditions (T = 25 and 37 °C), but it accelerates under thermophilic conditions (T = 55 °C). The apparent effect of elevated temperatures suggests that the polymer’s chain needs to be hydrolyzed before the microorganisms can act, which is similar to the PLA degradation in soil or compost. The fastest mineralization of PLA in the aerobic aquatic headspace test was reported at 60 °C, reaching 90% within 120 days. On the contrary, the anaerobic biodegradation of PLA under the same conditions was faster than aerobic one, and 60% mineralization of PLA was reached at 52 °C in just 40 days [197].
Musiol and collaborators [198] incubated PLA strips in the water at 70 °C for 70 days. The authors observed that the initially transparent samples became opaque after three days of incubation due to an increase in crystallinity degree during the degradation process. The disintegration of the samples was observed after 7 days of incubation. The authors observed bimodal mass distribution for the PLA samples incubated for 7 and 14 days, whereas the sample incubated for 21 days had a single peak distribution on the Gel Permeation Chromatography (GPC) chromatograms. This observation was due to the autocatalytic effect of hydrolysis degradation, whereas at a later stage of degradation, lactic acid and its dimer diffuse out of the samples’ interior, and the effect of autocatalytic degradation disappears after 21 days.
Deroine and collaborators [199] studied the degradation of PLA samples in seawater and distilled water for six months. Under both experimental conditions, PLA ageing was favored by increased temperatures. The Ea was lower in distilled water than in seawater. The degradation of PLA in distilled water at 50 °C was strongly accelerated, and the sample became considerably brittle only after one month of immersion. Furthermore, the immersion of samples at 40 and 50 °C in distilled water for six months resulted in an increase in melting enthalpy and degree of crystallinity. A significant reduction of the stress at break of PLA samples was observed in both environments after six months at 40 °C; however, it was more pronounced in distilled water (85% versus 70% in seawater). The lesser deterioration of PLA properties in seawater than in distilled water was attributed to the difference in water absorption. The accelerated aging tests in seawater and distilled water showed that the nature of the aqueous medium significantly affects the polymer properties. It was concluded that the degradation of PLA is faster in distilled water, probably due to the role of mineral salts, which facilitate the diffusion of seawater within the polymer and a lower pH of distilled water (≈6 against ≈8 for seawater), which can promote hydrolytic degradation.
Accelerated ageing of PLLA in seawater for three months revealed a decrease of almost 50% in molecular weight at 40 °C. Further, the elastic modulus was not affected by ageing, but the tensile strength significantly decreased. Furthermore, the failure strain increased with ageing at 20 °C but decreased at 40 °C [200].
Tsuji and Suzuyoshi [201] compared the degradation of PLA in static seawater in the laboratory and dynamic natural seawater [202]. In the natural seawater, mechanical stresses and strains (from waves and tides) are always loaded on the specimens together with UV radiation. These effects significantly accelerate the biodegradation of PLA. After one week of immersion in the dynamic seawater and 10 weeks in static seawater, there was no significant degradation by marine microbes. Moreover, an induction period of three weeks was observed in dynamic seawater, with a 65% weight loss obtained in 5 weeks.
In contrast, PLLA specimens appeared to be biodegradation-resistant in the static seawater as no significant weight loss or weight distribution changes were recorded up to ten weeks of immersion [201]. Moreover, the tensile strength of PLLA films in the natural dynamic seawater was lower than that in the controlled static seawater when compared at the same degradation time. The authors reported no UV irradiation influence on the chain cleavage in PLLA films during (bio)degradation in the natural dynamic seawater [202].
7. Control and Improvement of PLA Biodegradation
It is clear that PLA, although hydrolyzable, is very resistant to degradation under environmental conditions. In this sense, nanocomposites or blends represent an excellent solution to modify the initial characteristics of PLA. For example, the use of some of the nanoparticles, as “nanoreinforcements” of PLA, not only affects its mechanical properties but also dramatically influences its biodegradation behavior. Several different materials have been added to PLA, which can accelerate its decomposition, for example, clays, zinc oxide, TiO2, metal oxides, or natural polymers, such as chitosan. The development of PLA- based nanocomposites often involves a trade-off between the composites’ performance and their degradability at the end of their service life. This section will give a short overview of the most important works concerning the biodegradability of PLA.
Controlling the hydrolytic degradability of PLA is of great importance because, as mentioned before, hydrolysis is an obligatory step in PLA biodegradation in natural environments. Numerous studies have been undertaken to elucidate the effects of various features on the process. Hydrolysis of PLA composites containing nanoparticles is a complex phenomenon depending on the degradation temperature and pH, nature and dispersion of filler, type of bonding between components, dispersion of the nanofiller, and polymer crystallinity [203]. In comparison, the biodegradation rate of PLA composite in the compost or soil is more complicated. The composting process additionally depends on the environmental conditions (e.g., type of microbial population and nutrient supply) to which the material is subjected [204]. It is worth mentioning that since the biological contact in the compost or soil occurs at the material–environment interface, the exposed surface properties play the most crucial role. For example, a rough surface with a high number of polar hydrophilic functional groups is much more prone to biodegradation than a smooth, hydrophobic one.
Ho et al. [205] reported a faster degradation rate of PLA under composting conditions after incorporating bamboo charcoal particles. The effect was attributed to better adsorption of water by the composite materials than neat PLA, which resulted in a higher rate of the hydrolysis reaction. The authors pointed out that the composites’ higher degradation rates can be due to their lower crystallinity than a neat PLA.
Luzi and collaborators [178] showed that films based on PLA reinforced with cellulose nanocrystals were disintegrated in less than 14 days in compost conditions. The accelerating effect of the cellulose nanocrystals was assigned to the hydrophilicity of the surfactant used during the synthesis of the biopolymer. Additionally, several micro-holes in the composites’ structure improved the kinetics of the biocomposites’ degradation and aided water absorption. The developed material was proposed for application in food packaging with low environmental impact.
The addition of cellulose nanocrystals modified with a commercial surfactant improved the disintegration of PLA in composting conditions. The results were attributed to the hydrophilicity of the surfactant [178]. Enzymatic degradation of PLA was also accelerated in PLA reinforced with cellulose nanocrystals [206].
Biodegradation of the polyblends of PLA, PCL, and microcrystalline cellulose (MCC) was studied and compared with neat PLA and neat PCL. The blends with a higher concentration of PCL and MCC in the PLA matrix showed higher carbon mineralization percentages than the blends having low PCL and MCC components. The blend composition and filler concentration played a crucial role in the biodegradation behaviour of these composite materials [183].
Balaguer et al. [207] evaluated the influence of the addition of layered silicate, calcium carbonate nanoparticles, and nano silicon oxide to PLA films on the nanocomposites’ compostability. Results showed that films completely disintegrated into visually indistinguishable residues after 6–7 weeks of incubation in a composting environment. Further, biodegradation was higher in all the biocomposites than that of plain PLA after 130 days in composting, and the addition of calcium carbonate and clay obtained the most significant improvements. Furthermore, all the samples showed the typical PLA biodegradation pattern, which consists of an initial lag phase corresponding to hydrolysis of the ester bonds by extracellular hydrolytic enzymes and the subsequent biodegradation of the generated monomers and oligomers. The corresponding lag phase ranged from 9 days for the film PLA with clay to 17 days for the blank film and the film PLA with SiO2. The shortest lag phase was attributed to the high hydrophilicity of the nanoclay, which enhances the amount of water absorbed in the polymer matrix and thus prompts the hydrolytic degradation processes. Another study reported similar results on the addition of montmorillonite, or hectorite, and smectite-type layered silicates into the PLA matrix. The authors reported that these nanofillers increased the PLA matrix’s hydrophilicity and thus increased the polymer’s hydrolysis rate. Further, the clay particles’ morphology was found to influence water diffusion in the material, and a lower diffusion coefficient was found with the nanofillers’ pellets-like morphology [208]. Moreover, organically modified layered silicates such as mica or montmorillonite increased the enzymatic degradation of clay reinforced PLA/PBSA blends [88].
Fukushima et al. [209] studied PLA nanocomposites with the addition of organically modified montmorillonites. The degradation of nanocomposites was faster than that of neat PLA. The accelerated degradation was attributed to hydroxyl groups in the clays, which catalyzed the PLA matrix’s hydrolysis. Other studies underlined the importance of hydroxyl groups of the surfactants in clay-PLA composites, which improve the distribution of the clay in the polymer and promote adsorption and diffusion of water in the composites [182,209]. Another study dealing with PLA–clay nanocomposites reported that montmorillonite or fluorohectorite and sepiolite resulted in increased crystallinity of the nanocomposite compared to neat PLA. The hydrolysis of the samples in the phosphorous buffer solution at 58 °C showed no significant effect of clays on the degradation trend of PLA. The nanocomposites degraded similarly to the neat PLA, which was related to the easy access of water molecules to the bulk material, minimizing the effect of polymer crystallinity, clay nature, and aspect ratio on the polymer degradation [203].
A rational choice of the nanofiller’s chemical nature allows tuning both the degradation rate of PLA and its structure. Neppalli et al. [210] prepared nanocomposites of PLA with perkalite (anionic clay) and montmorillonite (cationic clay) and studied the relationship between the presence of clay, the crystallization behavior of the matrix, and the degradation rate of the nanocomposite. Perkalite induced higher crystallinity, a faster crystallization rate, and also a modification of the crystallization mechanism. Moreover, when perkalite was used, the lamellar framework of PLA was preserved. On the contrary, the cationic clay inflicted a disruption of the order of the lamellar stacks in PLA, causing less homogenous dispersion of the filler in the PLA matrix. After 125 days of immersion in a phosphate buffer solution at pH 7.4, PLA showed a negligible mass loss. On the other hand, PLA with cationic clays lost about 10% initial mass, whereas the sample containing perkalite showed degradation of 40%. Accelerated degradation of the PLA-perkalite composite was due to a high volume of the polymer matrix in contact with the nanoclay edges due to the excellent dispersion of perkalite in the PLA matrix. As a result, the water attack was more straightforward, and the rate of hydrolysis was accelerated compared to PLA- montmorillonite or neat PLA.
The degradation mechanism and kinetics of PLA nanocomposite films containing various commercially available native or organo-modified montmorillonites were studied under composting conditions in the process’s thermophilic phase and during abiotic hydrolysis. Biodegradation of PLA was enhanced in the presence of nanoclays, in accordance with other reports. In particular, the lag phase at the beginning of the process was shortened in the clays’ presence compared to neat PLA. The lag phase typical for biodegrading high molecular weight PLA [94] was discernible for all samples at the beginning of the process, but it was more pronounced for neat PLA. The authors also determined that the critical molecular weight for the hydrolysis of PLA is higher than the critical molecular weight for the onset of its mineralization, which confirmed that PLA chains must be shortened beyond the solubility limit before being assimilated by microorganisms [211].
Similarly to clays, the presence of graphene oxide (GO) in the PLA matrix also improves the material’s hydrophilicity. Duan et al. [212] carried out the hydrolytic degradation of PLLA-GO nanocomposites in three different environments, including alkaline, acidic and neutral solutions. The results demonstrated that the hydrolytic degradation of PLLA was greatly accelerated by adding GO in all media. Moreover, the acceleration effect was proportional to the amount of GO added. Furthermore, the hydrolytic degradation mechanism of the PLLA matrix was not affected by the presence of GO in the matrix.
Lizundia et al., studied the influence of ZnO on the thermal and hydrolytic degradation of PLLA. PLLA nanocomposites with five wt % ZnO nanoparticles were prepared involving solvent-precipitation method followed by compression molding. The thermal degradation tests were carried out in TG by heating the material to 500 °C. The results showed that ZnO nanoparticles catalyzed nanocomposites’ thermodegradation process, and the onset temperature was lowered by 100 °C. The catalytic activity of ZnO was also apparent in hydrolytic degradation tests, where nanocomposites with higher concentrations of ZnO underwent physical disintegration to a more considerable extent than neat PLLA. The analysis using the wide-angle X-ray diffraction technique revealed that the amounts of oxygen vacancies initially present in the ZnO lattice in nanocomposites were reduced upon degradation. The authors postulated that thermal and hydrolytic degradation of PLLA-ZnO nanocomposites were both initiated on ZnO surfaces. The accelerated thermal degradation of PLLA in the presence of ZnO nanoparticles was also reported by other authors [213,214,215]. The addition of ZnO or TiO2 has led to a significantly decreased activation energy for PLA pyrolysis and a substantially higher decomposition rate constant. The catalytic effect of ZnO was found to be more considerable than TiO2 [215].
On the other hand, TiO2 nanoparticles have been investigated in recent years because of their photocatalytic ability to decompose various organic chemicals, including PLA [216]. UV irradiation results in an electron (e−) in the TiO2 nanoparticles being removed into the conduction band, leaving a positive hole (h+) in the valence band, which produces active oxygen species. These species subsequently aid in the degradation of the polymer matrix. By adding photodegradability to PLA, its degradability can be efficiently promoted under any conditions, as was already mentioned. Studies indicated that the degradation efficiency of a PLA was improved by incorporating TiO2 nanoparticles [217].
Luo and coworkers [218] studied the biodegradability of PLA/TiO2 composites with different contents of TiO2 from 0.5 to 15.0 wt %. The biodegradation tests carried out under controlled composting conditions for three months indicated a considerable degradation of PLA/TiO2 composite compared to that of PLA. The degradation was evidenced by deep cracks and large voids on PLA/TiO2 composites’ surface due to the hydrolysis of PLA and microorganisms’ activity. Additionally, larger amounts of TiO2 in the PLA matrix accelerated the initial phase of degradation and enhanced the amount of CO2 generated at the end of incubation periods.
Another group reported that the presence of TiO2 increased the rate of hydrolytic degradation of PLA in a phosphate buffer solution of pH 7.4 at 37 °C [219]. The change in surface morphology of PLA-TiO2 nanocomposites after hydrolysis revealed that the degradation process was initiated at the PLA matrix and nanofillers interface. The water absorption and degradation rate of PLA-TiO2 were affected by nanofillers’ dispersion in the polymer matrix.
The catalytic effect of alkali earth metal oxides such as CaO and MgO on the thermal degradation of PLLA was previously reported by Fan et al. [220]. Metal oxides, especially MgO, suppressed oligomers’ production other than lactides and limited the racemization of lactide during thermal degradation of PLLA. At temperatures lower than 270 °C, the pyrolysis of PLLA/MgO (5 wt %) composite occurred smoothly via unzipping depolymerization mechanism and resulted in selective production of L-lactide.
PLA is often blended with starch to increase biodegradability and reduce costs. Lv and collaborators [221] studied the biodegradation behavior of PLA blended with starch and wood flour in soil. They reported that starch provided a biological fuel for the growth of microorganisms in the soil, which accelerated the composite’s degradation rate. Moreover, in the simulated soil, the growth of microorganisms was increased with the occurrence of glucose provided by thermoplastic starch (TPS), which was found to significantly accelerate the degradation of PLA and PLA/TPS blends [222].
Petinakis and collaborators [223] reported that both starch and wood-flour accelerated the thermal decomposition of PLA, and the former exhibited a relatively more substantial effect than the latter. The decomposition temperature of PLA was decreased by about 40 °C when the filler content reached 40%. Compared to wood-flour, the lower decomposition temperature of starch resulted in a lower decomposition temperature of PLA in the blends with starch. Concerning the impact of starch on biodegradation under composting conditions, it was observed that nanocomposites containing less than 40% of starch took a longer time to degrade due to the aforementioned lag phase. On the contrary, the degradation of blend PLA 60%–starch 40% initiated disintegration immediately under controlled compost conditions and achieved 80% biodegradation after 80 days in the compost. Furthermore, the improved compatibility between PLA and esterified starch enhanced the rate and degree of degradation of starch-PLA composites [224].
Plastic composites with natural fiber reinforcements result in lightweight structures having high stiffness and tailored properties for specific applications, thereby saving weight and reducing energy needs [225]. Aerobic biodegradation of PLA-TPS with 30% natural fiber was studied under controlled composting conditions. The degradation rate was influenced chiefly by starch, whereas the fibers showed only a marginal effect. Biofilm was observed on biodegraded materials due to bacterial and fungal fixation and growth [226].
Enhancement in the PLA’s degradation in a laboratory-scale simulated composting facility was found with the addition of natural biomass such as untreated soy and wheat straw [225]. Both nanocomposites showed similar rates of degradation irrespective of the type of biomass used. Compared to that of the neat PLA, the improved degradation rate was due to the presence of readily degradable natural substrate and reduced average molecular weight of the PLA.
Shinoda et al. [227] melt-blended PLA with a small amount of poly(aspartic acid-co-lactide) (PAL) or poly(sodium aspartate-co-lactide) (PALNa) and obtained homogeneous press films. They studied the degradation of these blends in compost and the soil. The results suggested that PAL and PALNa are useful additives for accelerating the hydrolysis of PLA. Interestingly, PAL did not accelerate the hydrolysis rate of PLA unless it came into contact with water. It was explained by the amphiphilic nature of the PAL, which would be hydrophilic in water and hydrophobic in air. Conversely, the PLA/PALNa blend degraded relatively quickly under the same ambient conditions. PALNa is water-soluble and relatively hydrophilic; its hygroscopic nature accelerated the rate of PLA hydrolysis. The strength of the blend films decreased faster than that of the pure PLA film under controlled composting conditions at 58 °C, with a water content of 60%.
The hydrolytic degradation of PLA can be controlled by improving the hydrophilicity of the polymer surface. The hydrolytic degradation of PLA grafted with acrylic acid (AA) at 37 °C was significantly accelerated compared with the neat PLA. Low molar mass compounds migrated from surface-grafted PLA after seven days of tests compared to 133 days for nongrafted PLA. Besides, the degradation product pattern of surface-grafted PLA showed significant variation as a function of hydrolysis time with the evolution of short and long AA-grafted lactic acid (LA) oligomers apart from plain lactic acid oligomers. The authors underlined the necessity to consider the degradation product patterns parallel with developing new PLA materials [228].
The biodegradation of PLA can be accelerated by treating the material with specific fungal strains (Aspergillus Niger, Chaetomium globosum, Paecilomyces variotti, Penicillium pinophilium and Trichoderma viride) to improve the kinetics of the process [172].
Finally, Pattanasuttichonlakul et al. [180] proposed the UV irradiation of PLA followed by burying in a soil mixture of bacteria from dairy wastewater sludge as an efficient method for accelerating the degradation of PLA waste.
8. Conclusions
Society’s primary challenges nowadays include environmental problems, increased pollution, plastic waste management issues, and consumer demands for environmentally friendly options. The use of biodegradable plastics is a way to solve the disposal problem of plastics in landfills and a valid alternative to replacing conventional polymers. Poly(l-lactide) (PLLA), which can be prepared from renewable resources, has been believed to be an ideal alternative to petroleum-based polymers due to its excellent comprehensive performance and biodegradable ability. Its performance characteristics, such as stiffness, strength, and gas permeability, are comparable with conventional petroleum-based plastics. PLA exhibits high strength, stiffness, biocompatibility, thermo-plasticity, and good processability. On the other side, the main drawbacks of PLA include its low toughness, high cost, and unacceptably slow degradation rate.
In general, PLA is more resistant to degradation under environmental conditions than other aliphatic biodegradable polymers. Thus, although compostable, PLA is not a readily degradable biopolymer.
Hydrolytic degradation is a critical first step in the biodegradation of PLLA. It can be affected by many features of the material itself (crystallinity, molecular weight etc.) and the media’s conditions (temperature, time, pH, etc.). Moreover, humidity, time, type of microorganisms, presence of oxygen and the supply of essential nutrients, population, and action potential of enzyme activity, etc. are some additional factors that should be taken into account if the PLA is meant to be degraded in compost at the end of its service life. A vast body of literature on PLA degradation in soil and compost suggested that the degradation behavior was a complex phenomenon, and the degradation rate of PLA is strongly affected by temperature. The primary condition to obtain substantial biodegradation of PLA is a temperature higher than the glass transition temperature of PLA.
When attempting to control the degradation of PLA, fine-tuning is needed to balance the necessity of maintaining suitable material properties during the entire service life in environmental conditions and the need for rapid degradation when the material becomes a waste. The degradation of PLA can be controlled by (i) addition of catalysts or organically modified nanoparticles; (ii) modifications by blending PLA with other easily degradable natural polymers such as chitosan, cellulose, or starch; (iii) combining with organic modifiers, or chain extenders, among others. The most common nanofillers favoring biodegradation of PLA are ZnO, clays, alkali earth metal oxides.
For the near future, new strategies are needed for the selective depolymerization of bioplastics such as PLA either to their constituent monomers or other useful intermediates that could be used as resources for other chemical processes. New strategies should be developed to recapture and reutilize plastic materials at the end of their useful life since their persistence in the environment causes pollution of the lands and oceans. New ways should be sought to decrease PLA production cost, improve its toughness, and boost its biodegradation behavior to make it more commercially attractive.
Although recycling could be more energetically feasible than composting for PLA products, composting seems to be more practical, as it does not require sorting and cleaning. Furthermore, compostable food packaging materials or agricultural products could trigger plastic waste diversion from landfills into composting facilities. Work should be done to make compostable materials compatible with the circular economy as immediate value is lost from the life-cycle.
Author Contributions
S.T., Conceptualization, Investigation, Writing—Original Draft; K.M.E., Conceptualization, Methodology, Validation, Investigation, Writing—Original Draft, Writing—Review and Editing; F.M., Investigation, Writing—Original Draft; M.F.R.P. Writing—Review and Editing, Funding Acquisition; J.L.F., Conceptualization, Writing—Review and Editing, Funding acquisition; S.T. and K.M.E. contributed equally to this work. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded Base Funding—UIDB/50020/2020 of the Associate Laboratory LSRE-LCM—funded by national funds through FCT/MCTES (PIDDAC). K.M.E. acknowledges FCT funding under DL57/2016 Transitory Norm Programme.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
This work was financially supported by Base Funding—UIDB/50020/2020 of the Associate Laboratory LSRE-LCM—funded by national funds through FCT/MCTES (PIDDAC). K.M.E. acknowledges FCT funding under DL57/2016 Transitory Norm Programme.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Alexis, F. Factors affecting the degradation and drug-release mechanism of poly(lactic acid) and poly[(lactic acid)-co-(glycolic acid)]. Polym. Int. 2004, 54, 36–46. [Google Scholar] [CrossRef]
- Araújo, A.; Oliveira, M.; Oliveira, R.; Botelho, G.; Machado, A.V. Biodegradation assessment of PLA and its nanocomposites. Environ. Sci. Pollut. Res. 2013, 21, 9477–9486. [Google Scholar] [CrossRef]
- Aso, Y.; Yoshioka, S.; Liwanpo, A.; Terao, T. Effect of temperature on mechanisms of drug release and matrix degradation of poly(d,l-lactide) microspheres. J. Control. Release 1994, 31, 33–39. [Google Scholar] [CrossRef]
- Castro-Aguirre, E.; Iñiguez-Franco, F.; Samsudin, H.; Fang, X.; Auras, R. Poly(lactic acid)—Mass production, processing, industrial applications, and end of life. Adv. Drug Deliv. Rev. 2016, 107, 333–366. [Google Scholar] [CrossRef] [Green Version]
- Jamshidian, M.; Tehrany, E.A.; Imran, M.; Jacquot, M.; Desobry, S. Poly-Lactic Acid: Production, Applications, Nanocomposites, and Release Studies. Compr. Rev. Food Sci. Food Saf. 2010, 9, 552–571. [Google Scholar] [CrossRef]
- Shah, A.A.; Hasan, F.; Hameed, A.; Ahmed, S. Biological degradation of plastics: A comprehensive review. Biotechnol. Adv. 2008, 26, 246–265. [Google Scholar] [CrossRef]
- Elsawy, M.A.; Kim, K.-H.; Park, J.-W.; Deep, A. Hydrolytic degradation of polylactic acid (PLA) and its composites. Renew. Sustain. Energy Rev. 2017, 79, 1346–1352. [Google Scholar] [CrossRef]
- Nampoothiri, K.M.; Nair, N.R.; John, R.P. An overview of the recent developments in polylactide (PLA) research. Bioresour. Technol. 2010, 101, 8493–8501. [Google Scholar] [CrossRef]
- Murariu, M.; Dubois, P. PLA composites: From production to properties. Adv. Drug Deliv. Rev. 2016, 107, 17–46. [Google Scholar] [CrossRef]
- Vink, E.T.; Rábago, K.R.; Glassner, D.A.; Gruber, P.R. Applications of life cycle assessment to NatureWorks™ polylactide (PLA) production. Polym. Degrad. Stab. 2003, 80, 403–419. [Google Scholar] [CrossRef]
- Platel, R.H.; Hodgson, L.M.; Williams, C.K. Biocompatible Initiators for Lactide Polymerization. Polym. Rev. 2008, 48, 11–63. [Google Scholar] [CrossRef]
- Malinconico, M. Soil Degradable Bioplastics for a Sustainable Modern Agriculture; Springer: New York, NY, USA, 2017. [Google Scholar]
- Wan, L.; Zhang, Y. Jointly modified mechanical properties and accelerated hydrolytic degradation of PLA by interface reinforcement of PLA-WF. J. Mech. Behav. Biomed. Mater. 2018, 88, 223–230. [Google Scholar] [CrossRef]
- Mehta, R.; Kumar, V.; Bhunia, H.; Upadhyay, S.N. Synthesis of Poly(Lactic Acid): A Review. J. Macromol. Sci. Part C 2005, 45, 325–349. [Google Scholar] [CrossRef]
- Lim, L.-T.; Auras, R.; Rubino, M. Processing technologies for poly(lactic acid). Prog. Polym. Sci. 2008, 33, 820–852. [Google Scholar] [CrossRef]
- Sin, L.T.; Rahmat, A.R.; Rahman, W.A.W.A. (Eds.) 3-Thermal Properties of Poly(lactic Acid). In Polylactic Acid; William Andrew Publishing: Oxford, UK, 2013; pp. 109–141. [Google Scholar]
- Fambri, L.; Migliaresi, C.; Kesenci, K.; Pişkin, E. Biodegradable Polymers. In Integrated Biomaterials Science; J.B. Metzler: Stuttgart, Germany, 2005; pp. 119–187. [Google Scholar]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [Green Version]
- Gupta, B.; Revagade, N.; Hilborn, J. Poly(lactic acid) fiber: An overview. Prog. Polym. Sci. 2007, 32, 455–482. [Google Scholar] [CrossRef]
- Södergård, A.; Stolt, M. Properties of lactic acid based polymers and their correlation with composition. Prog. Polym. Sci. 2002, 27, 1123–1163. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Preziosi, R.; Robson, G.D. Abiotic and biotic environmental degradation of the bioplastic polymer poly(lactic acid): A review. Polym. Degrad. Stab. 2017, 137, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Bergsma, J.E.; de Bruijn, W.C.; Rozema, F.R.; Bos, R.R.M.; Boering, G. Late degradation tissue response to poly(l-lactide) bone plates and screws. Biomaterials 1995, 16, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.pressreleasefinder.com/NatureWorks/NWPR005/en/ (accessed on 22 December 2020).
- Available online: https://www.natureworksllc.com/News-and-Events/Press-Releases/2020 (accessed on 12 November 2020).
- Momani, B. Assessment of the Impacts of Bioplastics: Energy Usage, Fossil Fuel Usage, Pollution, Health Effects, Effects on the Food Supply, and Economic Effects Compared to Petroleum Based Plastics. Available online: https://core.ac.uk/download/pdf/212988946.pdf (accessed on 28 November 2020).
- Momani, B. Assessment of the Impacts of Bioplastics: Energy Usage, Fossil Fuel Usage, Pollution, Health Effects, Effects on the Food Supply, and Economic Effects Compared to Petroleum Based Plastics; An Interactive Qualifying Project Report; Worcester Polytechnic Institute: Worcester, MA, USA, 2009. [Google Scholar]
- Zhang, Y.; Luo, H.; Kong, L.; Zhao, X.; Miao, G.; Zhu, L.; Li, S.; Sun, Y. Highly efficient production of lactic acid from xylose using Sn-beta catalysts. Green Chem. 2020, 22, 7333–7336. [Google Scholar] [CrossRef]
- Komesu, A.; Oliveira, J.A.R.d.; Martins, L.H.d.S.; Wolf Maciel, M.R.; Maciel Filho, R. Lactic Acid Production to Purification: A Review. BioResources 2017, 12, 20. [Google Scholar] [CrossRef]
- Sun, Y.; Xu, Z.; Zheng, Y.; Zhou, J.; Xiu, Z. Efficient production of lactic acid from sugarcane molasses by a newly microbial consortium CEE-DL15. Process. Biochem. 2019, 81, 132–138. [Google Scholar] [CrossRef]
- Budhavaram, N.K.; Fan, Z. Production of lactic acid from paper sludge using acid-tolerant, thermophilic Bacillus coagulan strains. Bioresour. Technol. 2009, 100, 5966–5972. [Google Scholar] [CrossRef]
- Schmidt, S.; Padukone, N. Production of lactic acid from wastepaper as a cellulosic feedstock. J. Ind. Microbiol. Biotechnol. 1997, 18, 10–14. [Google Scholar] [CrossRef]
- Gullón, B.; Yáñez, R.; Alonso, J.L.; Parajó, J. l-Lactic acid production from apple pomace by sequential hydrolysis and fermentation. Bioresour. Technol. 2008, 99, 308–319. [Google Scholar] [CrossRef]
- Garlotta, D. A Literature Review of Poly(Lactic Acid). J. Polym. Environ. 2001, 9, 63–84. [Google Scholar] [CrossRef]
- Rasal, R.M.; Janorkar, A.V.; Hirt, D.E. Poly(lactic acid) modifications. Prog. Polym. Sci. 2010, 35, 338–356. [Google Scholar] [CrossRef]
- Sin, L.T.; Rahmat, A.R.; Rahman, W.A.W.A. Synthesis and Production of Poly(lactic Acid). In Polylactic Acid; Elsevier BV: Amsterdam, The Netherlands, 2013; pp. 71–107. [Google Scholar]
- Drumright, R.E.; Gruber, P.R.; Henton, D.E. Polylactic Acid Technology. Adv. Mater. 2000, 12, 1841–1846. [Google Scholar] [CrossRef]
- Botvin, V.; Karaseva, S.; Salikova, D.; Dusselier, M. Syntheses and chemical transformations of glycolide and lactide as monomers for biodegradable polymers. Polym. Degrad. Stab. 2021, 183, 109427. [Google Scholar] [CrossRef]
- Hartmann, M.H. High Molecular Weight Polylactic Acid Polymers. In Biopolymers from Renewable Resources; Kaplan, D.L., Ed.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 367–411. [Google Scholar]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled Ring-Opening Polymerization of Lactide and Glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef]
- Bendix, D. Chemical synthesis of polylactide and its copolymers for medical applications. Polym. Degrad. Stab. 1998, 59, 129–135. [Google Scholar] [CrossRef]
- Qi, X.; Ren, Y.; Wang, X. New advances in the biodegradation of Poly(lactic) acid. Int. Biodeterior. Biodegradation 2017, 117, 215–223. [Google Scholar] [CrossRef]
- Metkar, S.; Sathe, V.; Rahman, I.; Idage, B.; Idage, S. Ring opening polymerization of lactide: Kinetics and modeling. Chem. Eng. Commun. 2019, 206, 1159–1167. [Google Scholar] [CrossRef]
- Huang, J.; Lisowski, M.S.; Runt, J.; Hall, E.S.; Kean, R.T.; Buehler, N.; Lin, J.S. Crystallization and Microstructure of Poly(l-lactide-co-meso-lactide) Copolymers. Macromolecules 1998, 31, 2593–2599. [Google Scholar] [CrossRef]
- Vasanthakumari, R.; Pennings, A. Crystallization kinetics of poly(l-lactic acid). Polymer 1983, 24, 175–178. [Google Scholar] [CrossRef]
- Marega, C.; Marigo, A.; Noto, V.; Zannetti, R.; Martorana, A.; Paganetto, G. Structure and crystallization kinetics of poly(L-Lactic Acid). Die Makromolekulare Chemie 1992, 193, 1599–1606. [Google Scholar] [CrossRef]
- Mazzullo, S.; Paganetto, G.; Celli, A. Regime III crystallization in poly-(l-lactic) acid. Solidif. Process. Polym. 2007, 34, 32–34. [Google Scholar] [CrossRef]
- Kolstad, J.J. Crystallization kinetics of poly(L-lactide-co-meso-lactide). J. Appl. Polym. Sci. 1996, 62, 1079–1091. [Google Scholar] [CrossRef]
- Zell, M.T.; Padden, B.E.; Paterick, A.J.; Hillmyer, M.A.; Kean, R.T.; Thakur, K.A.M.; Munson, E.J. Direct Observation of Stereodefect Sites in Semicrystalline Poly(lactide) Using 13C Solid-State NMR. J. Am. Chem. Soc. 1998, 120, 12672–12673. [Google Scholar] [CrossRef]
- Baratian, S.; Hall, E.S.; Lin, J.S.; Xu, R.; Runt, J. Crystallization and Solid-State Structure of Random Polylactide Copolymers: Poly(l-lactide-co-d-lactide)s. Macromolecules 2001, 34, 4857–4864. [Google Scholar] [CrossRef]
- Di Lorenzo, M.L. Crystallization behavior of poly(l-lactic acid). Eur. Polym. J. 2005, 41, 569–575. [Google Scholar] [CrossRef]
- Lopes, M.S.; Jardini, A.; Filho, R.M. Poly (Lactic Acid) Production for Tissue Engineering Applications. Procedia Eng. 2012, 42, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- Lunt, J. Large-scale production, properties and commercial applications of polylactic acid polymers. Polym. Degrad. Stab. 1998, 59, 145–152. [Google Scholar] [CrossRef]
- Eling, B.; Gogolewski, S.; Pennings, A. Biodegradable materials of poly(l-lactic acid): 1. Melt-spun and solution-spun fibres. Polymer 1982, 23, 1587–1593. [Google Scholar] [CrossRef]
- Available online: https://www.hitachi-hightech.com/file/global/pdf/products/science/appli/ana/thermal/application_TA_081e.pdf (accessed on 13 December 2020).
- Auras, R.; Harte, B.; Selke, S. An Overview of Polylactides as Packaging Materials. Macromol. Biosci. 2004, 4, 835–864. [Google Scholar] [CrossRef]
- Tsuji, H. Poly(lactide) Stereocomplexes: Formation, Structure, Properties, Degradation, and Applications. Macromol. Biosci. 2005, 5, 569–597. [Google Scholar] [CrossRef]
- Tsuji, H.; Horii, F.; Hyon, S.H.; Ikada, Y. Stereocomplex formation between enantiomeric poly(lactic acid)s. 2. Stereocomplex formation in concentrated solutions. Macromolecules 1991, 24, 2719–2724. [Google Scholar] [CrossRef]
- Jamshidi, K.; Hyon, S.-H.; Ikada, Y. Thermal characterization of polylactides. Polymer 1988, 29, 2229–2234. [Google Scholar] [CrossRef]
- Cheng, Y.; Deng, S.; Chen, P.; Ruan, R. Polylactic acid (PLA) synthesis and modifications: A review. Front. Chem. China 2009, 4, 259–264. [Google Scholar] [CrossRef]
- Gui, Z.; Xu, Y.; Gao, Y.; Lu, C.; Cheng, S. Novel polyethylene glycol-based polyester-toughened polylactide. Mater. Lett. 2012, 71, 63–65. [Google Scholar] [CrossRef]
- Yu, M.; Zheng, Y.; Tian, J. Study on the biodegradability of modified starch/polylactic acid (PLA) composite materials. RSC Adv. 2020, 10, 26298–26307. [Google Scholar] [CrossRef]
- Meaurio, E.; Hernandez-Montero, N.; Zuza, E.; Sarasua, J.R. Miscible Blends Based on Biodegradable Polymers. In Characterization of Polymer Blends: Miscibility, Morphology and Interfaces; Thomas, S., Grohens, Y., Jyotishkumar, P., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 7–86. [Google Scholar]
- Zhang, L.; Goh, S.; Lee, S. Miscibility and crystallization behaviour of poly(l-lactide)/poly(p-vinylphenol) blends. Polymer 1998, 39, 4841–4847. [Google Scholar] [CrossRef]
- Chen, H.-L.; Liu, H.-H.; Lin, J.S. Microstructure of Semicrystalline Poly(l-lactide)/Poly(4-vinylphenol) Blends Evaluated from SAXS Absolute Intensity Measurement. Macromolecules 2000, 33, 4856–4860. [Google Scholar] [CrossRef]
- Pitt, G.; Cha, Y.; Shah, S.; Zhu, K. Blends of PVA and PGLA: Control of the permeability and degradability of hydrogels by blending. J. Control. Release 1992, 19, 189–199. [Google Scholar] [CrossRef]
- Gajria, A.M.; Davé, V.; Gross, R.A.; McCarthy, S.P. Miscibility and biodegradability of blends of poly(lactic acid) and poly(vinyl acetate). Polymer 1996, 37, 437–444. [Google Scholar] [CrossRef]
- Riedl, B.; Prud’Homme, R.E. Thermodynamic study of poly(vinyl chloride)/polyester blends by inverse-phase gas chromatography at 120 °C. J. Polym. Sci. Part B Polym. Phys. 1986, 24, 2565–2582. [Google Scholar] [CrossRef]
- Wang, Y.; Hillmyer, M.A. Polyethylene-poly(L-lactide) diblock copolymers: Synthesis and compatibilization of poly(L-lactide)/polyethylene blends. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 2755–2766. [Google Scholar] [CrossRef]
- Nakafuku, C.; Sakoda, M. Melting and Crystallization of Poly(L-lactic acid) and Poly(ethylene oxide) Binary Mixture. Polym. J. 1993, 25, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Park, T.G.; Cohen, S.; Langer, R. Poly(L-lactic acid)/Pluronic blends: Characterization of phase separation behavior, degradation, and morphology and use as protein-releasing matrixes. Macromolecules 1992, 25, 116–122. [Google Scholar] [CrossRef]
- Yang, J.-M.; Chen, H.-L.; You, J.-W.; Hwang, J.C. Miscibility and Crystallization of Poly(L-lactide)/Poly(ethylene glycol) and Poly(L-lactide)/Poly(ε-caprolactone) Blends. Polym. J. 1997, 29, 657. [Google Scholar] [CrossRef]
- Yoon, J.-S.; Oh, S.-H.; Kim, M.-N.; Chin, I.-J.; Kim, Y.-H. Thermal and mechanical properties of poly(l-lactic acid)–poly (ethylene-co-vinyl acetate) blends1Dedicated to Professor Ick-Sam Noh on the occasion of his retirement from Inha University.1. Polymer 1999, 40, 2303–2312. [Google Scholar] [CrossRef]
- Jin, H.-J.; Chin, I.-J.; Kim, M.-N.; Kim, S.-H.; Yoon, J.-S. Blending of poly(L-lactic acid) with poly(cis-1,4-isoprene). Eur. Polym. J. 2000, 36, 165–169. [Google Scholar] [CrossRef]
- Mikos, A.G.; Thorsen, A.J.; Czerwonka, L.A.; Bao, Y.; Langer, R.; Winslow, D.N.; Vacanti, J.P. Preparation and characterization of poly(l-lactic acid) foams. Polymer 1994, 35, 1068–1077. [Google Scholar] [CrossRef]
- Avella, M.; Errico, M.; Immirzi, B.; Malinconico, M.; Martuscelli, E.; Paolillo, L.; Falcigno, L. Radical polymerization of poly(butyl acrylate) in the presence of poly(L-lactic acid), 1. Synthesis, characterization and properties of blends. Angew. Makromol. Chem. Appl. Macromol. Chem. Phys. 1997, 246, 49–63. [Google Scholar] [CrossRef]
- Eguiburu, J.; Iruin, J.; Fernandez-Berridi, M.; Román, J.S. Blends of amorphous and crystalline polylactides with poly(methyl methacrylate) and poly(methyl acrylate): A miscibility study. Polymer 1998, 39, 6891–6897. [Google Scholar] [CrossRef]
- Avella, M.; Errico, M.E.; Immirzi, B.; Malinconico, M.; Falcigno, L.; Paolillo, L. Radical polymerization of methyl methacrylate in the presence of biodegradable poly(L-lactic acid). Preparation of blends, chemical-physical characterization and morphology. Macromol. Chem. Phys. 2000, 201, 1295–1302. [Google Scholar] [CrossRef]
- Chen, G.-X.; Kim, H.-S.; Kim, E.-S.; Yoon, J.-S. Compatibilization-like effect of reactive organoclay on the poly(l-lactide)/poly(butylene succinate) blends. Polymer 2005, 46, 11829–11836. [Google Scholar] [CrossRef]
- Grijpma, D.W.; Van Hofslot, R.D.A.; Supèr, H.; Nijenhuis, A.J.; Pennings, A.J. Rubber toughening of poly(lactide) by blending and block copolymerization. Polym. Eng. Sci. 1994, 34, 1674–1684. [Google Scholar] [CrossRef]
- Ogata, N.; Jimenez, G.; Kawai, H.; Ogihara, T. Structure and thermal/mechanical properties of poly(l-lactide)-clay blend. J. Polym. Sci. Part B Polym. Phys. 1997, 35, 389–396. [Google Scholar] [CrossRef]
- Cha, Y.; Pitt, C. The biodegradability of polyester blends. Biomaterials 1990, 11, 108–112. [Google Scholar] [CrossRef]
- Joziasse, C.; Veenstra, H.; Topp, M.; Grijpma, D.; Pennings, A. Rubber toughened linear and star-shaped poly(d,l-lactide-co-glycolide): Synthesis, properties and in vitro degradation. Polymer 1998, 39, 467–474. [Google Scholar] [CrossRef]
- Gérard, T.; Budtova, T. PLA-PHA blends: Morphology, thermal and mechanical properties. In Proceedings of the International Conference on Biodegradable and Biobased Polymers—BIOPOL 2011, Strasbourg, France, 29–31 August 2011. [Google Scholar]
- Amass, W.; Amass, A.; Tighe, B. A review of biodegradable polymers: Uses, current developments in the synthesis and characterization of biodegradable polyesters, blends of biodegradable polymers and recent advances in biodegradation studies. Polym. Int. 1999, 47, 89–144. [Google Scholar] [CrossRef]
- Wang, L.; Ma, W.; Gross, R.; McCarthy, S. Reactive compatibilization of biodegradable blends of poly(lactic acid) and poly(ε-caprolactone). Polym. Degrad. Stab. 1998, 59, 161–168. [Google Scholar] [CrossRef]
- Nair, N.R.; Sekhar, V.C.; Nampoothiri, K.M.; Pandey, A. 32-Biodegradation of Biopolymers. In Current Developments in Biotechnology and Bioengineering; Pandey, A., Negi, S., Soccol, C.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 739–755. [Google Scholar]
- Zaaba, N.F.; Jaafar, M. A review on degradation mechanisms of polylactic acid: Hydrolytic, photodegradative, microbial, and enzymatic degradation. Polym. Eng. Sci. 2020, 60, 2061–2075. [Google Scholar] [CrossRef]
- Malwela, T.; Ray, S.S. Enzymatic degradation behavior of nanoclay reinforced biodegradable PLA/PBSA blend composites. Int. J. Biol. Macromol. 2015, 77, 131–142. [Google Scholar] [CrossRef]
- Bikiaris, D.N. Nanocomposites of aliphatic polyesters: An overview of the effect of different nanofillers on enzymatic hydrolysis and biodegradation of polyesters. Polym. Degrad. Stab. 2013, 98, 1908–1928. [Google Scholar] [CrossRef]
- Huang, Q.; Hiyama, M.; Kabe, T.; Kimura, S.; Iwata, T. Enzymatic Self-Biodegradation of Poly(l-lactic acid) Films by Embedded Heat-Treated and Immobilized Proteinase K. Biomacromolecules 2020, 21, 3301–3307. [Google Scholar] [CrossRef] [PubMed]
- de Jong, S.; Arias, E.; Rijkers, D.; van Nostrum, C.; Bosch, J.K.-V.D.; Hennink, W. New insights into the hydrolytic degradation of poly(lactic acid): Participation of the alcohol terminus. Polymer 2001, 42, 2795–2802. [Google Scholar] [CrossRef]
- Engineer, C.; Parikh, J.; Raval, A. Review on hydrolytic degradation behavior of biodegradable polymers from controlled drug delivery system. Trends. Biomater. Artif. Organs 2011, 25, 79–85. [Google Scholar]
- Laycock, B.; Nikolić, M.; Colwell, J.M.; Gauthier, E.; Halley, P.; Bottle, S.; George, G. Lifetime prediction of biodegradable polymers. Prog. Polym. Sci. 2017, 71, 144–189. [Google Scholar] [CrossRef] [Green Version]
- Husárová, L.; Pekařová, S.; Stloukal, P.; Kucharzcyk, P.; Verney, V.; Commereuc, S.; Ramone, A.; Koutny, M. Identification of important abiotic and biotic factors in the biodegradation of poly(l-lactic acid). Int. J. Biol. Macromol. 2014, 71, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Blasco, L.S.; Ribes-Greus, A.; Alamo, R. Comparative thermal, biological and photodegradation kinetics of polylactide and effect on crystallization rates. Polym. Degrad. Stab. 2013, 98, 771–784. [Google Scholar] [CrossRef]
- Kale, G.; Kijchavengkul, T.; Auras, R.; Rubino, M.; Selke, S.E.; Singh, S.P. Compostability of Bioplastic Packaging Materials: An Overview. Macromol. Biosci. 2007, 7, 255–277. [Google Scholar] [CrossRef]
- Tasaka, F.; Miyazaki, H.; Ohya, A.Y.; Ouchi, T. Synthesis of Comb-Type Biodegradable Polylactide through Depsipeptide−Lactide Copolymer Containing Serine Residues. Macromolecules 1999, 32, 6386–6389. [Google Scholar] [CrossRef]
- Polman, E.M.; Gruter, G.-J.M.; Parsons, J.R.; Tietema, A. Comparison of the aerobic biodegradation of biopolymers and the corresponding bioplastics: A review. Sci. Total Environ. 2021, 753, 141953. [Google Scholar] [CrossRef]
- Siparsky, G.L.; Voorhees, K.J.; Miao, F. Hydrolysis of Polylactic Acid (PLA) and Polycaprolactone (PCL) in Aqueous Acetonitrile Solutions: Autocatalysis. J. Polym. Environ. 1998, 6, 31–41. [Google Scholar] [CrossRef]
- Hirao, K.; Nakatsuchi, Y.; Ohara, H. Alcoholysis of Poly(l-lactic acid) under microwave irradiation. Polym. Degrad. Stab. 2010, 95, 925–928. [Google Scholar] [CrossRef]
- Iñiguez-Franco, F.; Auras, R.; Burgess, G.; Holmes, D.; Fang, X.; Rubino, M.; Soto-Valdez, H. Concurrent solvent induced crystallization and hydrolytic degradation of PLA by water-ethanol solutions. Polymer 2016, 99, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Gondo, D.; Wada, T.; Kanehashi, S.; Nagai, K. Effects of various liquid organic solvents on solvent-induced crystallization of amorphous poly(lactic acid) film. J. Appl. Polym. Sci. 2012, 129, 1607–1617. [Google Scholar] [CrossRef]
- McNeill, I.; Leiper, H. Degradation studies of some polyesters and polycarbonates—2. Polylactide: Degradation under isothermal conditions, thermal degradation mechanism and photolysis of the polymer. Polym. Degrad. Stab. 1985, 11, 309–326. [Google Scholar] [CrossRef]
- Tsuji, H.; Ikada, Y. Properties and morphology of poly(L-lactide). II. hydrolysis in alkaline solution. J. Polym. Sci. Part A Polym. Chem. 1998, 36, 59–66. [Google Scholar] [CrossRef]
- Chen, H.-M.; Shen, Y.; Yang, J.-H.; Huang, T.; Zhang, N.; Wang, Y.; Zhou, Z.-W. Molecular ordering and α′-form formation of poly(l-lactide) during the hydrolytic degradation. Polymer 2013, 54, 6644–6653. [Google Scholar] [CrossRef]
- Suuronen, R.; Pohjonen, T.; Hietanen, J.; Lindqvist, C. A 5-year in vitro and in vivo study of the biodegradation of polylactide plates. J. Oral Maxillofac. Surg. 1998, 56, 604–614. [Google Scholar] [CrossRef]
- da Silva, D.; Kaduri, M.; Poley, M.; Adir, O.; Krinsky, N.; Shainsky-Roitman, J.; Schroeder, A. Biocompatibility, biodegradation and excretion of polylactic acid (PLA) in medical implants and theranostic systems. Chem. Eng. J. 2018, 340, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.H. Biodegradable Polymers as Drug Delivery Systems. In Biomedical Polymers; Woodhead Publishing: Sawston, UK, 2007. [Google Scholar]
- Lyu, S.; Schley, J.; Loy, B.; Lind, D.; Hobot, C.; Sparer, R.; Untereker, D. Kinetics and Time−Temperature Equivalence of Polymer Degradation. Biomacromolecules 2007, 8, 2301–2310. [Google Scholar] [CrossRef] [PubMed]
- Schliecker, G.; Schmidt, C.; Fuchs, S.; Kissel, T. Characterization of a homologous series of d,l-lactic acid oligomers; a mechanistic study on the degradation kinetics in vitro. Biomaterials 2003, 24, 3835–3844. [Google Scholar] [CrossRef]
- Shih, C. Chain-end scission in acid catalyzed hydrolysis of poly (d,l-lactide) in solution. J. Control. Release 1995, 34, 9–15. [Google Scholar] [CrossRef]
- Pitt, C.G.; Chasalow, F.I.; Hibionada, Y.M.; Klimas, D.M.; Schindler, A. Aliphatic polyesters. I. The degradation of poly(ϵ-caprolactone) in vivo. J. Appl. Polym. Sci. 1981, 26, 3779–3787. [Google Scholar] [CrossRef]
- Li, S.M.; Garreau, H.; Vert, M. Structure-property relationships in the case of the degradation of massive poly(α-hydroxy acids) in aqueous media. J. Mater. Sci. Mater. Med. 1990, 1, 131–139. [Google Scholar] [CrossRef]
- Henton, D.; Gruber, P.; Lunt, J.; Randall, J. Polylactic Acid Technology. In Natural Fibers, Biopolymers, and Biocomposites; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Gajjar, C.R.; King, M.W. Resorbable Fiber-Forming Polymers for Biotextile Applications; Springer: New York, NY, USA, 2014. [Google Scholar]
- Kronenthal, R.L. Biodegradable Polymers in Medicine and Surgery. In Polymers in Medicine and Surgery; Kronenthal, R.L., Oser, Z., Martin, E., Eds.; Springer: Boston, MA, USA, 1975; pp. 119–137. [Google Scholar]
- Göpferich, A. Mechanisms of polymer degradation and erosion. Biomaterials 1996, 17, 103–114. [Google Scholar] [CrossRef]
- Lyu, S.; Sparer, R.; Untereker, D. Analytical solutions to mathematical models of the surface and bulk erosion of solid polymers. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 383–397. [Google Scholar] [CrossRef]
- Kale, G.; Auras, R.; Singh, S.P. Comparison of the degradability of poly(lactide) packages in composting and ambient exposure conditions. Packag. Technol. Sci. 2007, 20, 49–70. [Google Scholar] [CrossRef]
- Siepmann, J.; Göpferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliv. Rev. 2001, 48, 229–247. [Google Scholar] [CrossRef]
- Shah, S.; Cha, Y.; Pitt, C. Poly (glycolic acid-co-dl-lactic acid): Diffusion or degradation controlled drug delivery? J. Control. Release 1992, 18, 261–270. [Google Scholar] [CrossRef]
- von Burkersroda, F.; Schedl, L.; Göpferich, A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials 2002, 23, 4221–4231. [Google Scholar] [CrossRef]
- Goto, T.; Kishita, M.; Sun, Y.; Sako, T.; Okajima, I. Degradation of Polylactic Acid Using Sub-Critical Water for Compost. Polymer 2020, 12, 2434. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Pan, J.; Buchanan, F.; Weir, N.; Farrar, D. Analysis of degradation data of poly(l-lactide–co-l,d-lactide) and poly(l-lactide) obtained at elevated and physiological temperatures using mathematical models. Acta Biomater. 2010, 6, 3882–3889. [Google Scholar] [CrossRef]
- Han, X.; Pan, J. A model for simultaneous crystallisation and biodegradation of biodegradable polymers. Biomaterials 2009, 30, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Coszach, P.; Bogaert, J.-C.; Willocq, J. Chemical Recycling of Pla by Hydrolysis; GALACTIC, S.A.: Milwaukee, WI, USA, 2012; p. 7. [Google Scholar]
- Gorrasi, G.; Pantani, P. Hydrolysis and Biodegradation of Poly(lactic acid). In Synthesis, Structure and Properties of Poly(lactic acid); Springer International Publishing: Cham, Switzerlands, 2017. [Google Scholar]
- Lyu, S.; Untereker, D. Degradability of Polymers for Implantable Biomedical Devices. Int. J. Mol. Sci. 2009, 10, 4033–4065. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.H.; Ree, M.; Kim, H. Acid- and base-catalyzed hydrolyses of aliphatic polycarbonates and polyesters. Catal. Today 2006, 115, 283–287. [Google Scholar] [CrossRef]
- Rocca-Smith, J.; Chau, N.; Champion, D.; Brachais, C.-H.; Marcuzzo, E.; Sensidoni, A.; Piasente, F.; Karbowiak, T.; Debeaufort, F. Effect of the state of water and relative humidity on ageing of PLA films. Food Chem. 2017, 236, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Höglund, A.; Odelius, K.; Albertsson, A.-C. Crucial Differences in the Hydrolytic Degradation between Industrial Polylactide and Laboratory-Scale Poly(L-lactide). ACS Appl. Mater. Interfaces 2012, 4, 2788–2793. [Google Scholar] [CrossRef]
- Limsukon, W.; Auras, R.; Selke, S. Hydrolytic degradation and lifetime prediction of poly(lactic acid) modified with a multifunctional epoxy-based chain extender. Polym. Test. 2019, 80, 106108. [Google Scholar] [CrossRef]
- Gupta, M.C.; Deshmukh, V.G. Thermal oxidative degradation of poly-lactic acid Part I. Colloid Polym. Sci. 1982, 260, 308–311. [Google Scholar] [CrossRef]
- Oliveira, M.; Santos, E.; Araújo, A.; Fechine, G.J.; Machado, A.V.; Botelho, G. The role of shear and stabilizer on PLA degradation. Polym. Test. 2016, 51, 109–116. [Google Scholar] [CrossRef]
- Cuadri, A.; Martín-Alfonso, J. Thermal, thermo-oxidative and thermomechanical degradation of PLA: A comparative study based on rheological, chemical and thermal properties. Polym. Degrad. Stab. 2018, 150, 37–45. [Google Scholar] [CrossRef]
- Lucas, N.; Bienaime, C.; Belloy, C.; Queneudec, M.; Silvestre, F.; Nava-Saucedo, J.-E. Polymer biodegradation: Mechanisms and estimation techniques—A review. Chemosphere 2008, 73, 429–442. [Google Scholar] [CrossRef]
- Zou, H.; Yi, C.; Wang, L.; Liu, H.; Xu, W. Thermal degradation of poly(lactic acid) measured by thermogravimetry coupled to Fourier transform infrared spectroscopy. J. Therm. Anal. Calorim. 2009, 97, 929–935. [Google Scholar] [CrossRef]
- Kopinke, F.-D.; Mackenzie, K. Mechanistic aspects of the thermal degradation of poly(lactic acid) and poly(β-hydroxybutyric acid). J. Anal. Appl. Pyrolysis 1997, 40–41, 43–53. [Google Scholar] [CrossRef]
- Kopinke, F.-D.; Remmler, M.; Mackenzie, K.; Möder, M.; Wachsen, O. Thermal decomposition of biodegradable polyesters—II. Poly(lactic acid). Polym. Degrad. Stab. 1996, 53, 329–342. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Lüderwald, I. Strukturuntersuchung von polyestern durch direkten abbau im massenspektrometer, 3. Poly-β-propiolacton, poly-β-pivalolacton und poly-δ-valerolacton. Die Makromol. Chem. 1978, 179, 421–427. [Google Scholar] [CrossRef]
- Auras, R.; Lim, L.-T.; Selke, S.; Tsuji, H. Poly (lactic acid): Synthesis, Structures, Properties, Processing, and Applications; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Carrasco, F.; Pagès, P.; Gámez-Pérez, J.; Santana, O.; Maspoch, M. Kinetics of the thermal decomposition of processed poly(lactic acid). Polym. Degrad. Stab. 2010, 95, 2508–2514. [Google Scholar] [CrossRef]
- Babanalbandi, A.; Hill, D.; O’Donnell, J.; Pomery, P.; Whittaker, A. An electron spin resonance study on γ-irradiated poly(l-lactic acid) and poly(d,l-lactic acid). Polym. Degrad. Stab. 1995, 50, 297–304. [Google Scholar] [CrossRef]
- Nishida, H. Thermal Degradation, Poly (lactic acid): Synthesis, Structures, Properties, Processing, and Applications; Auras, R.A., Lim, T.-K., Selke, S.E.M., Tsuji, H., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 401–412. [Google Scholar]
- Meng, Q.; Heuzey, M.-C.; Carreau, P.J. Control of thermal degradation of polylactide/clay nanocomposites during melt processing by chain extension reaction. Polym. Degrad. Stab. 2012, 97, 2010–2020. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.; Carreau, P.; Wood-Adams, P.M. Control of thermal degradation of polylactide (PLA)-clay nanocomposites using chain extenders. Polym. Degrad. Stab. 2012, 97, 554–565. [Google Scholar] [CrossRef]
- Cam, D.; Marucci, M. Influence of residual monomers and metals on poly (l-lactide) thermal stability. Polymer 1997, 38, 1879–1884. [Google Scholar] [CrossRef]
- Abe, H.; Takahashi, N.; Kim, K.J.; Mochizuki, M.; Doi, Y. Thermal Degradation Processes of End-Capped Poly(l-lactide)s in the Presence and Absence of Residual Zinc Catalyst. Biomacromolecules 2004, 5, 1606–1614. [Google Scholar] [CrossRef]
- Kricheldorf, H.R.; Serra, A. Polylactones. Polym. Bull. 1985, 14, 497–502. [Google Scholar] [CrossRef]
- Nijenhuis, A.J.; Grijpma, D.W.; Pennings, A.J. Lewis acid catalyzed polymerization of L-lactide. Kinetics and mechanism of the bulk polymerization. Macromolecules 1992, 25, 6419–6424. [Google Scholar] [CrossRef]
- Amorin, N.S.Q.S.; Rosa, G.; Alves, J.F.; Gonçalves, S.P.C.; Franchetti, S.M.M.; Fechine, G.J.M. Study of thermodegradation and thermostabilization of poly(lactide acid) using subsequent extrusion cycles. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Cameron, G.; Kerr, G. Thermal degradation of polystyrene—I. Chain scission at low temperatures. Eur. Polym. J. 1968, 4, 709–717. [Google Scholar] [CrossRef]
- Rasselet, D.; Ruellan, A.; Guinault, A.; Miquelard-Garnier, G.; Sollogoub, C.; Fayolle, B. Oxidative degradation of polylactide (PLA) and its effects on physical and mechanical properties. Eur. Polym. J. 2014, 50, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zou, Y.; Li, W.; Cao, G.; Chen, W. Kinetics of thermo-oxidative and thermal degradation of poly(d,l-lactide) (PDLLA) at processing temperature. Polym. Degrad. Stab. 2006, 91, 3259–3265. [Google Scholar] [CrossRef]
- Gupta, M.C.; Deshmukh, V.G. Thermal oxidative degradation of poly-lactic acid part II. Colloid Polym. Sci. 1982, 260, 514–517. [Google Scholar] [CrossRef]
- Jeon, H.J.; Kim, M.N. Biodegradation of poly(l-lactide) (PLA) exposed to UV irradiation by a mesophilic bacterium. Int. Biodeterior. Biodegrad. 2013, 85, 289–293. [Google Scholar] [CrossRef]
- Wataru Sakai, N. Tsutsumi, Photodegradation and Radiation Degradation, Poly (lactic acid); John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 413–422. [Google Scholar]
- Ikada, E. Photo- and Bio-degradable Polyesters. Photodegradation Behaviors of Aliphatic Polyesters. J. Photopolym. Sci. Technol. 1997, 10, 265–270. [Google Scholar] [CrossRef]
- Tsuji, H.; Echizen, Y.; Saha, S.K.; Nishimura, Y. Photodegradation of Poly(L-lactic acid): Effects of Photosensitizer. Macromol. Mater. Eng. 2005, 290, 1192–1203. [Google Scholar] [CrossRef]
- Tsuji, H.; Echizen, Y.; Nishimura, Y. Photodegradation of biodegradable polyesters: A comprehensive study on poly(l-lactide) and poly(ɛ-caprolactone). Polym. Degrad. Stab. 2006, 91, 1128–1137. [Google Scholar] [CrossRef]
- Janorkar, A.V.; Metters, A.T.; Hirt, D.E. Degradation of poly(L-lactide) films under ultraviolet-induced photografting and sterilization conditions. J. Appl. Polym. Sci. 2007, 106, 1042–1047. [Google Scholar] [CrossRef]
- Bocchini, S.; Fukushima, K.; Di Blasio, A.; Fina, A.; Frache, A.; Geobaldo, F. Polylactic Acid and Polylactic Acid-Based Nanocomposite Photooxidation. Biomacromolecules 2010, 11, 2919–2926. [Google Scholar] [CrossRef]
- Benyathiar, P.; Selke, S.E.; Harte, B.R.; Mishra, D.K. The Effect of Irradiation Sterilization on Poly(Lactic) Acid Films. J. Polym. Environ. 2021, 29, 460–471. [Google Scholar] [CrossRef]
- Kantoǧlu, Ö.; Güven, O. Radiation induced crystallinity damage in poly(L-lactic acid). Nucl. Instruments Methods Phys. Res. Sect. B Beam Interactions Mater. Atoms 2002, 197, 259–264. [Google Scholar] [CrossRef]
- Birkinshaw, C.; Buggy, M.; Henn, G.; Jones, E. Irradiation of poly-d,l-lactide. Polym. Degrad. Stab. 1992, 38, 249–253. [Google Scholar] [CrossRef]
- Vargas, L.F.; Welt, B.A.; Pullammanappallil, P.; Teixeira, A.A.; Balaban, M.O.; Beatty, C.L. Effect of electron beam treatments on degradation kinetics of polylactic acid (PLA) plastic waste under backyard composting conditions. Packag. Technol. Sci. 2009, 22, 97–106. [Google Scholar] [CrossRef]
- Tsuji, H.; Sasaki, H.; Sato, H.; Gotoh, Y.; Ishikawa, J. Neuron attachment properties of carbon negative-ion implanted bioabsorbable polymer of poly-lactic acid. Nuclear Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2002, 191, 815–819. [Google Scholar] [CrossRef]
- Koo, G.-H.; Jang, J. Surface modification of poly(lactic acid) by UV/Ozone irradiation. Fibers Polym. 2008, 9, 674–678. [Google Scholar] [CrossRef]
- Alves, C.M.; Yang, Y.; Marton, D.; Carnes, D.L.; Ong, J.L.; Sylvia, V.L.; Dean, D.D.; Reis, R.L.; Agrawal, C.M. Plasma surface modification of poly(D,L-lactic acid) as a tool to enhance protein adsorption and the attachment of different cell types. J. Biomed. Mater. Res. Part B Appl. Biomater. 2008, 87, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, N.; Kaneda, A.; Kanazawa, S.; Yagi, T.; Mitomo, H.; Yoshii, F.; Tamada, M. Application of poly(lactic acid) modified by radiation crosslinking. Nuclear Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms. 2005, 236, 611–616. [Google Scholar] [CrossRef]
- Ma, Z.; Gao, C.; Ji, J.; Shen, J. Protein immobilization on the surface of poly-L-lactic acid films for improvement of cellular interactions. Eur. Polym. J. 2002, 38, 2279–2284. [Google Scholar] [CrossRef]
- Saadi, Z.; Rasmont, A.; Cesar, G.; Bewa, H.; Benguigui, L. Fungal Degradation of Poly(l-lactide) in Soil and in Compost. J. Polym. Environ. 2011, 20, 273–282. [Google Scholar] [CrossRef]
- Machado, A.V.; Araújo, A.; Oliveira, M. Assessment of polymer-based nanocomposites biodegradability. In Biodegradable Polymers: Advancement in Biodegradation Study and Applications; Chu, C.-C., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2015; p. 26. [Google Scholar]
- Tomita, K.; Tsuji, H.; Nakajima, T.; Kikuchi, Y.; Ikarashi, K.; Ikeda, N. Degradation of poly(d-lactic acid) by a thermophile. Polym. Degrad. Stab. 2003, 81, 167–171. [Google Scholar] [CrossRef]
- Butbunchu, N.; Pathom-Aree, W. Actinobacteria as Promising Candidate for Polylactic Acid Type Bioplastic Degradation. Front. Microbiol. 2019, 10, 2834. [Google Scholar] [CrossRef]
- Sankhla, I.S.; Sharma, G.; Tak, A. Chapter 4—Fungal degradation of bioplastics: An overview. In New and Future Developments in Microbial Biotechnology and Bioengineering; Singh, J., Gehlot, P., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 35–47. [Google Scholar]
- Maqbool, Z.; Hussain, S.; Imran, M.; Mahmood, F.; Shahzad, T.; Ahmed, Z.; Azeem, F.; Muzammil, S. Perspectives of using fungi as bioresource for bioremediation of pesticides in the environment: A critical review. Environ. Sci. Pollut. Res. 2016, 23, 16904–16925. [Google Scholar] [CrossRef] [PubMed]
- Luzi, F.; Fortunati, E.; Puglia, D.; Petrucci, R.; Kenny, J.; Torre, L. Study of disintegrability in compost and enzymatic degradation of PLA and PLA nanocomposites reinforced with cellulose nanocrystals extracted from Posidonia Oceanica. Polym. Degrad. Stab. 2015, 121, 105–115. [Google Scholar] [CrossRef]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Pattanasuttichonlakul, W.; Sombatsompop, N.; Prapagdee, B. Accelerating biodegradation of PLA using microbial consortium from dairy wastewater sludge combined with PLA-degrading bacterium. Int. Biodeterior. Biodegrad. 2018, 132, 74–83. [Google Scholar] [CrossRef]
- Sangwan, P.; Wu, D.Y. New Insights into Polylactide Biodegradation from Molecular Ecological Techniques. Macromol. Biosci. 2008, 8, 304–315. [Google Scholar] [CrossRef]
- Ghorpade, V.M.; Gennadios, A.; Hanna, M.A. Laboratory composting of extruded poly(lactic acid) sheets. Bioresour. Technol. 2001, 76, 57–61. [Google Scholar] [CrossRef]
- Kalita, N.K.; Bhasney, S.M.; Mudenur, C.; Kalamdhad, A.; Katiyar, V. End-of-life evaluation and biodegradation of Poly(lactic acid) (PLA)/Polycaprolactone (PCL)/Microcrystalline cellulose (MCC) polyblends under composting conditions. Chemosphere 2020, 247, 125875. [Google Scholar] [CrossRef]
- Gajalakshmi, S.; Abbasi, S.A. Solid Waste Management by Composting: State of the Art. Crit. Rev. Environ. Sci. Technol. 2008, 38, 311–400. [Google Scholar] [CrossRef]
- Cooperband, L.R. Composting: Art and Science of Organic Waste Conversion to a Valuable Soil Resource. Lab. Med. 2000, 31, 283–290. [Google Scholar] [CrossRef]
- Lv, S.; Liu, X.; Gu, J.; Jiang, Y.; Tan, H.; Zhang, Y. Microstructure analysis of polylactic acid-based composites during degradation in soil. Int. Biodeterior. Biodegrad. 2017, 122, 53–60. [Google Scholar] [CrossRef]
- Ho, K.-L.G.; Iii, A.L.P.; Gadea-Rivas, A.; Briceño, J.A.; Rojas, A. Degradation of Polylactic Acid (PLA) Plastic in Costa Rican Soil and Iowa State University Compost Rows. J. Polym. Environ. 1999, 7, 173–177. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Robson, G.D. The influence of biotic and abiotic factors on the rate of degradation of poly(lactic) acid (PLA) coupons buried in compost and soil. Polym. Degrad. Stab. 2013, 98, 2063–2071. [Google Scholar] [CrossRef]
- Calmon, A.; Guillaume, S.; Bellon-Maurel, V.; Feuilloley, P.; Silvestre, F. Evaluation of Material Biodegradability in Real Conditions–Development of a Burial Test and an Analysis Methodology Based on Numerical Vision. J. Polym. Environ. 1999, 7, 157–166. [Google Scholar] [CrossRef]
- Karamanlioglu, M.; Houlden, A.; Robson, G.D. Isolation and characterisation of fungal communities associated with degradation and growth on the surface of poly(lactic) acid (PLA) in soil and compost. Int. Biodeterior. Biodegrad. 2014, 95, 301–310. [Google Scholar] [CrossRef]
- Shogren, R.; Doane, W.; Garlotta, D.; Lawton, J.; Willett, J. Biodegradation of starch/polylactic acid/poly(hydroxyester-ether) composite bars in soil. Polym. Degrad. Stab. 2003, 79, 405–411. [Google Scholar] [CrossRef]
- Kolstad, J.J.; Vink, E.T.; De Wilde, B.; Debeer, L. Assessment of anaerobic degradation of Ingeo™ polylactides under accelerated landfill conditions. Polym. Degrad. Stab. 2012, 97, 1131–1141. [Google Scholar] [CrossRef]
- Rudnik, E.; Briassoulis, D. Comparative Biodegradation in Soil Behaviour of two Biodegradable Polymers Based on Renewable Resources. J. Polym. Environ. 2011, 19, 18–39. [Google Scholar] [CrossRef]
- Vasile, C.; Pamfil, D.; Râpă, M.; Darie-Niţă, R.N.; Mitelut, A.C.; Popa, E.E.; Popescu, P.A.; Draghici, M.C.; Popa, M.E. Study of the soil burial degradation of some PLA/CS biocomposites. Compos. Part B Eng. 2018, 142, 251–262. [Google Scholar] [CrossRef]
- Santonja-Blasco, L.; Moriana, R.; Badía, J.; Ribes-Greus, A. Thermal analysis applied to the characterization of degradation in soil of polylactide: I. Calorimetric and viscoelastic analyses. Polym. Degrad. Stab. 2010, 95, 2185–2191. [Google Scholar] [CrossRef] [Green Version]
- Weng, Y.-X.; Jin, Y.-J.; Meng, Q.-Y.; Wang, L.; Zhang, M.; Wang, Y.-Z. Biodegradation behavior of poly(butylene adipate-co-terephthalate) (PBAT), poly(lactic acid) (PLA), and their blend under soil conditions. Polym. Test. 2013, 32, 918–926. [Google Scholar] [CrossRef]
- Itävaara, M.; Karjomaa, S.; Selin, J.-F. Biodegradation of polylactide in aerobic and anaerobic thermophilic conditions. Chemosphere 2002, 46, 879–885. [Google Scholar] [CrossRef]
- Musioł, M.; Sikorska, W.; Adamus, G.; Janeczek, H.; Richert, J.; Malinowski, R.; Jiang, G.; Kowalczuk, M. Forensic engineering of advanced polymeric materials. Part III—Biodegradation of thermoformed rigid PLA packaging under industrial composting conditions. Waste Manag. 2016, 52, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Deroiné, M.; le Duigou, A.; Corre, Y.-M.; le Gac, P.-Y.; Davies, P.; César, G.; Bruzaud, S. Accelerated ageing of polylactide in aqueous environments: Comparative study between distilled water and seawater. Polym. Degrad. Stab. 2014, 108, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Le Duigou, A.; Davies, P.; Baley, C. Seawater ageing of flax/poly(lactic acid) biocomposites. Polym. Degrad. Stab. 2009, 94, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, H.; Suzuyoshi, K. Environmental degradation of biodegradable polyesters 1. Poly(ε-caprolactone), poly[(R)-3-hydroxybutyrate], and poly(L-lactide) films in controlled static seawater. Polym. Degrad. Stab. 2002, 75, 347–355. [Google Scholar] [CrossRef]
- Tsuji, H.; Suzuyoshi, K. Environmental degradation of biodegradable polyesters 2. Poly(ε-caprolactone), poly[(R)-3-hydroxybutyrate], and poly(L-lactide) films in natural dynamic seawater. Polym. Degrad. Stab. 2002, 75, 357–365. [Google Scholar] [CrossRef]
- Fukushima, K.; Tabuani, D.; Dottori, M.; Armentano, I.; Kenny, J.; Camino, G. Effect of temperature and nanoparticle type on hydrolytic degradation of poly(lactic acid) nanocomposites. Polym. Degrad. Stab. 2011, 96, 2120–2129. [Google Scholar] [CrossRef]
- Brebu, M. Environmental Degradation of Plastic Composites with Natural Fillers—A Review. Polymer 2020, 12, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.-P.; Lau, K.-T.; Wang, H.; Hui, D. Improvement on the properties of polylactic acid (PLA) using bamboo charcoal particles. Compos. Part B Eng. 2015, 81, 14–25. [Google Scholar] [CrossRef]
- Hegyesi, N.; Zhang, Y.; Kohári, A.; Polyák, P.; Sui, X.; Pukánszky, B. Enzymatic degradation of PLA/cellulose nanocrystal composites. Ind. Crops Prod. 2019, 141, 111799. [Google Scholar] [CrossRef] [Green Version]
- Balaguer, M.P.; Aliaga, C.; Fito, C.; Hortal, M. Compostability assessment of nano-reinforced poly(lactic acid) films. Waste Manag. 2016, 48, 143–155. [Google Scholar] [CrossRef]
- Ray, S.S.; Bousmina, M. Biodegradable polymers and their layered silicate nanocomposites: In greening the 21st century materials world. Prog. Mater. Sci. 2005, 50, 962–1079. [Google Scholar] [CrossRef]
- Fukushima, K.; Abbate, C.; Tabuani, D.; Gennari, M.; Camino, G. Biodegradation of poly(lactic acid) and its nanocomposites. Polym. Degrad. Stab. 2009, 94, 1646–1655. [Google Scholar] [CrossRef]
- Neppalli, R.; Causin, V.; Marega, C.; Modesti, M.; Adhikari, R.; Scholtyssek, S.; Ray, S.S.; Marigo, A. The effect of different clays on the structure, morphology and degradation behavior of poly(lactic acid). Appl. Clay Sci. 2014, 87, 278–284. [Google Scholar] [CrossRef]
- Stloukal, P.; Pekařová, S.; Kalendova, A.; Mattausch, H.; Laske, S.; Holzer, C.; Chitu, L.; Bodner, S.; Maier, G.; Slouf, M.; et al. Kinetics and mechanism of the biodegradation of PLA/clay nanocomposites during thermophilic phase of composting process. Waste Manag. 2015, 42, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Xie, Y.-N.; Yang, J.-H.; Huang, T.; Zhang, N.; Wang, Y.; Zhang, J.-H. Graphene oxide induced hydrolytic degradation behavior changes of poly(l-lactide) in different mediums. Polym. Test. 2016, 56, 220–228. [Google Scholar] [CrossRef]
- Anžlovar, A.; Kržan, A.; Žagar, E. Degradation of PLA/ZnO and PHBV/ZnO composites prepared by melt processing. Arab. J. Chem. 2018, 11, 343–352. [Google Scholar] [CrossRef]
- Sun, X.; Xue, B.; Yang, S.; Huo, K.; Liao, X.; Li, X.; Xie, L.; Qin, S.; Zheng, Q. Structural conversion of PLLA/ZnO composites facilitated by interfacial crystallization to potential application in oil-water separation. Appl. Surface Sci. 2020, 517, 146135. [Google Scholar] [CrossRef]
- Wang, X.-J.; Huang, Z.; Wei, M.-Y.; Lu, T.; Nong, D.-D.; Zhao, J.-X.; Gao, X.-Y.; Teng, L.-J. Catalytic effect of nanosized ZnO and TiO2 on thermal degradation of poly(lactic acid) and isoconversional kinetic analysis. Thermochim. Acta 2019, 672, 14–24. [Google Scholar] [CrossRef]
- Nakayama, N.; Hayashi, T. Preparation and characterization of poly(l-lactic acid)/TiO2 nanoparticle nanocomposite films with high transparency and efficient photodegradability. Polym. Degrad. Stab. 2007, 92, 1255–1264. [Google Scholar] [CrossRef]
- Kaseem, M.; Hamad, K.; Rehman, Z.U. Review of Recent Advances in Polylactic Acid/TiO2 Composites. Materials 2019, 12, 3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Lin, Z.; Guo, G. Biodegradation Assessment of Poly (Lactic Acid) Filled with Functionalized Titania Nanoparticles (PLA/TiO2) under Compost Conditions. Nanoscale Res. Lett. 2019, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.-B.; Wang, X.-L.; Wang, Y.-Z. Effect of TiO2 nanoparticles on the long-term hydrolytic degradation behavior of PLA. Polym. Degrad. Stab. 2012, 97, 721–728. [Google Scholar] [CrossRef]
- Fan, Y.; Nishida, H.; Mori, T.; Shirai, Y.; Endo, T. Thermal degradation of poly(l-lactide): Effect of alkali earth metal oxides for selective l,l-lactide formation. Polymer 2004, 45, 1197–1205. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, Y.; Gu, J.; Tan, H. Biodegradation behavior and modelling of soil burial effect on degradation rate of PLA blended with starch and wood flour. Colloids Surfaces B Biointerfaces 2017, 159, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, C.A.; Tofanello, A.; Nantes, I.L.; Rosa, D.S. Biological Oxidative Mechanisms for Degradation of Poly(lactic acid) Blended with Thermoplastic Starch. ACS Sustain. Chem. Eng. 2015, 3, 2756–2766. [Google Scholar] [CrossRef]
- Petinakis, E.; Liu, X.; Yu, L.; Way, C.; Sangwan, P.; Dean, K.; Bateman, S.; Edward, G. Biodegradation and thermal decomposition of poly(lactic acid)-based materials reinforced by hydrophilic fillers. Polym. Degrad. Stab. 2010, 95, 1704–1707. [Google Scholar] [CrossRef]
- Zuo, Y.F.; Gu, J.; Qiao, Z.; Tan, H.; Cao, J.; Zhang, Y. Effects of dry method esterification of starch on the degradation characteristics of starch/polylactic acid composites. Int. J. Biol. Macromol. 2015, 72, 391–402. [Google Scholar] [CrossRef]
- Pradhan, R.; Misra, M.; Erickson, L.; Mohanty, A. Compostability and biodegradation study of PLA–wheat straw and PLA–soy straw based green composites in simulated composting bioreactor. Bioresour. Technol. 2010, 101, 8489–8491. [Google Scholar] [CrossRef] [PubMed]
- Iovino, R.; Zullo, R.; Rao, M.; Cassar, L.; Gianfreda, L. Biodegradation of poly(lactic acid)/starch/coir biocomposites under controlled composting conditions. Polym. Degrad. Stab. 2008, 93, 147–157. [Google Scholar] [CrossRef]
- Shinoda, H.; Asou, Y.; Kashima, T.; Kato, T.; Tseng, Y.; Yagi, T. Amphiphilic biodegradable copolymer, poly(aspartic acid-co-lactide): Acceleration of degradation rate and improvement of thermal stability for poly(lactic acid), poly(butylene succinate) and poly(ε-caprolactone). Polym. Degrad. Stab. 2003, 80, 241–250. [Google Scholar] [CrossRef]
- Höglund, A.; Hakkarainen, M.; Edlund, U.; Albertsson, A.-C. Surface Modification Changes the Degradation Process and Degradation Product Pattern of Polylactide. Langmuir 2010, 26, 378–383. [Google Scholar] [CrossRef]
Figure 1.
Chemical structure of lactic acid stereoisomers: L-(+)-lactic acid is also known as (S)-lactic acid and D-(−)-Lactic acid, also known as (R)-lactic acid, adapted from [4].
Figure 1.
Chemical structure of lactic acid stereoisomers: L-(+)-lactic acid is also known as (S)-lactic acid and D-(−)-Lactic acid, also known as (R)-lactic acid, adapted from [4].

Figure 2.
Available routes to obtain PLA, ring-opening polymerization (ROP), and direct polymerization (DP). Polylactide is a synonym for poly(lactic)acid (PLA), and the names can be used interchangeably.
Figure 2.
Available routes to obtain PLA, ring-opening polymerization (ROP), and direct polymerization (DP). Polylactide is a synonym for poly(lactic)acid (PLA), and the names can be used interchangeably.

Figure 3.
The stereoisomers of lactic acid and the resulting lactides, together with their optical activity adapted from [9].
Figure 3.
The stereoisomers of lactic acid and the resulting lactides, together with their optical activity adapted from [9].

Figure 4.
Metastable phases of high Mw amorphous PLA adapted from [55].
Figure 4.
Metastable phases of high Mw amorphous PLA adapted from [55].

Figure 5.
Scheme of nucleophilic substitution reaction, adapted from [109].
Figure 5.
Scheme of nucleophilic substitution reaction, adapted from [109].

Figure 7.
Effect of residual catalyst presence on PLA thermal degradation [147]. Reproduced with permission from Elsevier, 2021.
Figure 7.
Effect of residual catalyst presence on PLA thermal degradation [147]. Reproduced with permission from Elsevier, 2021.

Figure 8.
Excited states of PLA molecule adapted from [159] (a) the excited state of the C=O group via n-pi* transition exhibits an antibonding and biradical property, (b) photoexcitation of C=O tends to cause some chemical reactions, such as bond cleavage or (c) radical addition, electron abstraction or electron transfer.
Figure 8.
Excited states of PLA molecule adapted from [159] (a) the excited state of the C=O group via n-pi* transition exhibits an antibonding and biradical property, (b) photoexcitation of C=O tends to cause some chemical reactions, such as bond cleavage or (c) radical addition, electron abstraction or electron transfer.

Figure 9.
Mc Neil and Leiper mechanism adapted from [157].
Figure 9.
Mc Neil and Leiper mechanism adapted from [157].

Figure 10.
Scheme of photodegradation via Norish II mechanism adapted from [4].
Figure 10.
Scheme of photodegradation via Norish II mechanism adapted from [4].

Figure 11.
Basic mechanisms proposed to predict the degradation products of PLA adapted from [161].
Figure 11.
Basic mechanisms proposed to predict the degradation products of PLA adapted from [161].

Figure 12.
Mechanism of enzymatic biodegradation of plastics under aerobic conditions, adapted from [173].
Figure 12.
Mechanism of enzymatic biodegradation of plastics under aerobic conditions, adapted from [173].

Figure 13.
General steps involved in biodegradation of a PLA bottle under composting conditions.


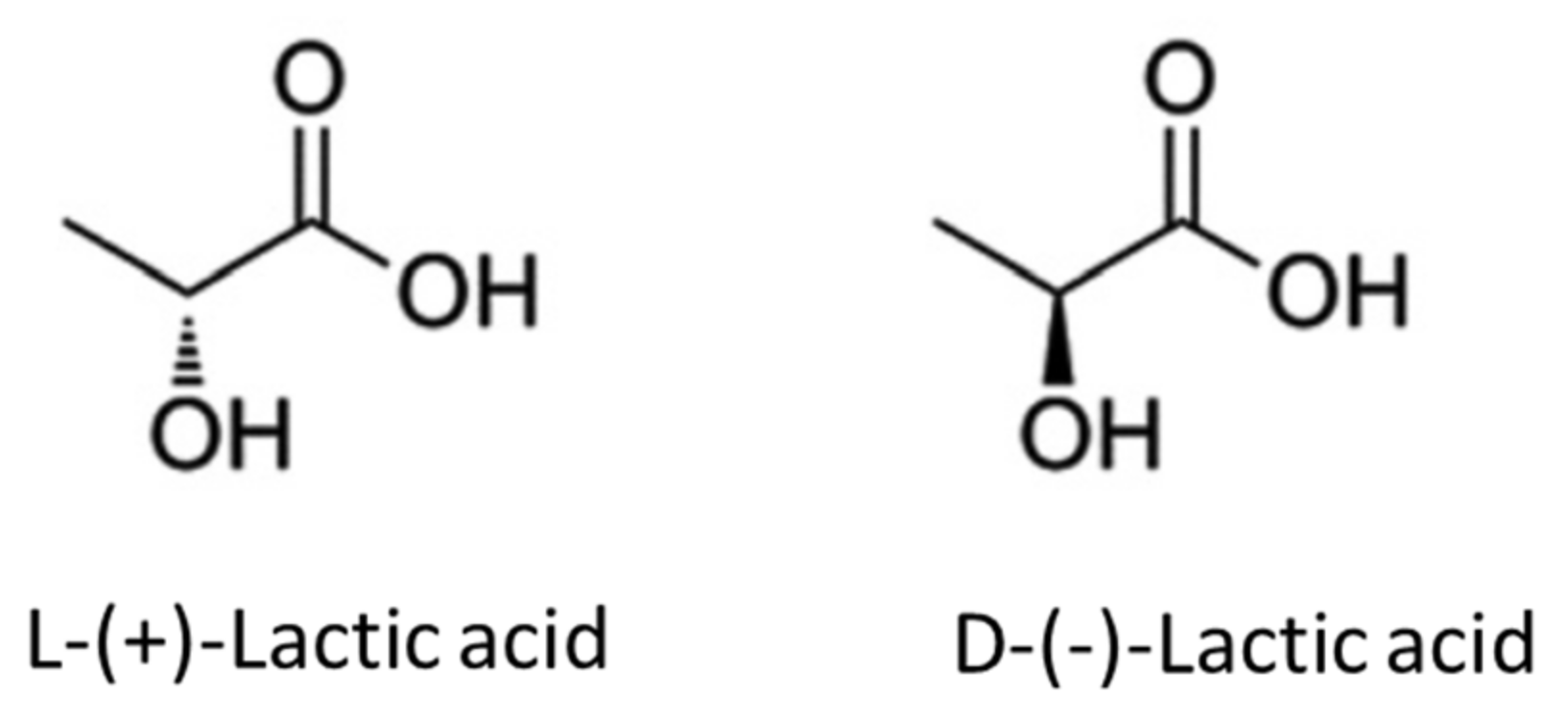{kind=link}
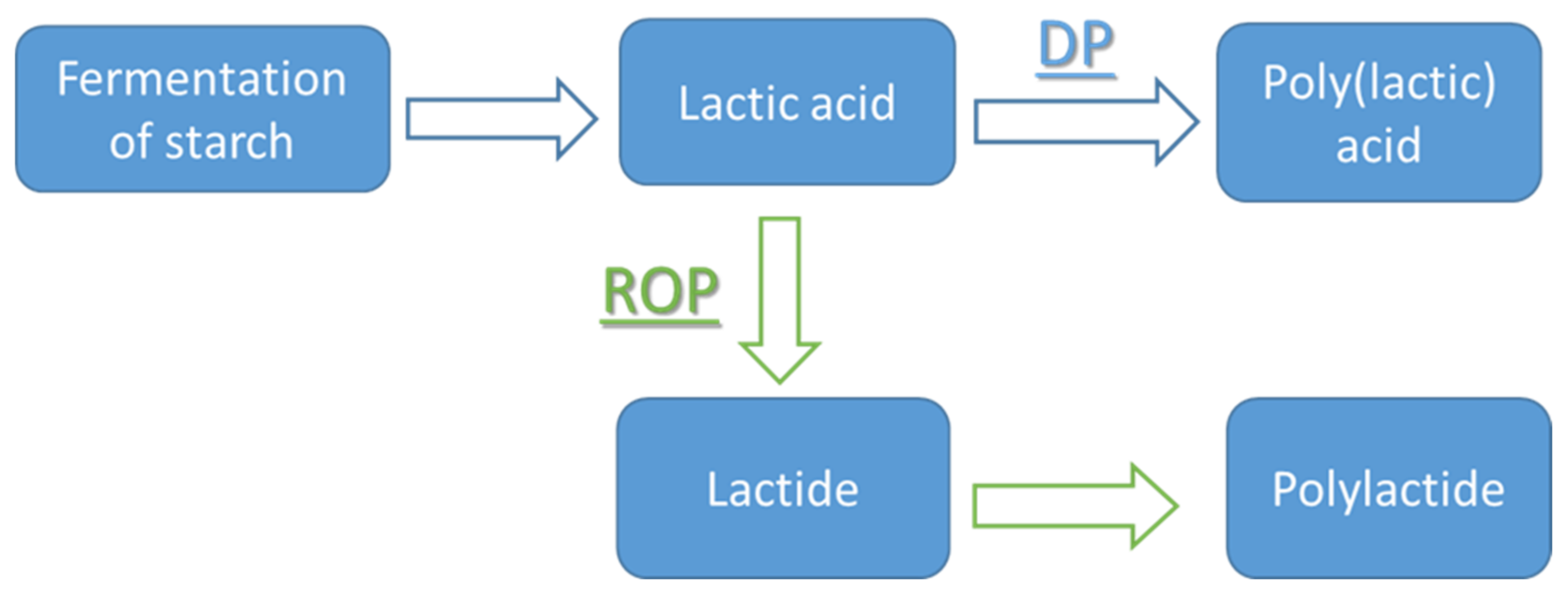{kind=link}
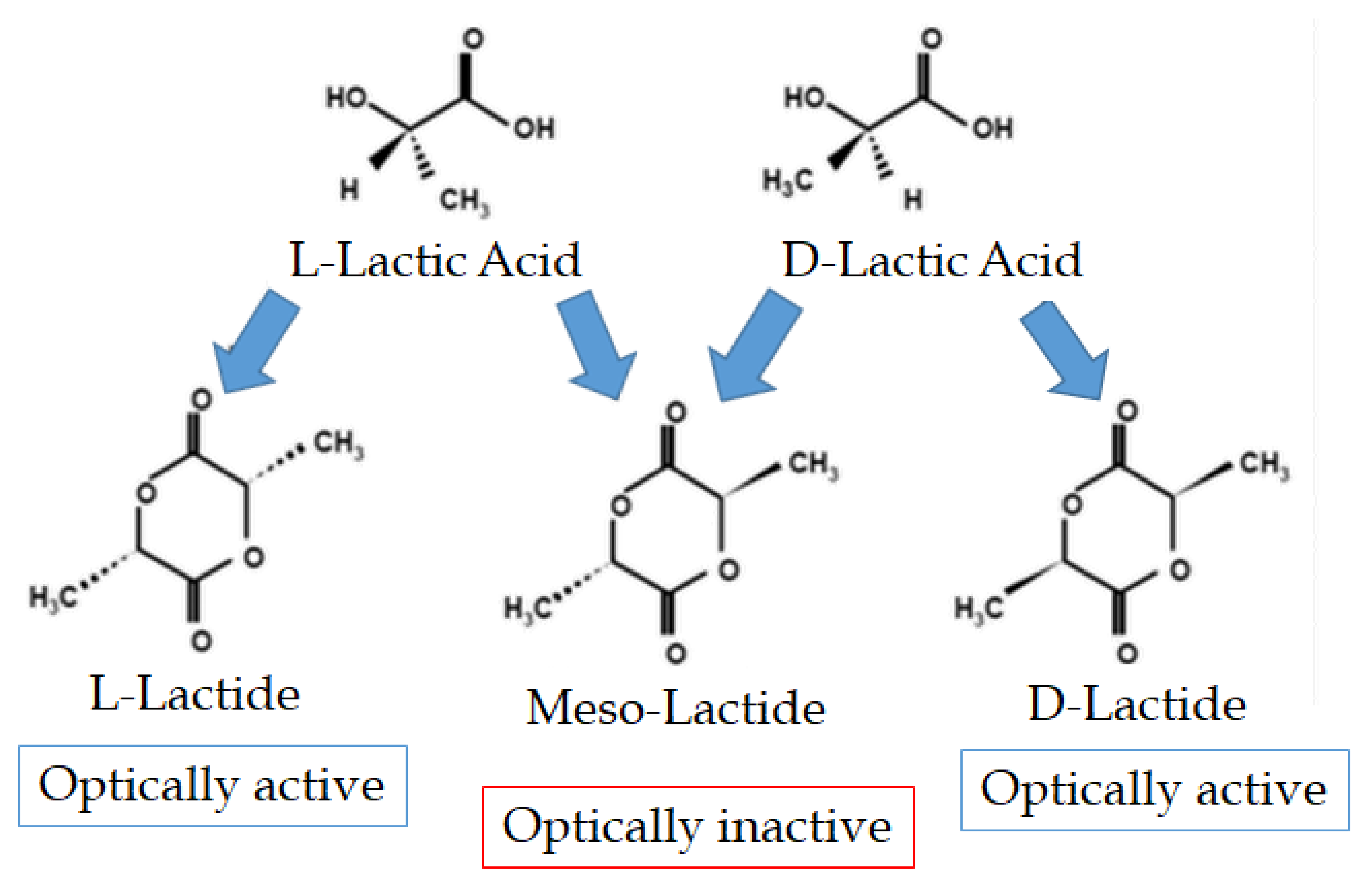{kind=link}
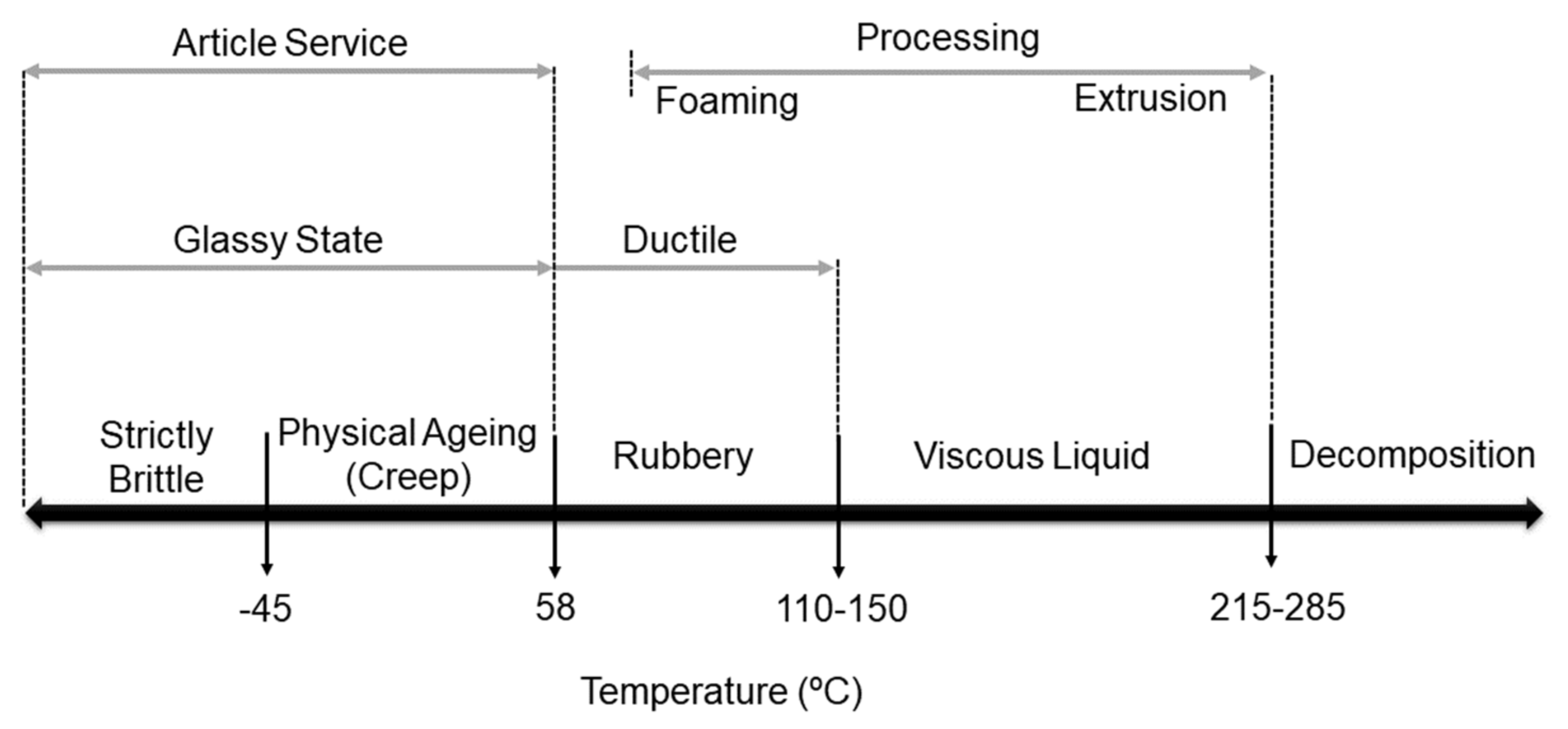{kind=link}
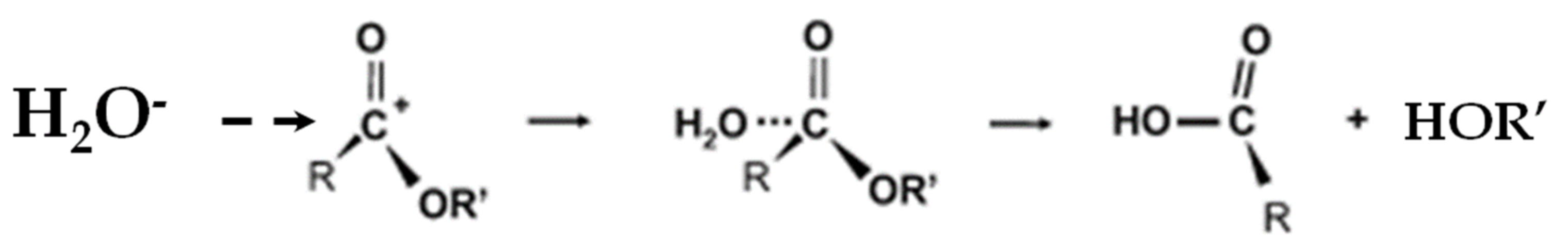{kind=link}
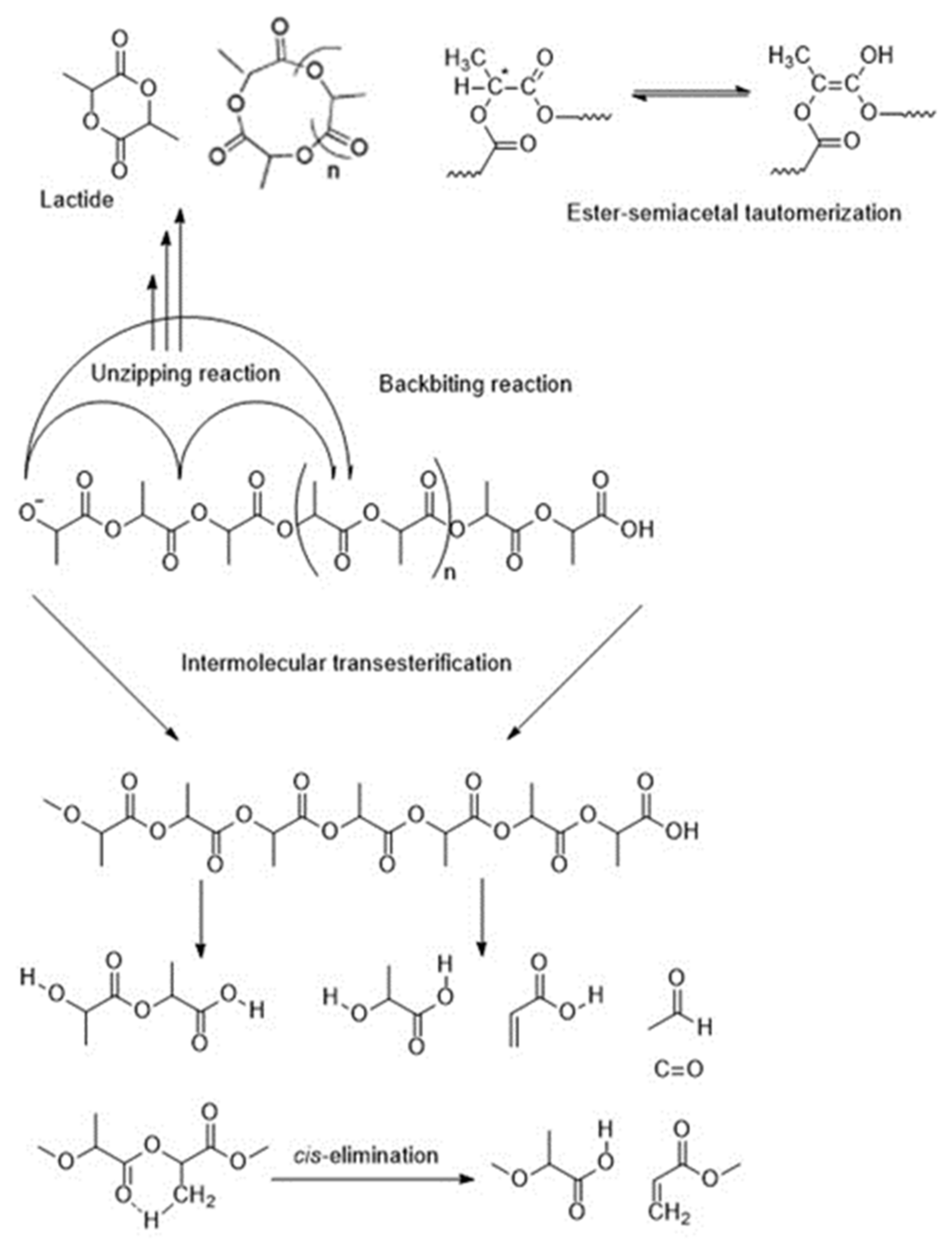{kind=link}
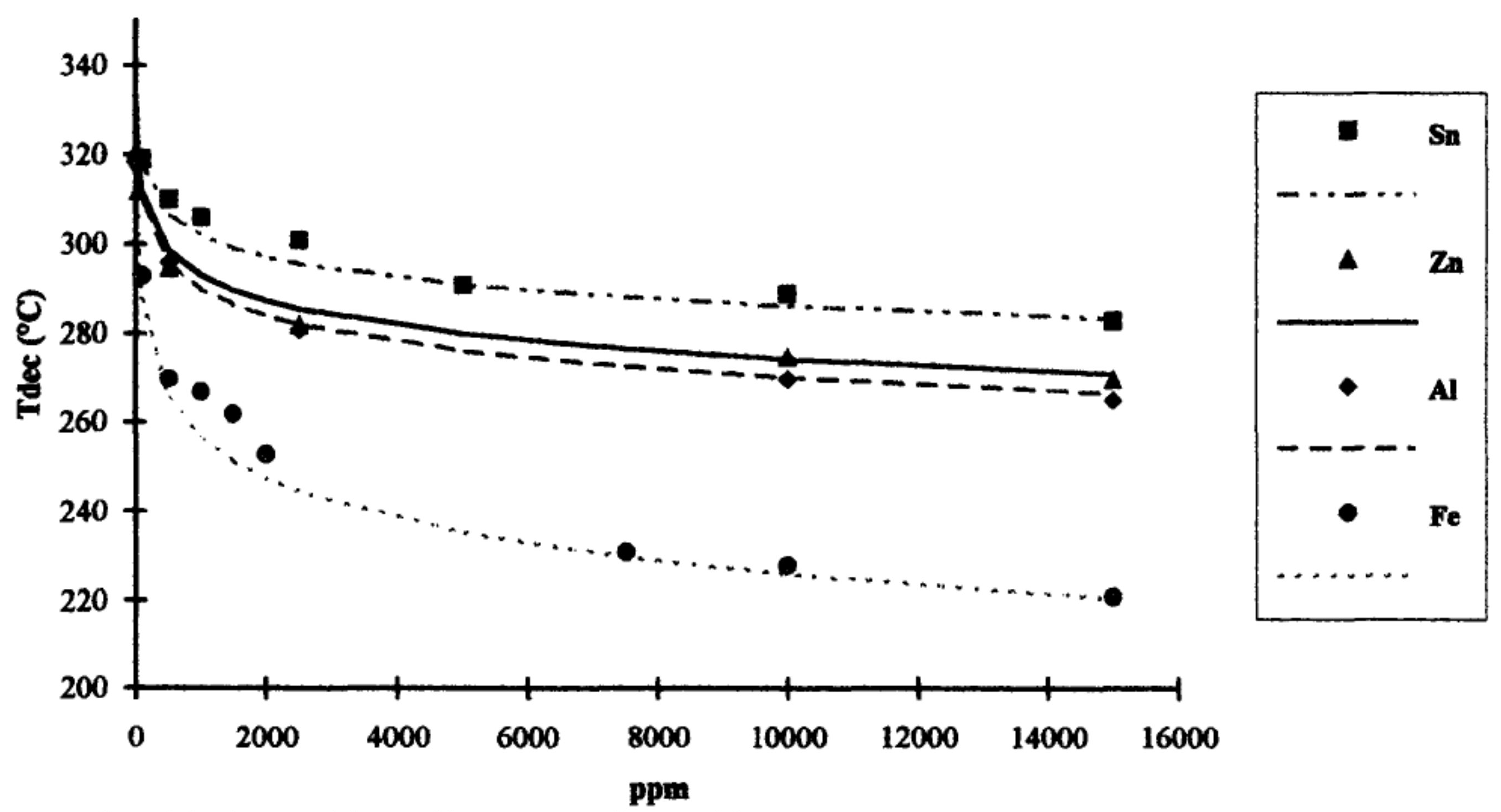{kind=link}
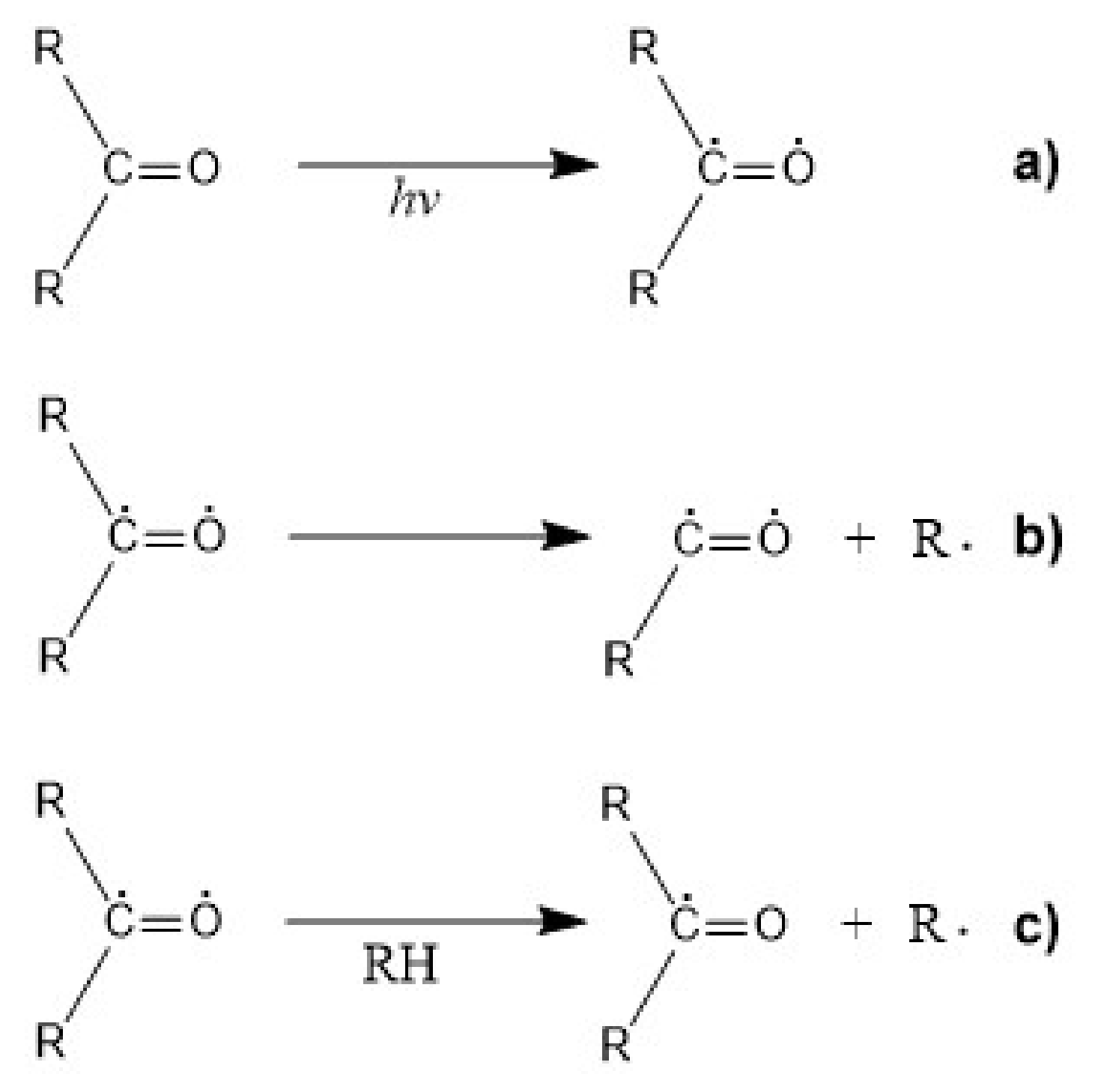{kind=link}
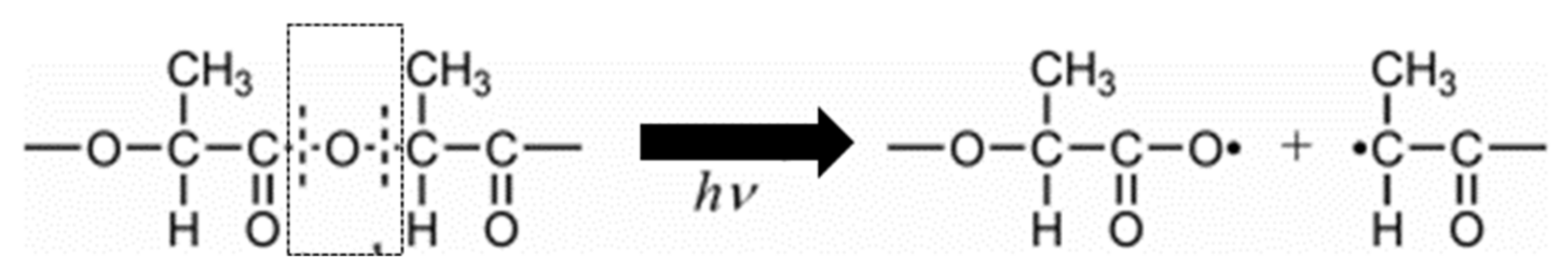{kind=link}
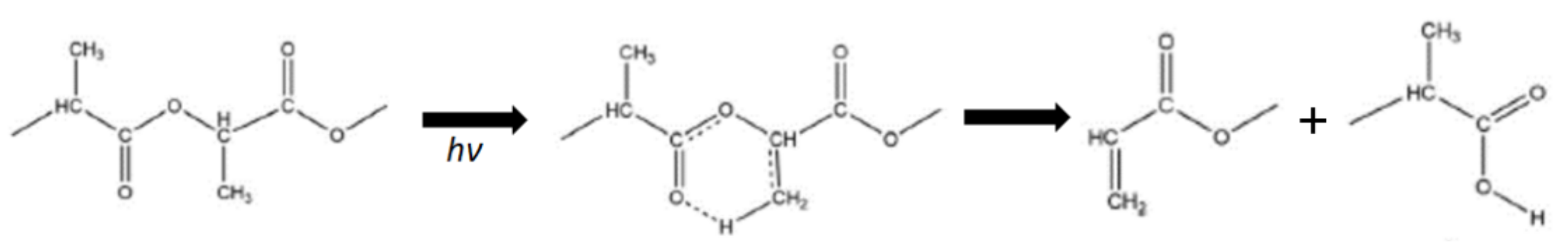{kind=link}
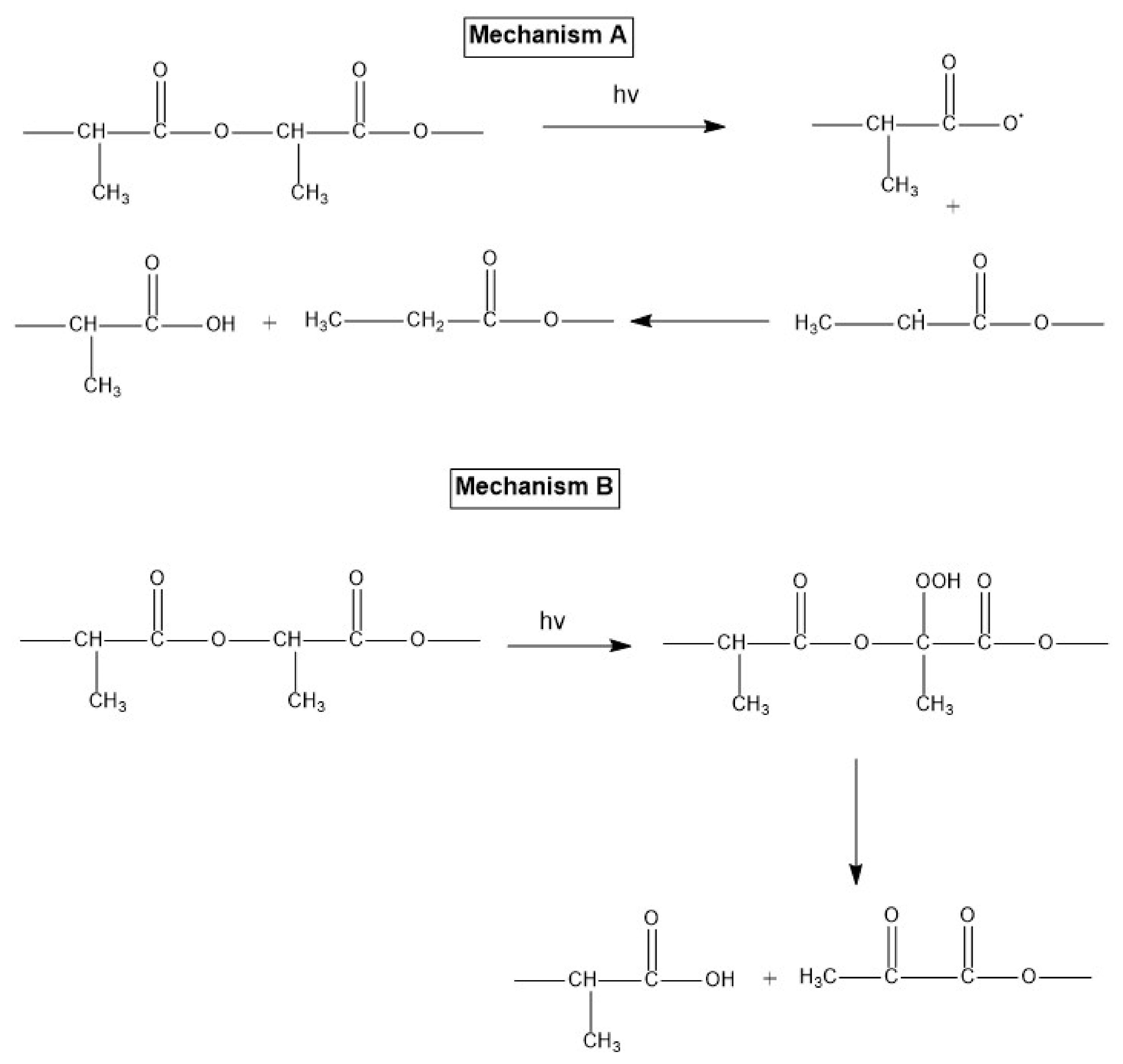{kind=link}
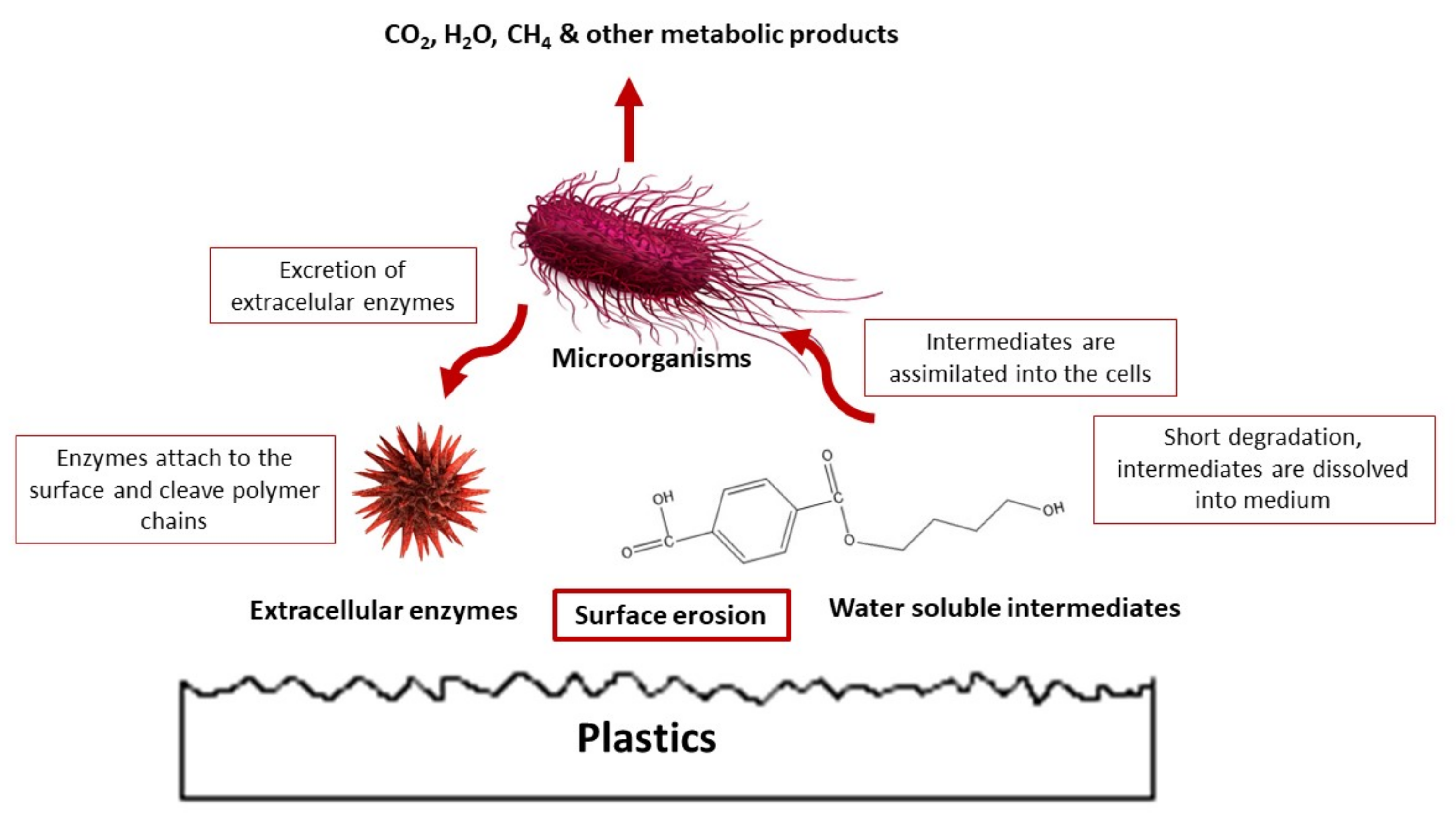{kind=link}
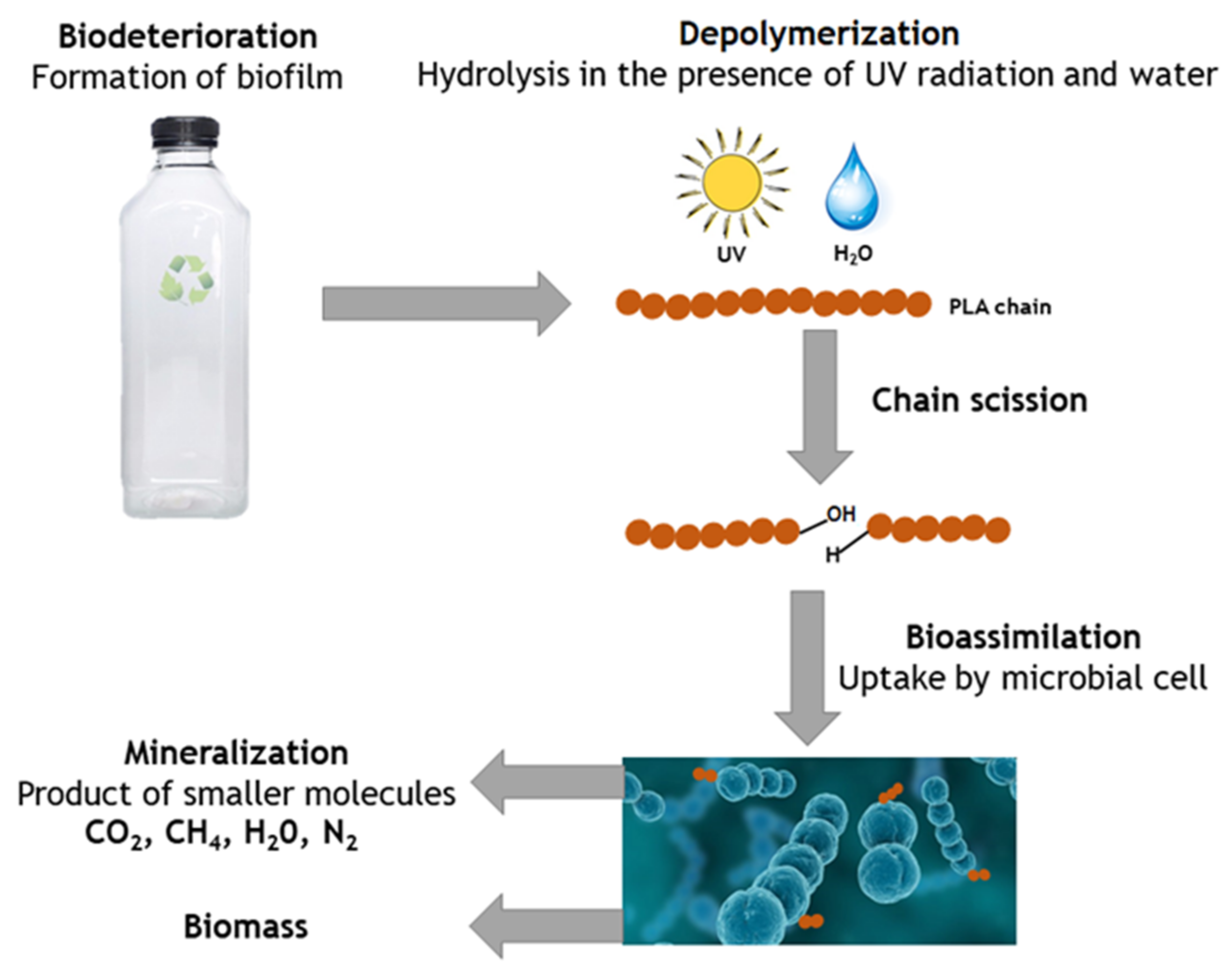{kind=link}
Table 1.
Microorganisms and enzymes degrading PLA with the temperature of the mineralization process [41].
Table 1.
Microorganisms and enzymes degrading PLA with the temperature of the mineralization process [41].
| Microorganism | Representative Species | Enzyme | Temperature of Digestion (°C) |
|---|---|---|---|
| Actinomycetes | Amycolotopsis strain K104-1 | Protease | 55–60 |
| Amycolotopsis strain 41 | 37–45 | ||
| Amycolotopsis strain orientalis | 30 | ||
| Actinomadura strain T16-1 | 70 | ||
| Bacteria | Bacillus smithii strain PL21 | Esterase | 60 |
| Alcanivorax borkumenesis ABO2449 | 30–37 | ||
| Rhodopseudomonas palustris RPA1511 | 55–60 | ||
| Paenibacillus amylolyticus strain TB-13 | Lipase | 50 | |
| Alcaligenes sp. | 55 | ||
| Pseudomonas tamsuii TKU015 | 60 | ||
| Fungus | Tritirachium album ATCC 22563 | Protease | 37 |
| Cryptococcus sp. strain S-2 | Cutinase | 37 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Teixeira, S.; Eblagon, K.M.; Miranda, F.; R. Pereira, M.F.; Figueiredo, J.L. Towards Controlled Degradation of Poly(lactic) Acid in Technical Applications. C 2021, 7, 42. https://0-doi-org.brum.beds.ac.uk/10.3390/c7020042
AMA Style
Teixeira S, Eblagon KM, Miranda F, R. Pereira MF, Figueiredo JL. Towards Controlled Degradation of Poly(lactic) Acid in Technical Applications. C. 2021; 7(2):42. https://0-doi-org.brum.beds.ac.uk/10.3390/c7020042
Chicago/Turabian StyleTeixeira, Stefanie, Katarzyna Morawa Eblagon, Filipa Miranda, M. Fernando R. Pereira, and José Luis Figueiredo. 2021. "Towards Controlled Degradation of Poly(lactic) Acid in Technical Applications" C 7, no. 2: 42. https://0-doi-org.brum.beds.ac.uk/10.3390/c7020042
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.