
Facile Synthesis and Characterization of a Bromine-Substituted (Chloromethyl)Pyridine Precursor towards the Immobilization of Biomimetic Metal Ion Chelates on Functionalized Carbons

 , and
, and
Abstract
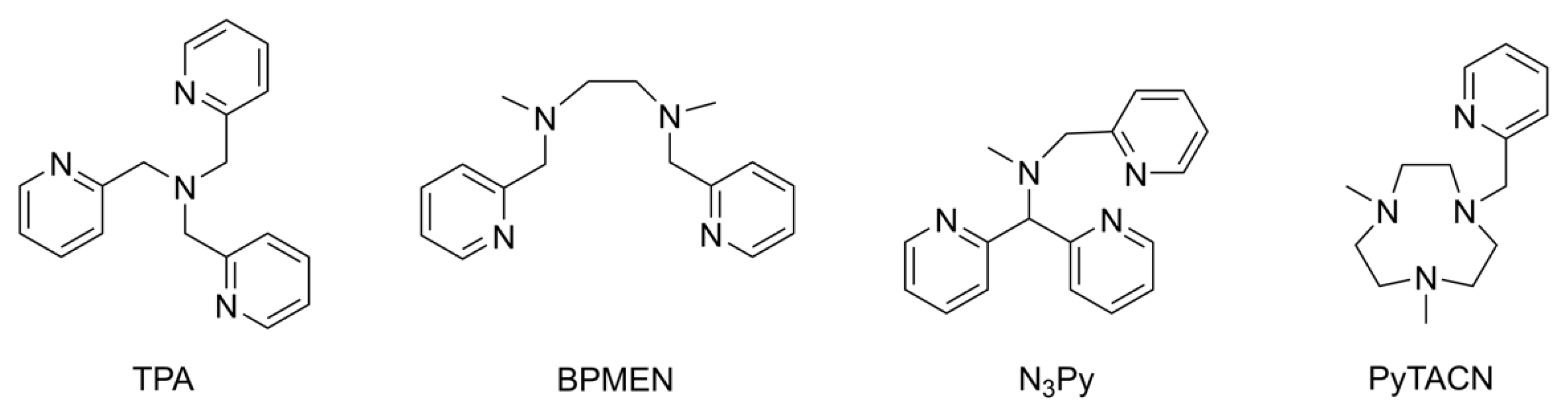
:1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Materials
2.3. Synthetic Procedures
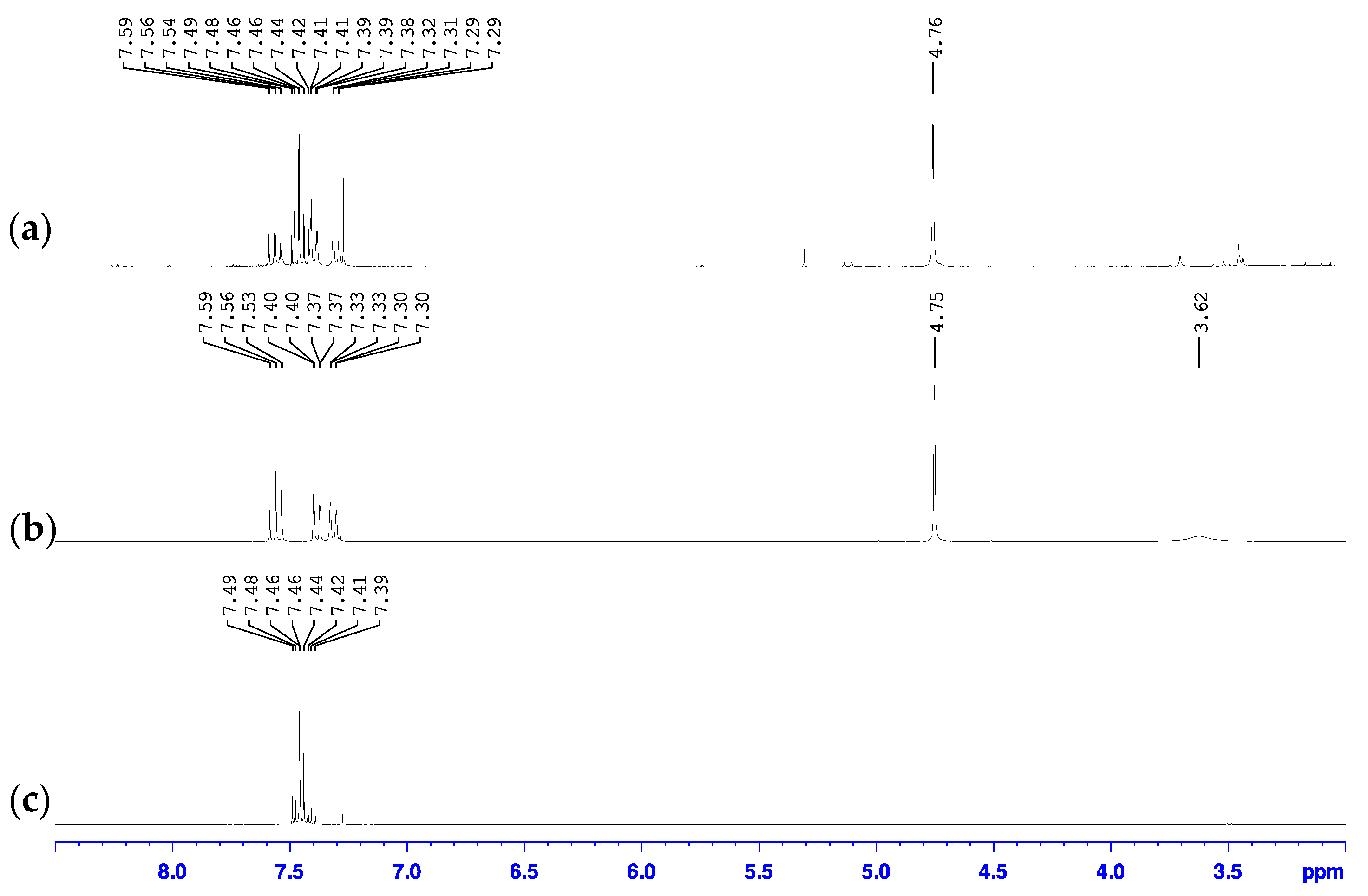
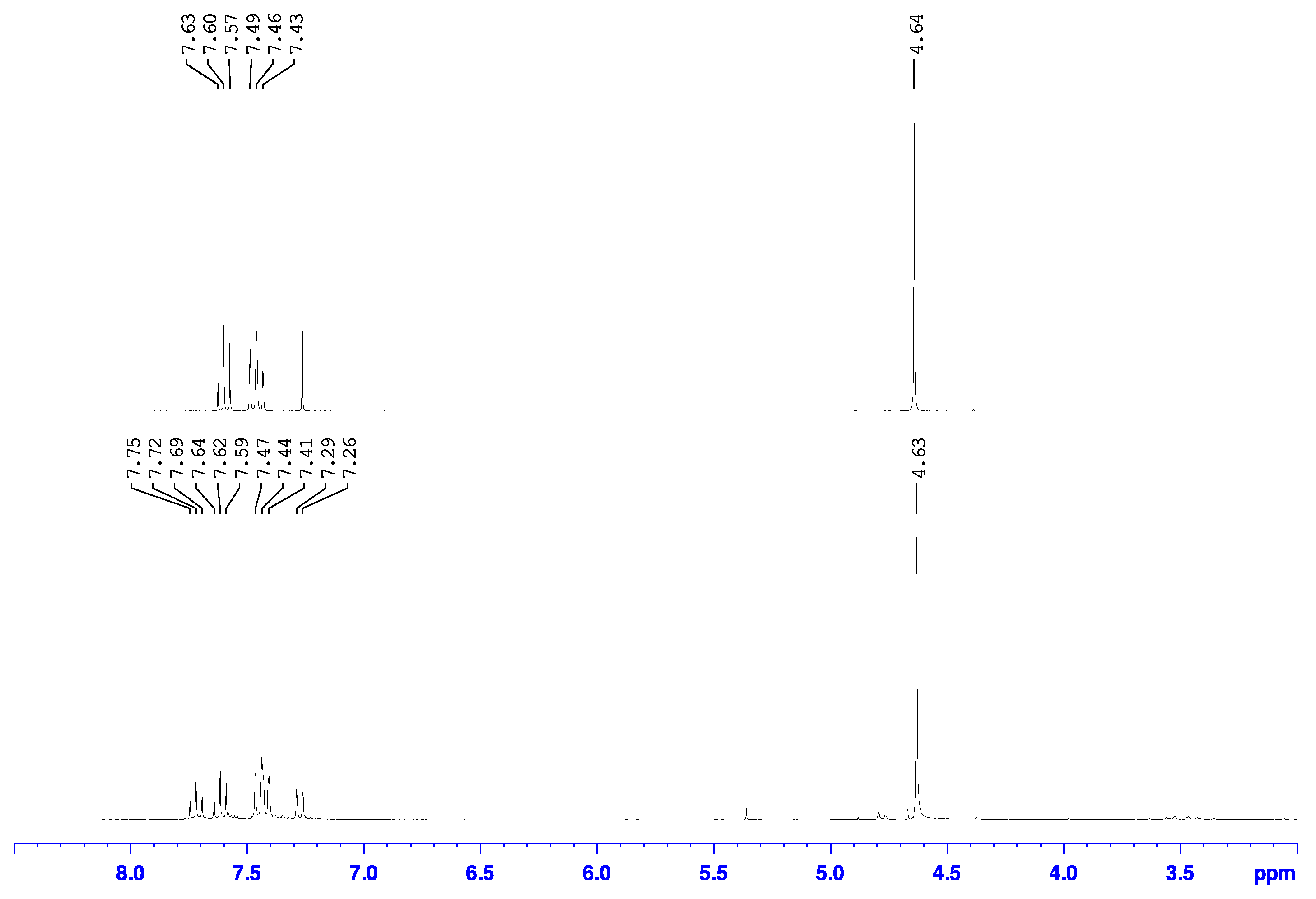
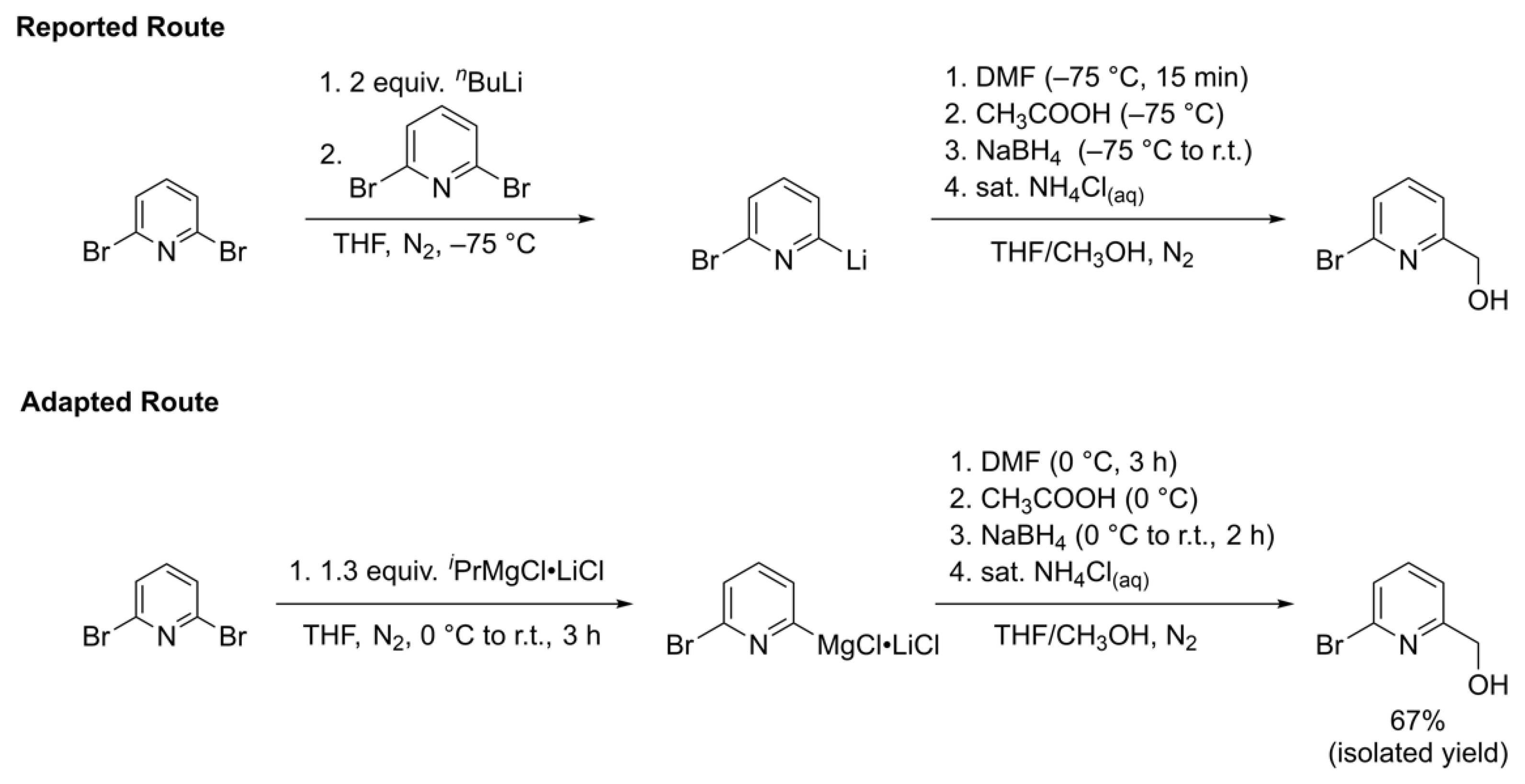
2.3.1. 2-bromo-6-hydroxymethylpyridine
2.3.2. 2-bromo-6-chloromethylpyridine
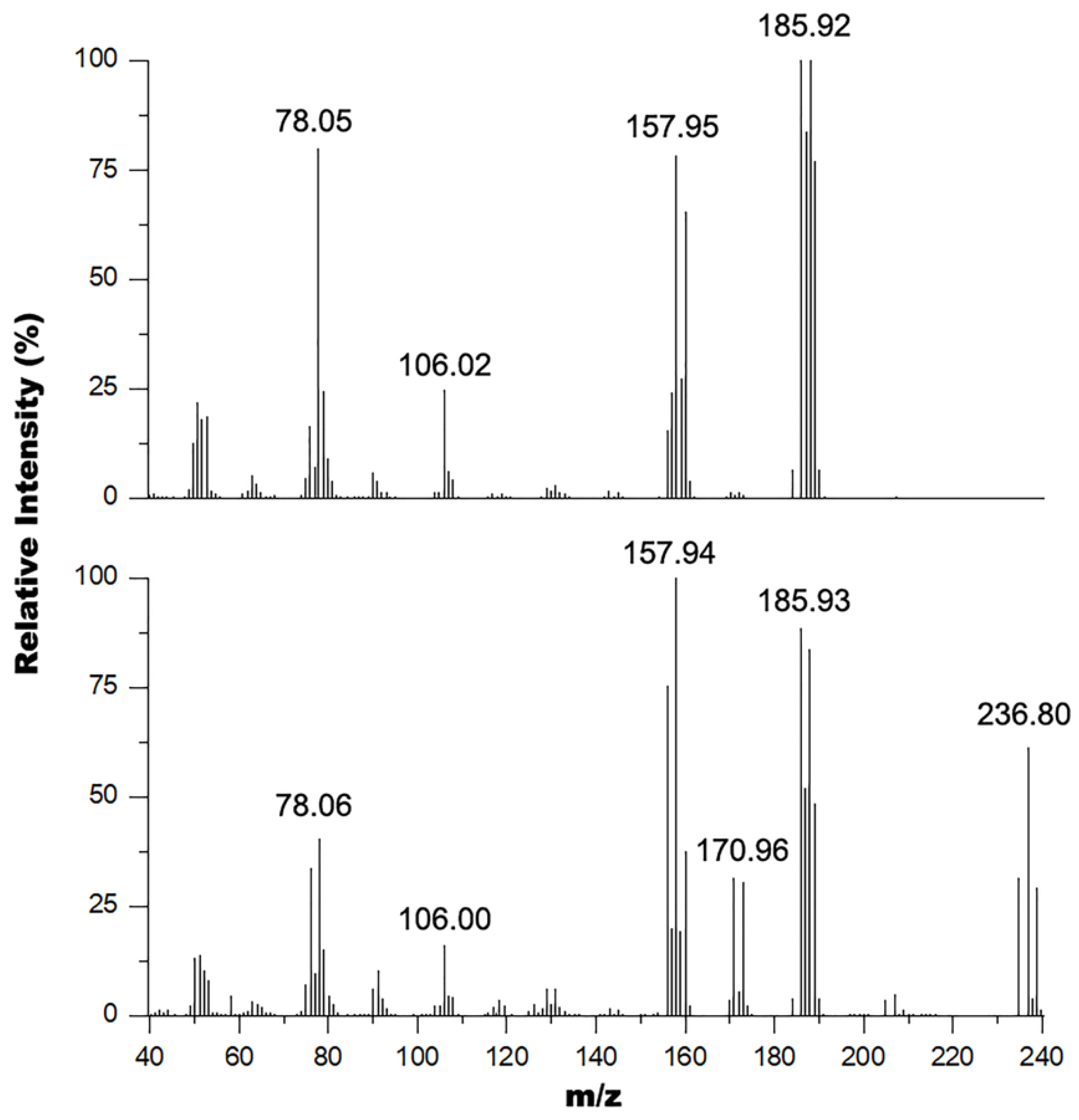
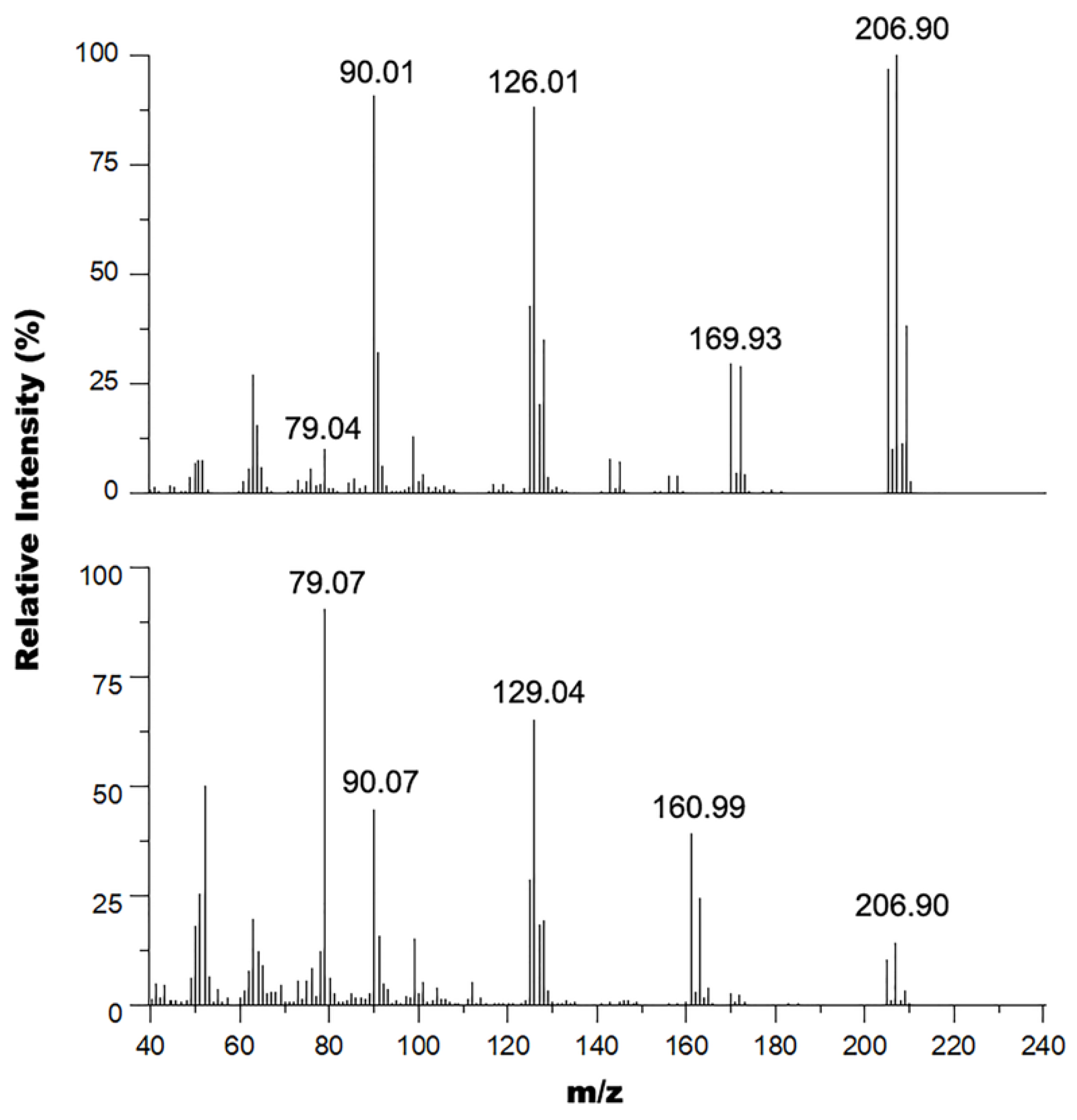
2.4. GC-MS
3. Results and Discussion
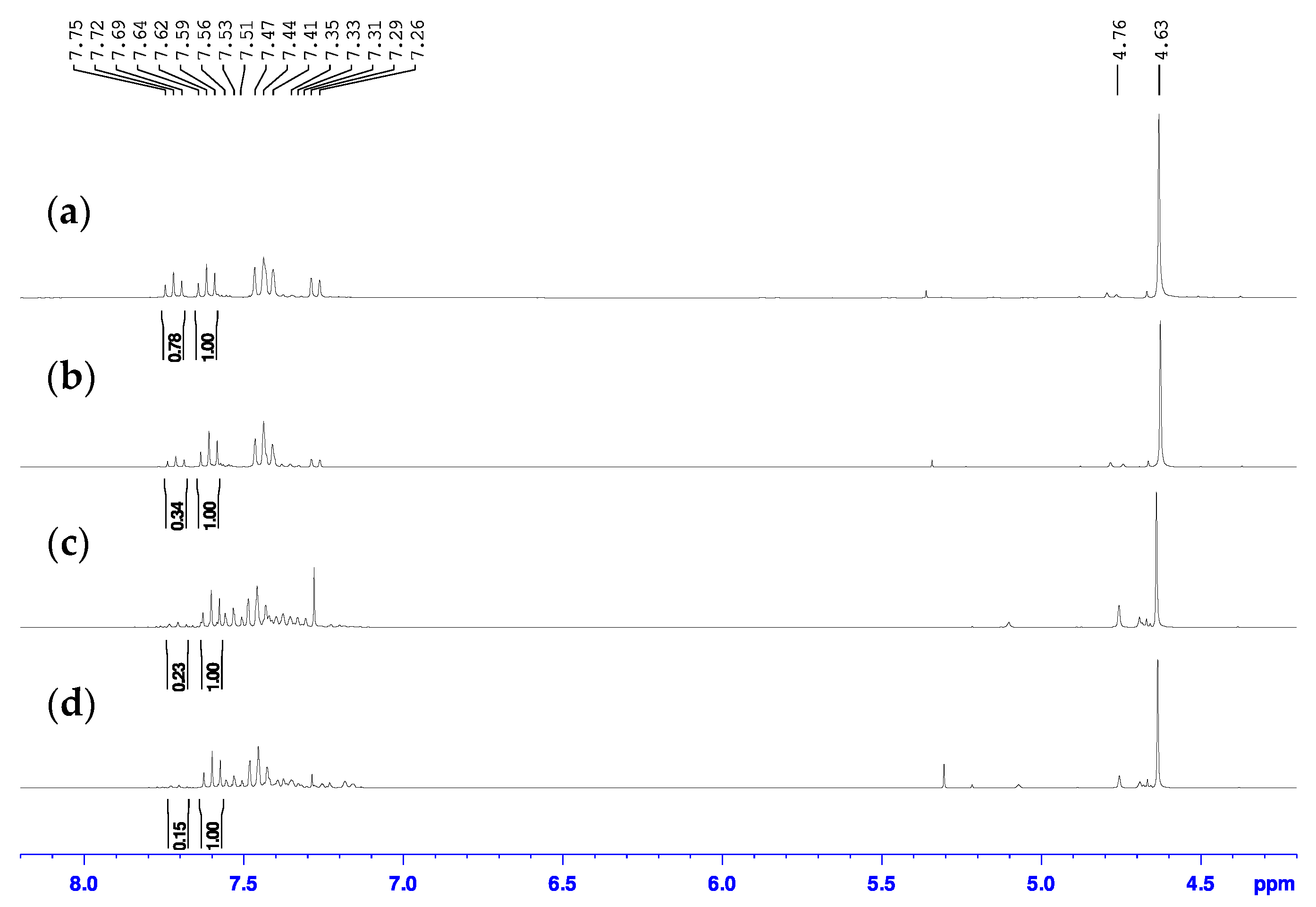
3.1. Metal Halogen Exchange using Turbo Grignard
3.2. Chlorine Transfer Using Cyanuric Chloride versus Thionyl Chloride
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, W.; Sun, Q. Bioinspired Manganese and Iron Complexes for Enantioselective Oxidation Reactions: Ligand Design, Catalytic Activity, and Beyond. Acc. Chem. Res. 2019, 52, 2370–2381. [Google Scholar] [CrossRef]
- Costas, M.; Tipton, A.K.; Chen, K.; Jo, D.-H.; Que, L. Modeling Rieske Dioxygenases: The First Example of Iron-Catalyzed Asymmetric Cis-Dihydroxylation of Olefins. J. Am. Chem. Soc. 2001, 123, 6722–6723. [Google Scholar] [CrossRef]
- White, M.C.; Doyle, A.G.; Jacobsen, E.N. A Synthetically Useful, Self-Assembling MMO Mimic System for Catalytic Alkene Epoxidation with Aqueous H2O2. J. Am. Chem. Soc. 2001, 123, 7194–7195. [Google Scholar] [CrossRef]
- Oddon, F.; Girgenti, E.; Lebrun, C.; Marchi-Delapierre, C.; Pécaut, J.; Ménage, S. Iron Coordination Chemistry of N2Py2 Ligands Substituted by Carboxylic Moieties and Their Impact on Alkene Oxidation Catalysis. Eur. J. Inorg. Chem. 2012, 2012, 85–96. [Google Scholar] [CrossRef]
- Chen, M.S.; White, M.C. A Predictably Selective Aliphatic C–H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [Google Scholar] [CrossRef]
- Gómez, L.; Garcia-Bosch, I.; Company, A.; Benet-Buchholz, J.; Polo, A.; Sala, X.; Ribas, X.; Costas, M. Stereospecific C-H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem. Int. Ed. 2009, 48, 5720–5723. [Google Scholar] [CrossRef]
- Bukowski, M.R.; Comba, P.; Lienke, A.; Limberg, C.; de Laorden, C.L.; Mas-Ballesté, R.; Merz, M.; Que, L., Jr. Catalytic Epoxidation and 1,2-Dihydroxylation of Olefins with Bispidine–Iron(II)/H2O2 Systems. Angew. Chem. Int. Ed. 2006, 45, 3446–3449. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef] [PubMed]
- Tshuva, E.Y.; Lippard, S.J. Synthetic Models for Non-Heme Carboxylate-Bridged Diiron Metalloproteins: Strategies and Tactics. Chem. Rev. 2004, 104, 987–1012. [Google Scholar] [CrossRef]
- Keown, W.; Gary, J.B.; Stack, T.D.P. High-Valent Copper in Biomimetic and Biological Oxidations. JBIC J. Biol. Inorg. Chem. 2017, 22, 289–305. [Google Scholar] [CrossRef]
- Itoh, S.; Abe, T.; Morimoto, Y.; Sugimoto, H. 2-(2-Pyridyl)Ethylamine (Pye) Ligands in Copper(I)-Dioxygen Chemistry. Inorg. Chim. Acta 2018, 481, 38–46. [Google Scholar] [CrossRef]
- Karlin, K.D.; Hayes, J.C.; Gultneh, Y.; Cruse, R.W.; McKown, J.W.; Hutchinson, J.P.; Zubieta, J. Copper-Mediated Hydroxylation of an Arene: Model System for the Action of Copper Monooxygenases. Structures of a Binuclear Copper(I) Complex and Its Oxygenated Product. J. Am. Chem. Soc. 1984, 106, 2121–2128. [Google Scholar] [CrossRef]
- Karlin, K.D.; Haka, M.S.; Cruse, R.W.; Meyer, G.J.; Farooq, A.; Gultneh, Y.; Hayes, J.C.; Zubieta, J. Dioxygen-Copper Reactivity Models for Hemocyanin: Reversible O2 and CO Binding to Structurally Characterized Dicopper(I) Complexes Containing Hydrocarbon-Linked Bis[2-(2-Pyridyl)Ethyl]Amine Units. J. Am. Chem. Soc. 1988, 110, 1196–1207. [Google Scholar] [CrossRef]
- Koehntop, K.D.; Emerson, J.P.; Que, L. The 2-His-1-Carboxylate Facial Triad: A Versatile Platform for Dioxygen Activation by Mononuclear Non-Heme Iron(II) Enzymes. JBIC J. Biol. Inorg. Chem. 2005, 10, 87–93. [Google Scholar] [CrossRef]
- Diebold, A.R.; Neidig, M.L.; Moran, G.R.; Straganz, G.D.; Solomon, E.I. The Three-His Triad in Dke1: Comparisons to the Classical Facial Triad. Biochemistry 2010, 49, 6945–6952. [Google Scholar] [CrossRef] [Green Version]
- Monkcom, E.C.; de Bruin, D.; de Vries, A.J.; Lutz, M.; Ye, S.; Gebbink, R.J.M.K. Structurally Modelling the 2-His-1-Carboxylate Facial Triad with a Bulky N,N,O Phenolate Ligand. Chem. Eur. J. 2021, 27, 5191–5204. [Google Scholar] [CrossRef]
- Monkcom, E.C.; Ghosh, P.; Folkertsma, E.; Negenman, H.A.; Lutz, M.; Gebbink, R.J.M.K. Bioinspired Non-Heme Iron Complexes: The Evolution of Facial N, N, O Ligand Design. Chim. Int. J. Chem. 2020, 74, 450–466. [Google Scholar] [CrossRef]
- Lindhorst, A.C.; Haslinger, S.; Kühn, F.E. Molecular Iron Complexes as Catalysts for Selective C–H Bond Oxygenation Reactions. Chem. Commun. 2015, 51, 17193–17212. [Google Scholar] [CrossRef] [Green Version]
- Rohde, J.-U.; Stubna, A.; Bominaar, E.L.; Münck, E.; Nam, W.; Que, L. Nonheme Oxoiron(IV) Complexes of Tris(2-Pyridylmethyl)Amine with Cis-Monoanionic Ligands. Inorg. Chem. 2006, 45, 6435–6445. [Google Scholar] [CrossRef]
- Andris, E.; Jašík, J.; Gómez, L.; Costas, M.; Roithová, J. Spectroscopic Characterization and Reactivity of Triplet and Quintet Iron(IV) Oxo Complexes in the Gas Phase. Angew. Chem. Int. Ed. 2016, 55, 3637–3641. [Google Scholar] [CrossRef] [Green Version]
- Dantignana, V.; Serrano-Plana, J.; Draksharapu, A.; Magallón, C.; Banerjee, S.; Fan, R.; Gamba, I.; Guo, Y.; Que, L.; Costas, M.; et al. Spectroscopic and Reactivity Comparisons between Nonheme Oxoiron(IV) and Oxoiron(V) Species Bearing the Same Ancillary Ligand. J. Am. Chem. Soc. 2019, 141, 15078–15091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lutz, M.; Milan, M.; Costas, M.; Otte, M.; Gebbink, R.J.M.K. Non-Heme Iron Catalysts with a Rigid Bis-Isoindoline Backbone and Their Use in Selective Aliphatic C−H Oxidation. Adv. Synth. Catal. 2017, 359, 2590–2595. [Google Scholar] [CrossRef]
- Singh, R.; Ganguly, G.; Malinkin, S.O.; Demeshko, S.; Meyer, F.; Nordlander, E.; Paine, T.K. A Mononuclear Nonheme Iron(IV)-Oxo Complex of a Substituted N4Py Ligand: Effect of Ligand Field on Oxygen Atom Transfer and C–H Bond Cleavage Reactivity. Inorg. Chem. 2019, 58, 1862–1876. [Google Scholar] [CrossRef]
- Park, H.; Lee, D. Ligand Taxonomy for Bioinorganic Modeling of Dioxygen-Activating Non-Heme Iron Enzymes. Chem. Eur. J. 2020, 26, 5916–5926. [Google Scholar] [CrossRef] [PubMed]
- Karimov, R.R.; Kazhkenov, Z.-G.M.; Modjewski, M.J.; Peterson, E.M.; Zhdankin, V.V. Preparation and Reactivity of Polymer-Supported 2-Iodylphenol Ethers, an Efficient Recyclable Oxidizing System. J. Org. Chem. 2007, 72, 8149–8151. [Google Scholar] [CrossRef]
- McNamara, C.A.; Dixon, M.J.; Bradley, M. Recoverable Catalysts and Reagents Using Recyclable Polystyrene-Based Supports. Chem. Rev. 2002, 102, 3275–3300. [Google Scholar] [CrossRef] [PubMed]
- Radi, S.; Tighadouini, S.; Bacquet, M.; Degoutin, S.; Janus, L.; Mabkhot, Y.N. Fabrication and Covalent Modification of Highly Chelated Hybrid Material Based on Silica-Bipyridine Framework for Efficient Adsorption of Heavy Metals: Isotherms, Kinetics and Thermodynamics Studies. RSC Adv. 2016, 6, 82505–82514. [Google Scholar] [CrossRef]
- Smith, V.A.; Rivera, J.F.A.; Bello, R.; Rodríguez-Aguado, E.; Elshaer, M.R.; Wodzinski, R.L.; Bashkova, S. The Role of Surface Chemistry and Polyethylenimine Grafting in the Removal of Cr (VI) by Activated Carbons from Cashew Nut Shells. C 2021, 7, 27. [Google Scholar] [CrossRef]
- Chuang, C.-L.; Dos Santos, O.; Xu, X.; Canary, J.W. Synthesis and Cyclic Voltammetry Studies of Copper Complexes of Bromo- and Alkoxyphenyl-Substituted Derivatives of Tris(2-Pyridylmethyl)Amine: Influence of Cation-Alkoxy Interactions on Copper Redox Potentials. Inorg. Chem. 1997, 36, 1967–1972. [Google Scholar] [CrossRef]
- Tanaka, K.; Ewing, W.R.; Yu, J.-Q. Hemilabile Benzyl Ether Enables γ-C(Sp3)–H Carbonylation and Olefination of Alcohols. J. Am. Chem. Soc. 2019, 141, 15494–15497. [Google Scholar] [CrossRef]
- Kmentova, I.; Sutherland, H.S.; Palmer, B.D.; Blaser, A.; Franzblau, S.G.; Wan, B.; Wang, Y.; Ma, Z.; Denny, W.A.; Thompson, A.M. Synthesis and Structure−Activity Relationships of Aza- and Diazabiphenyl Analogues of the Antitubercular Drug (6S)-2-Nitro-6-{[4-(Trifluoromethoxy)Benzyl]Oxy}-6,7-Dihydro-5H-Imidazo[2,1-b][1,3]Oxazine (PA-824). J. Med. Chem. 2010, 53, 8421–8439. [Google Scholar] [CrossRef]
- Cai, D.; Hughes, D.L.; Verhoeven, T.R. A Study of the Lithiation of 2,6-Dibromopyridine with Butyllithium, and Its Application to Synthesis of L-739,010. Tetrahedron Lett. 1996, 37, 2537–2540. [Google Scholar] [CrossRef]
- Meth-Cohn, O.; Jiang, H. Ligands Containing Alternating 2,6-Linked Pyridine and 2,5-Linked Thiophene Units 1. J. Chem. Soc. Perkin Trans. 1998, 22, 3737–3746. [Google Scholar] [CrossRef]
- Li, X.; Gibb, C.L.D.; Kuebel, M.E.; Gibb, B.C. Two New Ligands for Carbonic Anhydrase Mimicry. Tetrahedron 2001, 57, 1175–1182. [Google Scholar] [CrossRef]
- Landa, A.; Minkkilä, A.; Blay, G.; Jørgensen, K.A. Bis(Oxazoline) Lewis Acid Catalyzed Aldol Reactions of Pyridine N-Oxide Aldehydes—Synthesis of Optically Active 2-(1-Hydroxyalkyl)Pyridine Derivatives: Development, Scope, and Total Synthesis of an Indolizine Alkaloid. Chem. Eur. J. 2006, 12, 3472–3483. [Google Scholar] [CrossRef]
- Tsukube, H.; Uenishi, J.; Higaki, H.; Kikkawa, K.; Tanaka, T.; Wakabayashi, S.; Oae, S. Side Arm Effects on Cation Binding, Extraction, and Transport Functions of Oligopyridine-Functionalized Aza-Crown Ethers. J. Org. Chem. 1993, 58, 4389–4397. [Google Scholar] [CrossRef]
- Ulrich, S.; Buhler, E.; Lehn, J.-M. Reversible Constitutional Switching between Macrocycles and Polymers Induced by Shape Change in a Dynamic Covalent System. New J. Chem. 2009, 33, 271–292. [Google Scholar] [CrossRef]
- Maindron, N.; Poupart, S.; Hamon, M.; Langlois, J.-B.; Plé, N.; Jean, L.; Romieu, A.; Renard, P.-Y. Synthesis and Luminescence Properties of New Red-Shifted Absorption Lanthanide(Iii) Chelates Suitable for Peptide and Protein Labelling. Org. Biomol. Chem. 2011, 9, 2357–2370. [Google Scholar] [CrossRef] [PubMed]
- Krasovskiy, A.; Knochel, P. A LiCl-Mediated Br/Mg Exchange Reaction for the Preparation of Functionalized Aryl- and Heteroarylmagnesium Compounds from Organic Bromides. Angew. Chem. Int. Ed. 2004, 43, 3333–3336. [Google Scholar] [CrossRef]
- De Luca, L.; Giacomelli, G.; Porcheddu, A. An Efficient Route to Alkyl Chlorides from Alcohols Using the Complex TCT/DMF. Org. Lett. 2002, 4, 553–555. [Google Scholar] [CrossRef]
- Lin, H.-S.; Paquette, L.A. A Convenient Method for Determining the Concentration of Grignard Reagents. Synth. Commun. 1994, 24, 2503–2506. [Google Scholar] [CrossRef]
- Bolm, C.; Schlingloff, G.; Harms, K. Catalyzed Enantioselective Alkylation of Aldehydes. Chem. Ber. 1992, 125, 1191–1203. [Google Scholar] [CrossRef]
- Do, H.-Q.; Daugulis, O. A Simple Base-Mediated Halogenation of Acidic Sp2 C−H Bonds under Noncryogenic Conditions. Org. Lett. 2009, 11, 421–423. [Google Scholar] [CrossRef] [Green Version]
- Popov, I.; Do, H.-Q.; Daugulis, O. In Situ Generation and Trapping of Aryllithium and Arylpotassium Species by Halogen, Sulfur, and Carbon Electrophiles. J. Org. Chem. 2009, 74, 8309–8313. [Google Scholar] [CrossRef] [Green Version]
- Pesti, J.A.; Huhn, G.F.; Yin, J.; Xing, Y.; Fortunak, J.M.; Earl, R.A. Efficient Pyridinylmethyl Functionalization: Synthesis of 10,10-Bis[(2-Fluoro-4-Pyridinyl)Methyl]-9(10H)-Anthracenone (DMP 543), an Acetylcholine Release Enhancing Agent. J. Org. Chem. 2000, 65, 7718–7722. [Google Scholar] [CrossRef]
- Shawcross, A.P.; Stanforth, S.P. Preparation of Some Substituted Imidazo[1,2-a]Pyridine Derivatives. J. Heterocycl. Chem. 1993, 30, 563–565. [Google Scholar] [CrossRef]
- Huang, S.; Hong, X.; Cui, H.-Z.; Zhan, B.; Li, Z.-M.; Hou, X.-F. Bimetallic Bis-NHC-Ir(III) Complex Bearing 2-Arylbenzo[d]Oxazolyl Ligand: Synthesis, Catalysis, and Bimetallic Effects. Organometallics 2020, 39, 3514–3523. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions of SOCl2 Addition |  | ||
|---|---|---|---|
| x = Br | x = Cl | ||
| Attempt 1 | neat at 40 °C | 56% | 44% |
| Attempt 2 | neat at r.t | 75% | 25% |
| Attempt 3 | neat at 0 °C | ~81% | ~19% |
| Attempt 4 | to DCM solution at 0 °C | ~87% | ~13% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Handlovic, T.T.; Moreira, T.; Khan, A.; Saeed, H.; Khan, Y.; Elshaer, M.R.; Bogart, J.A. Facile Synthesis and Characterization of a Bromine-Substituted (Chloromethyl)Pyridine Precursor towards the Immobilization of Biomimetic Metal Ion Chelates on Functionalized Carbons. C 2021, 7, 54. https://0-doi-org.brum.beds.ac.uk/10.3390/c7030054
Handlovic TT, Moreira T, Khan A, Saeed H, Khan Y, Elshaer MR, Bogart JA. Facile Synthesis and Characterization of a Bromine-Substituted (Chloromethyl)Pyridine Precursor towards the Immobilization of Biomimetic Metal Ion Chelates on Functionalized Carbons. C. 2021; 7(3):54. https://0-doi-org.brum.beds.ac.uk/10.3390/c7030054
Chicago/Turabian StyleHandlovic, Troy T., Tyler Moreira, Anoshia Khan, Haroon Saeed, Yousuf Khan, Mohammed R. Elshaer, and Justin A. Bogart. 2021. "Facile Synthesis and Characterization of a Bromine-Substituted (Chloromethyl)Pyridine Precursor towards the Immobilization of Biomimetic Metal Ion Chelates on Functionalized Carbons" C 7, no. 3: 54. https://0-doi-org.brum.beds.ac.uk/10.3390/c7030054