
Green Extraction of Volatile Fatty Acids from Fermented Wastewater Using Hydrophobic Deep Eutectic Solvents

 , , and
, , and
Abstract
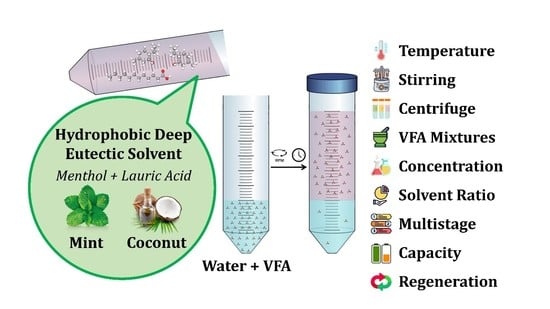
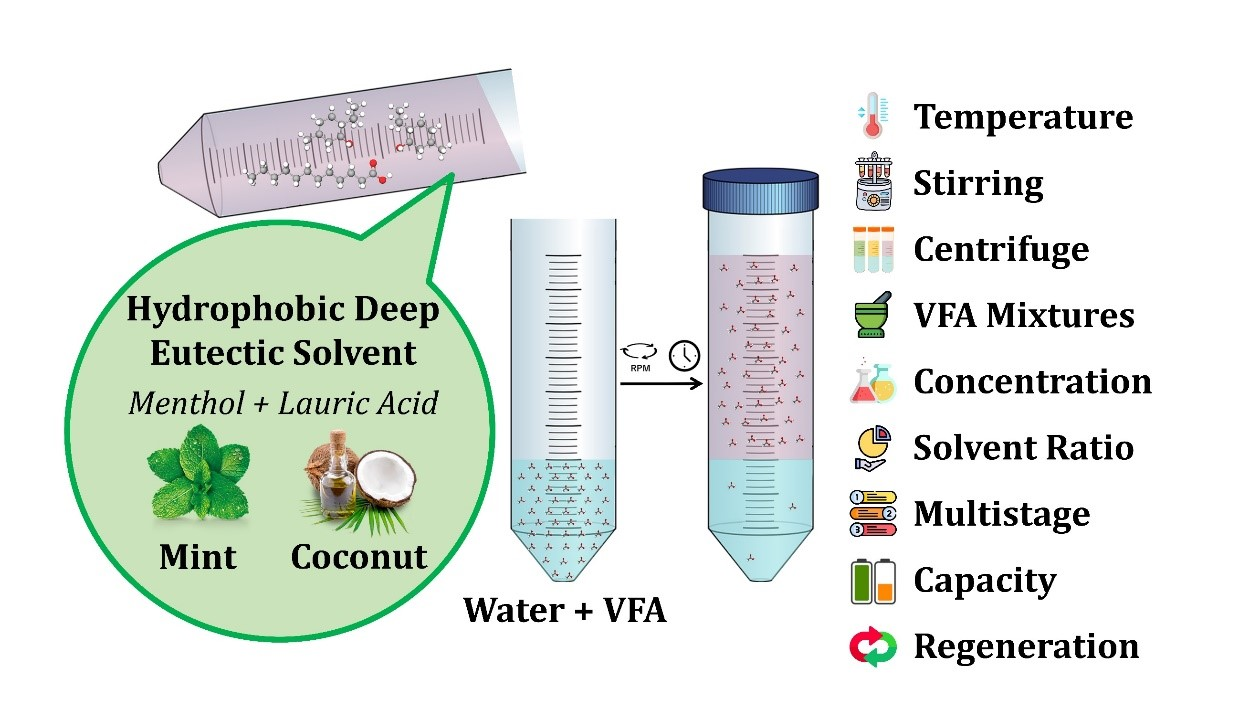
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Extraction Experiments
2.3. Solvent Preparation and Selection
2.4. HDES Characterization
2.5. Solubility Test
2.6. Parametric Extraction Experiments
3. Results and Discussion
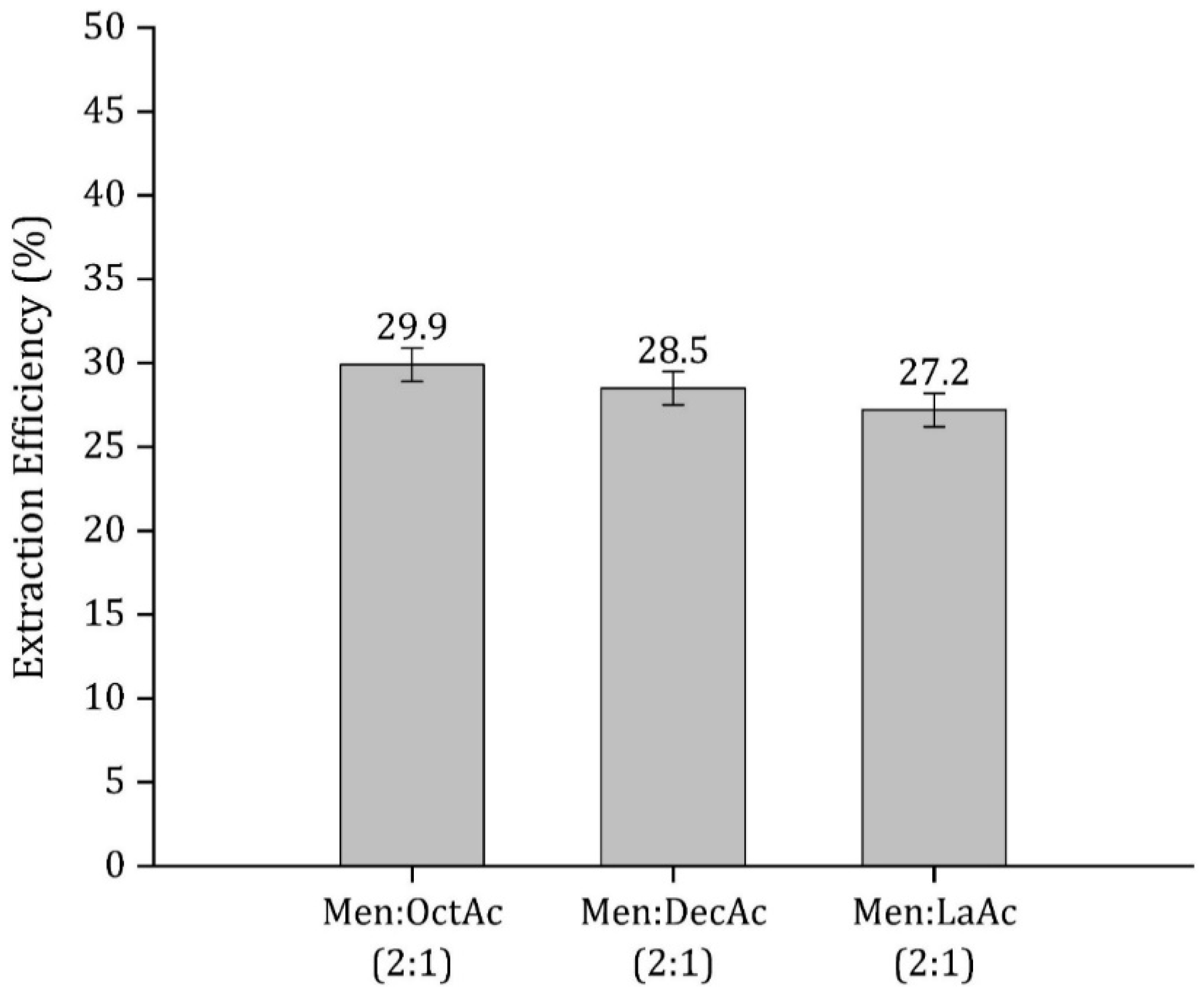
3.1. HDES Selection
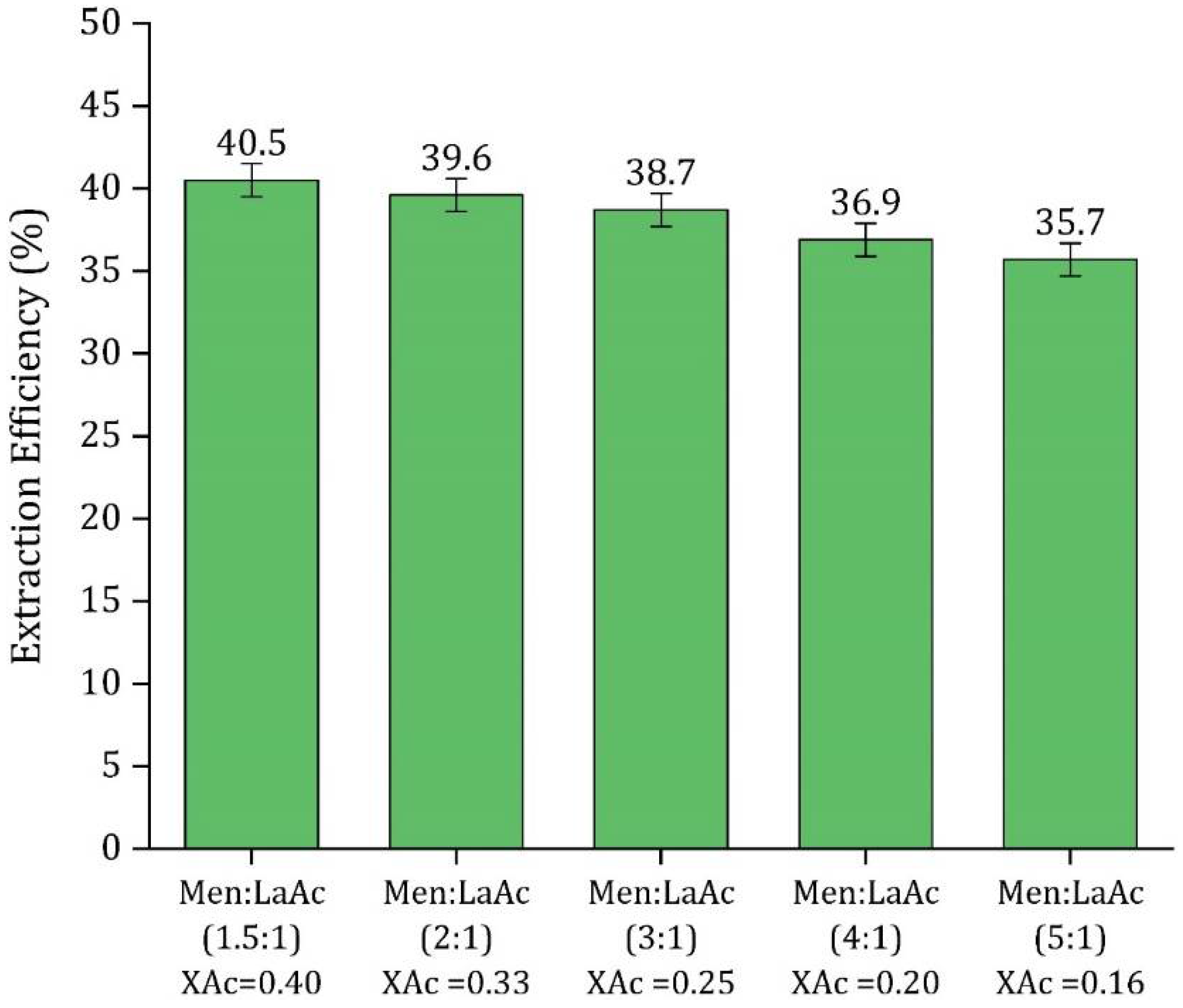
3.2. HDES Molar Ratio Optimization
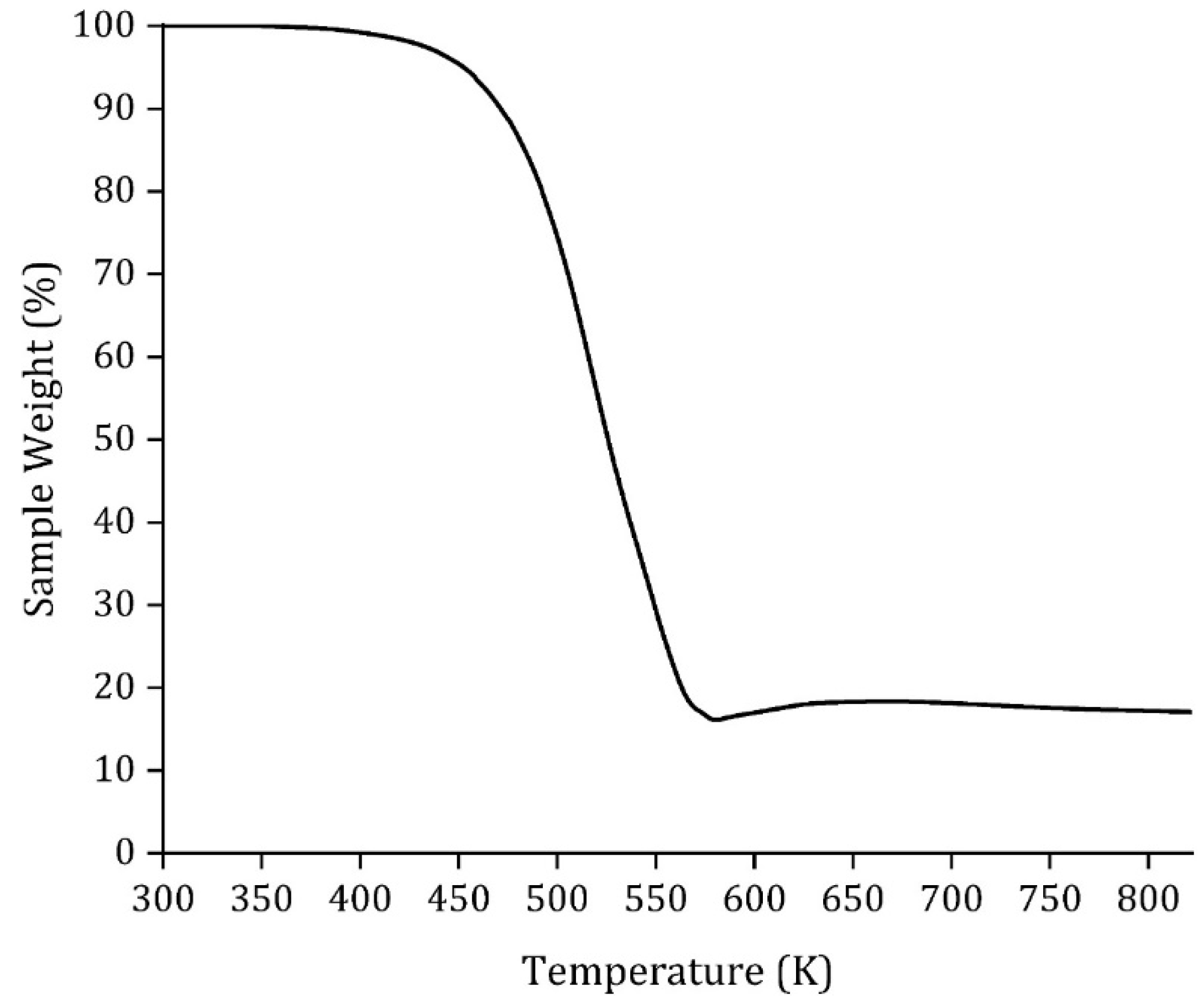
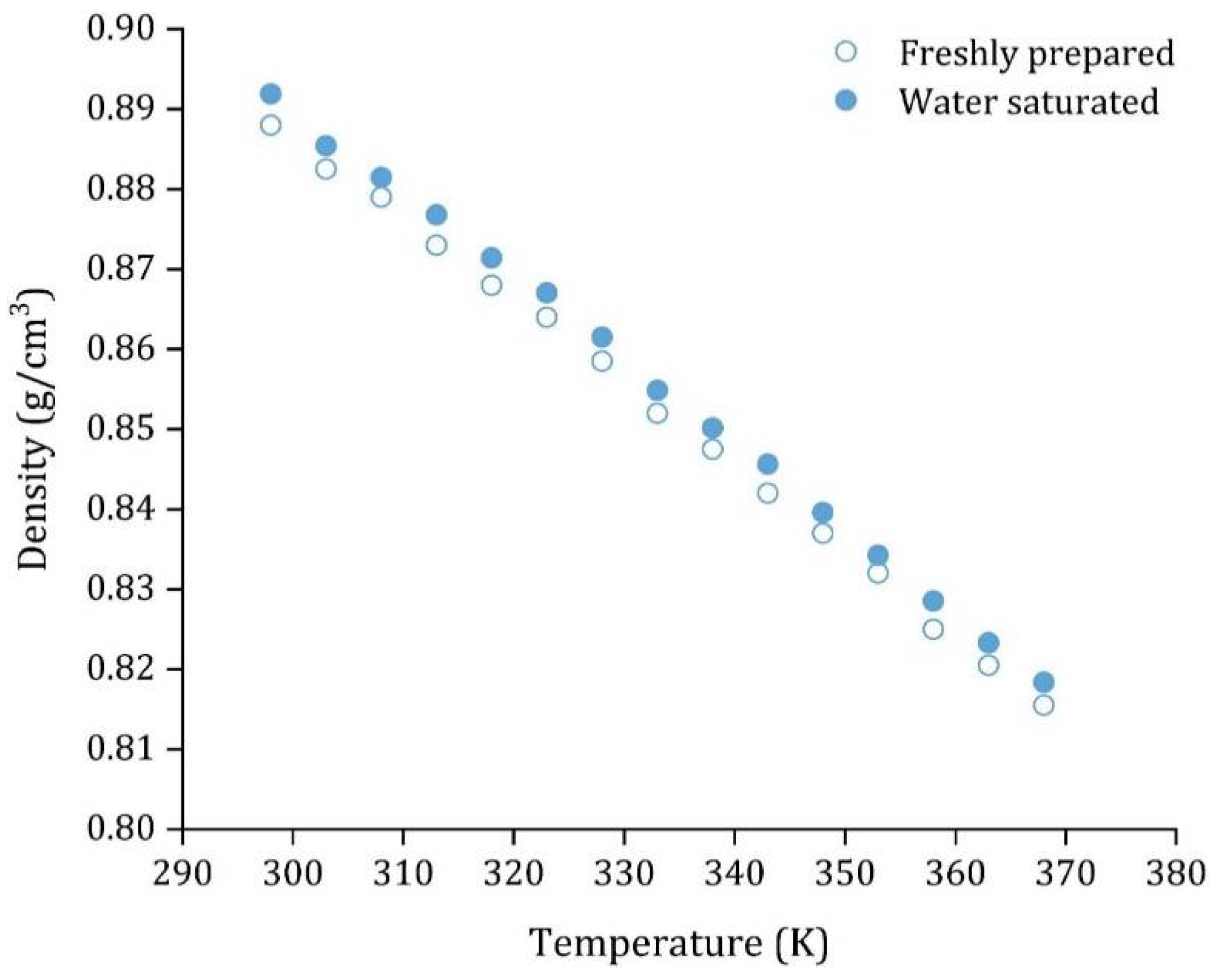
3.3. HDES Characterization
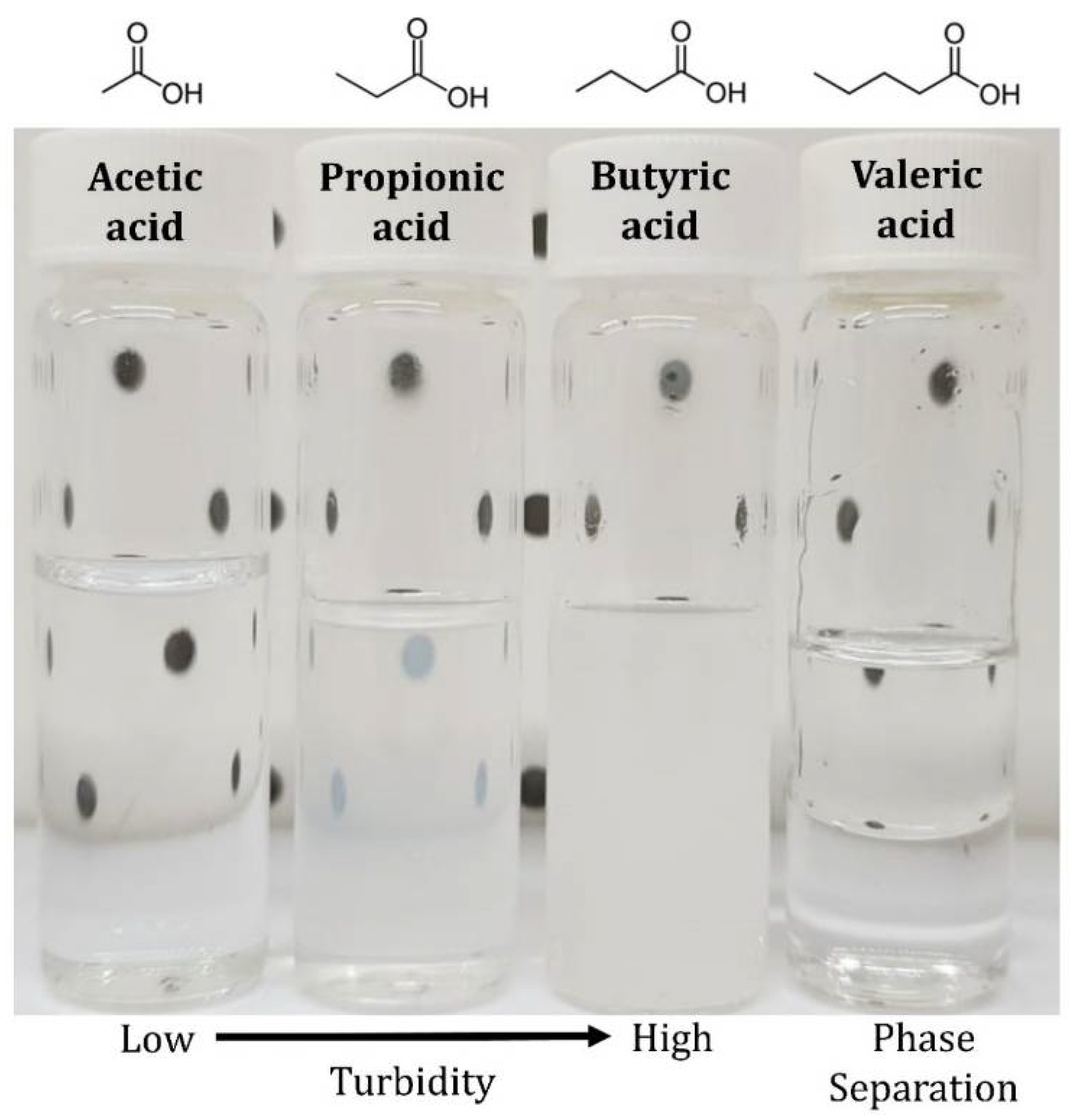
3.4. Solubility Data
3.5. Parametric Study
3.5.1. Effect of Stirring Time
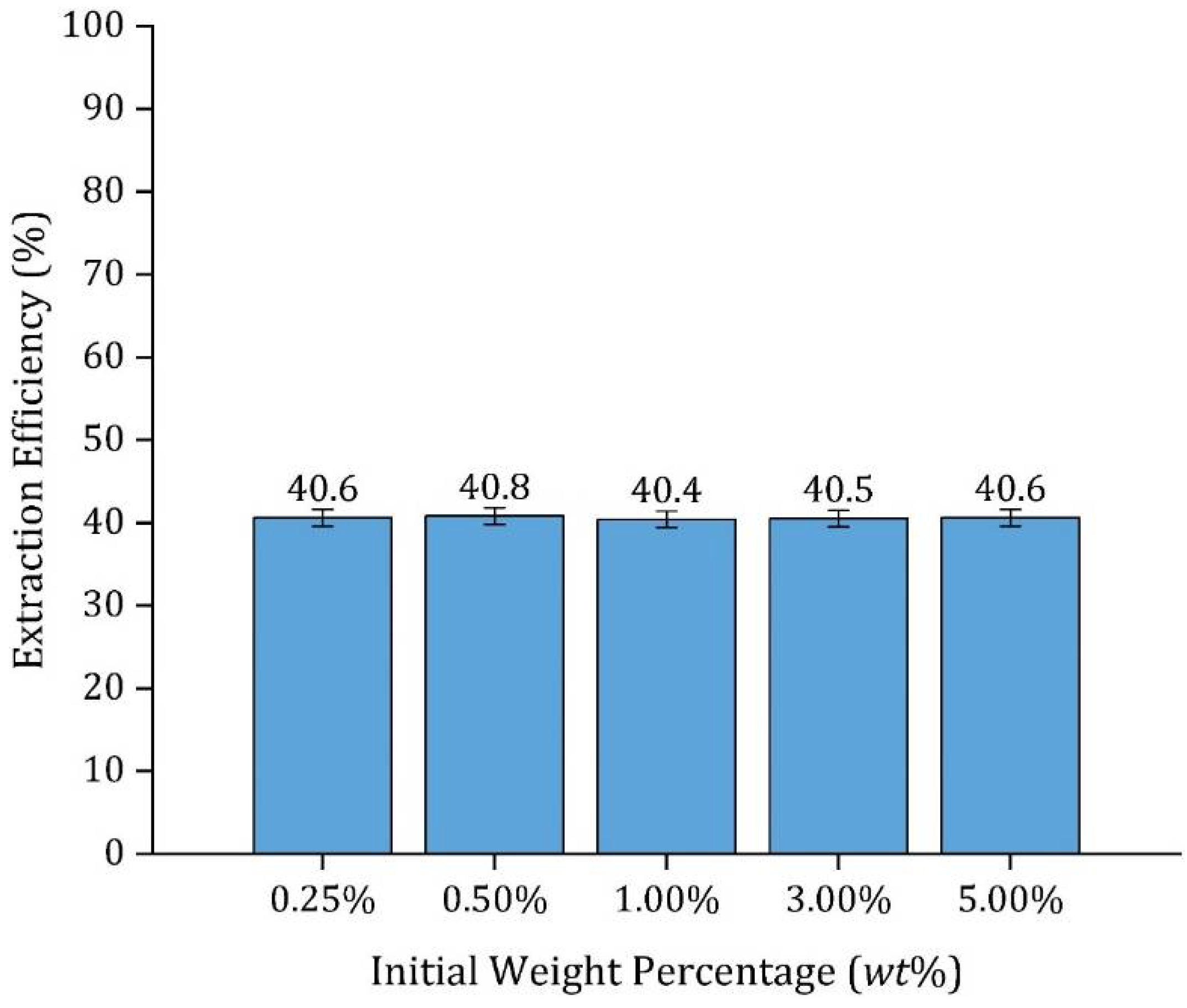
3.5.2. Effect of VFA Initial Concentration
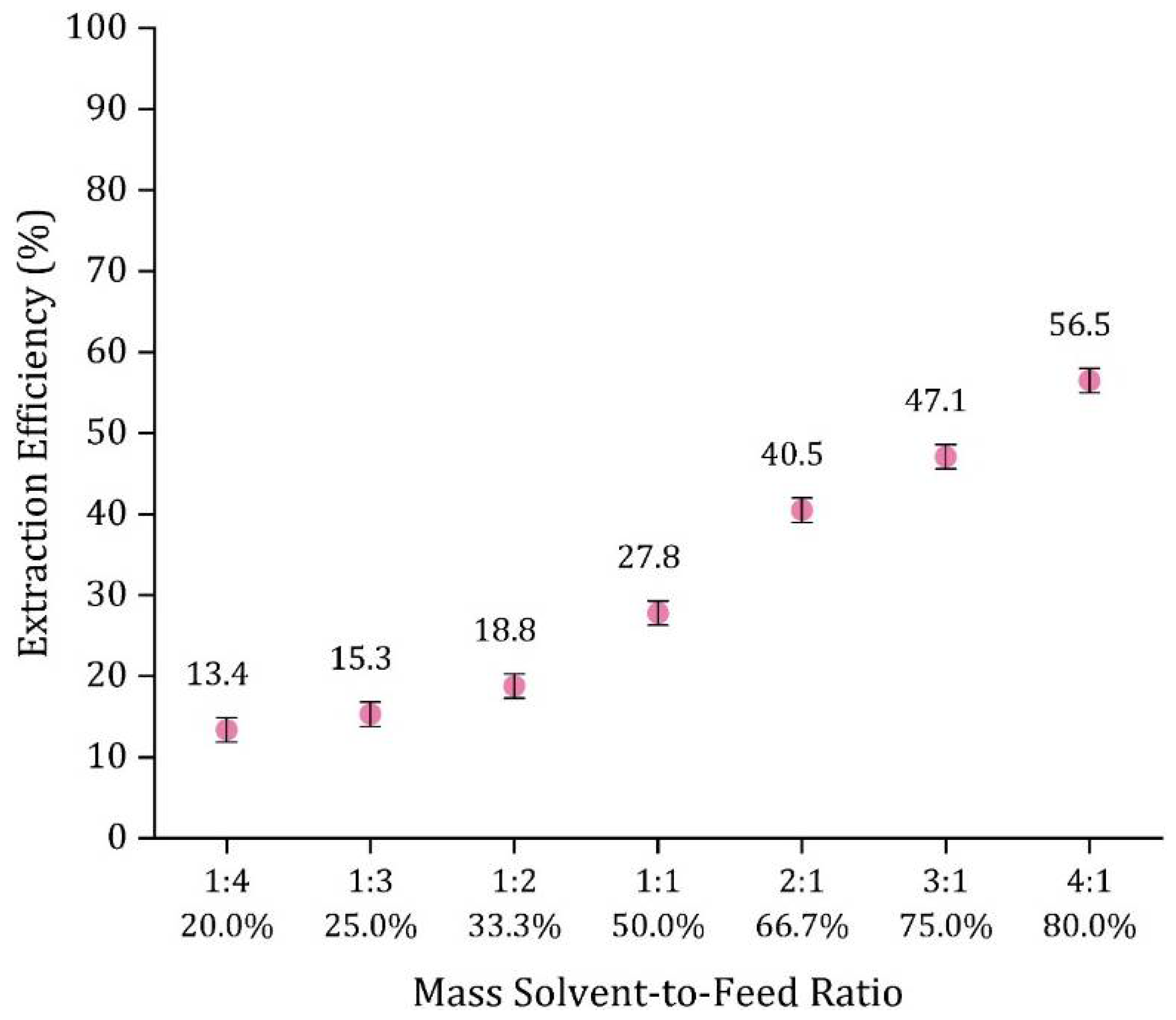
3.5.3. Effect of Solvent-to-Feed Ratio
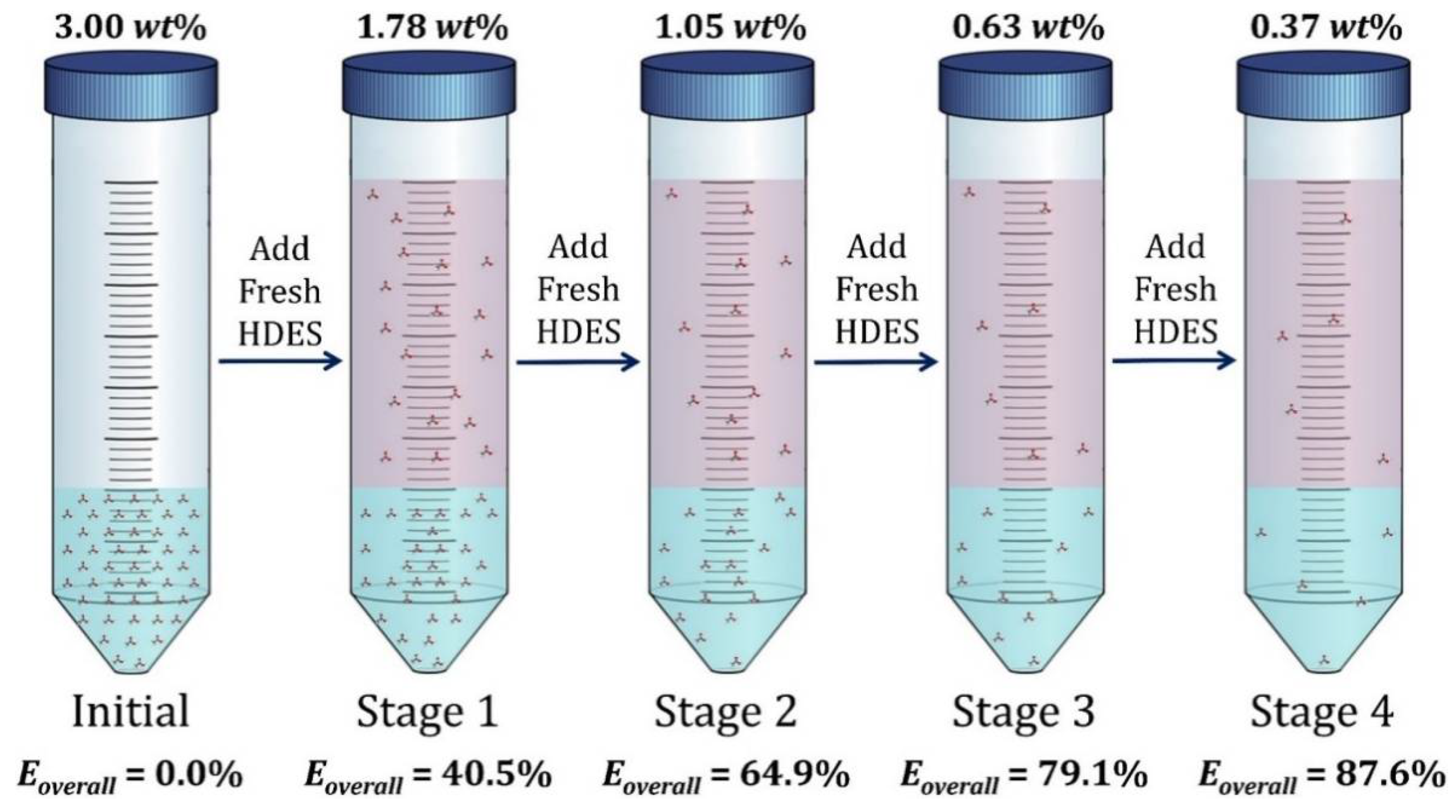
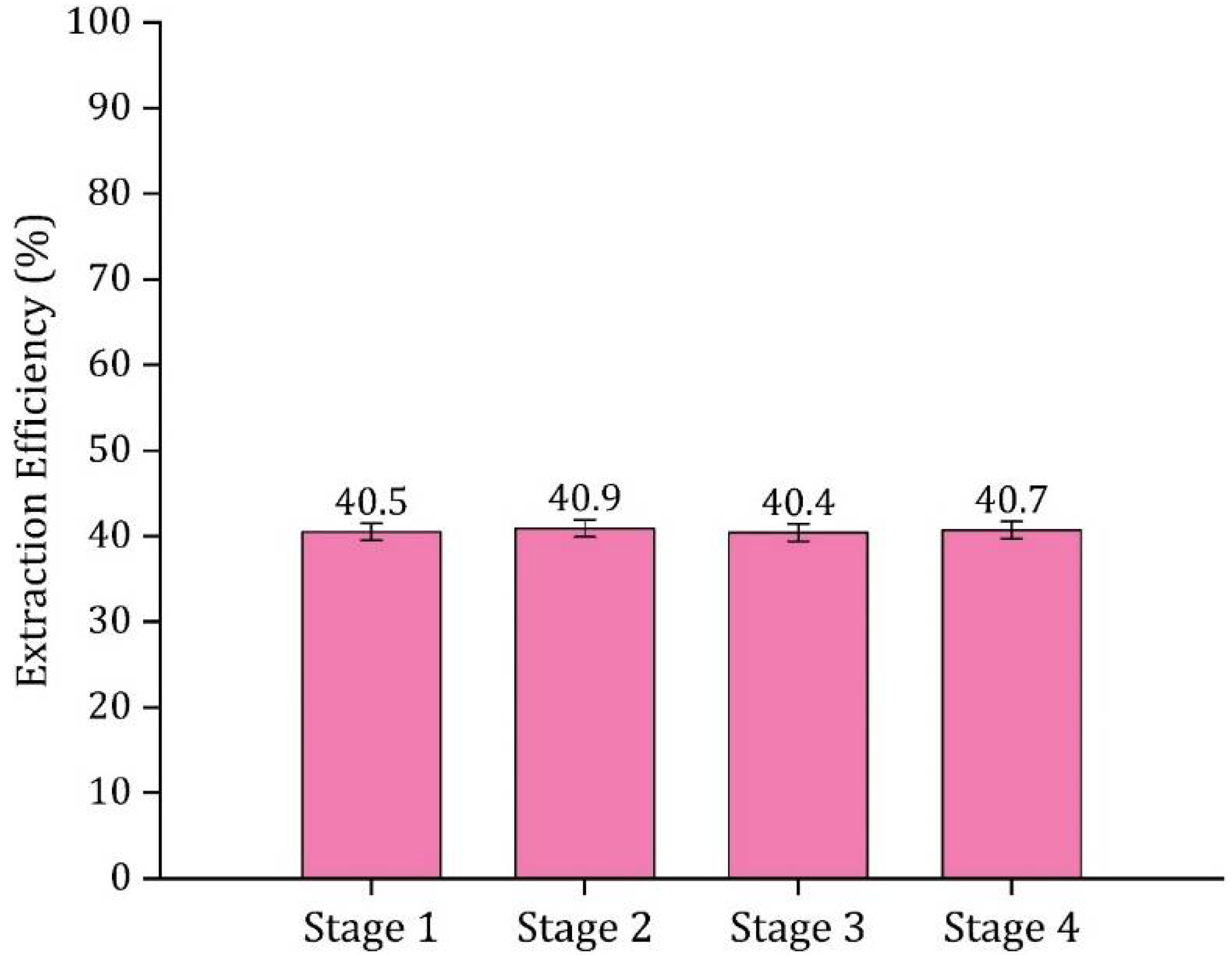
3.6. Multi-Stage Extraction
3.7. Extraction Capacity of HDES
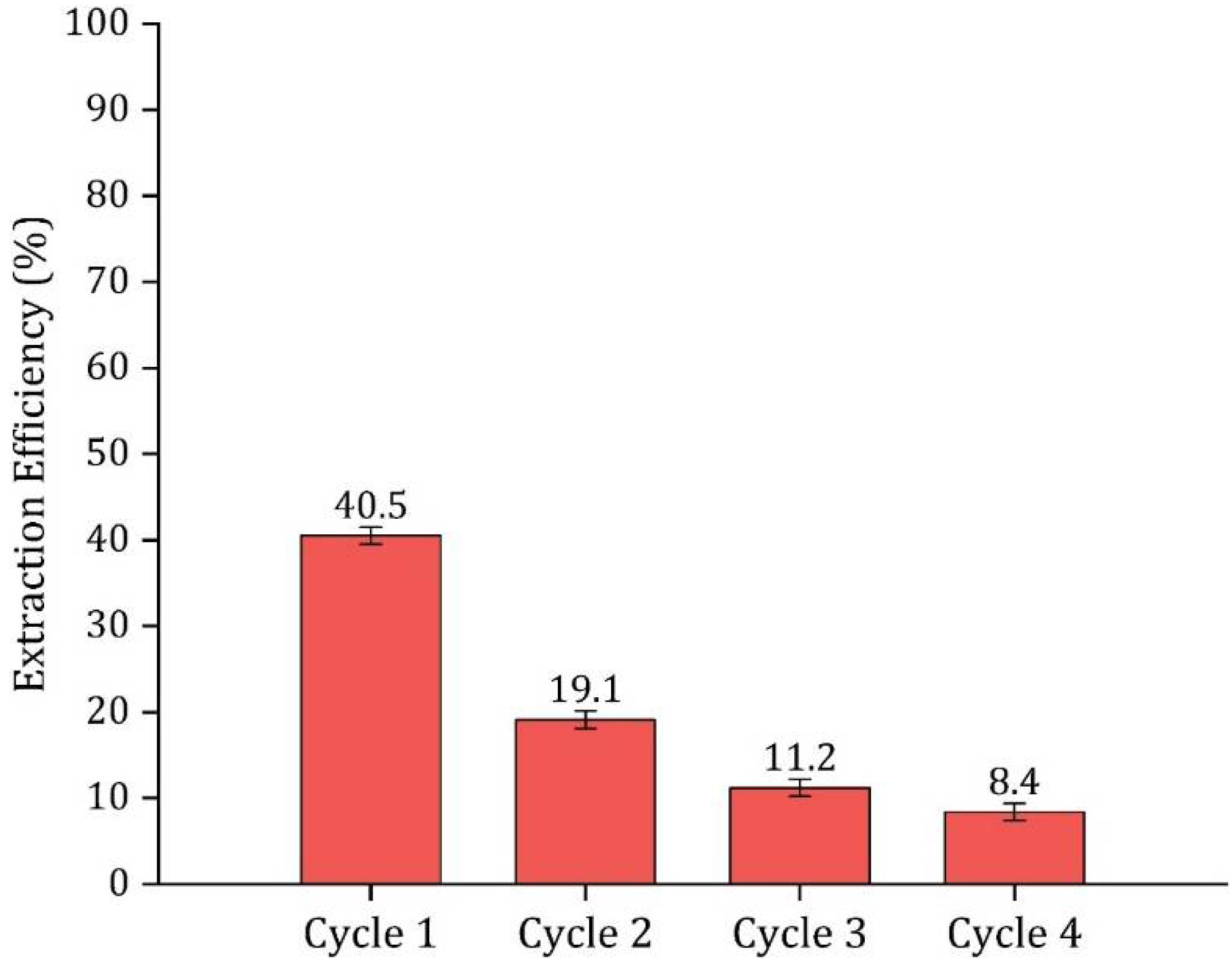
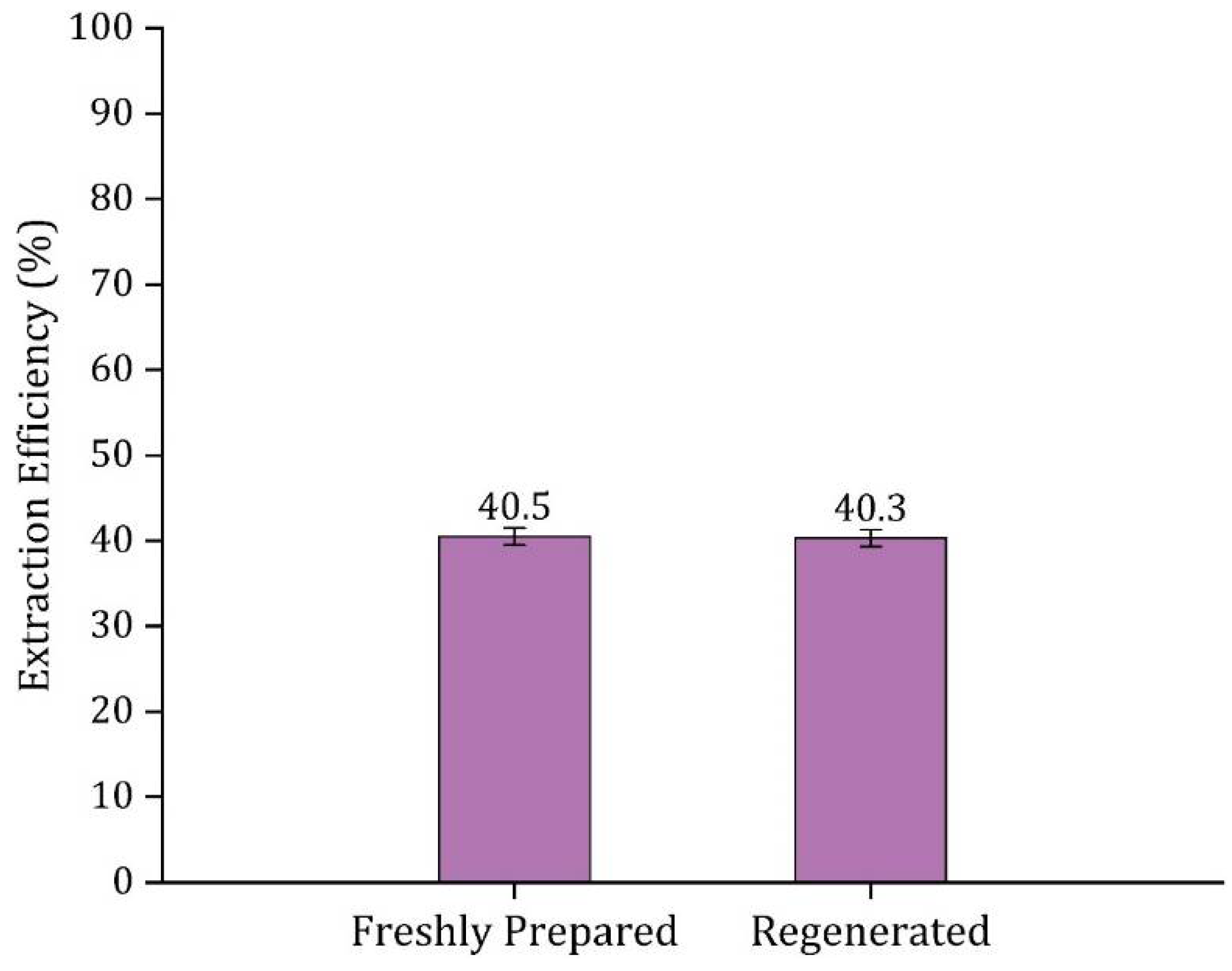
3.8. HDES Regeneration
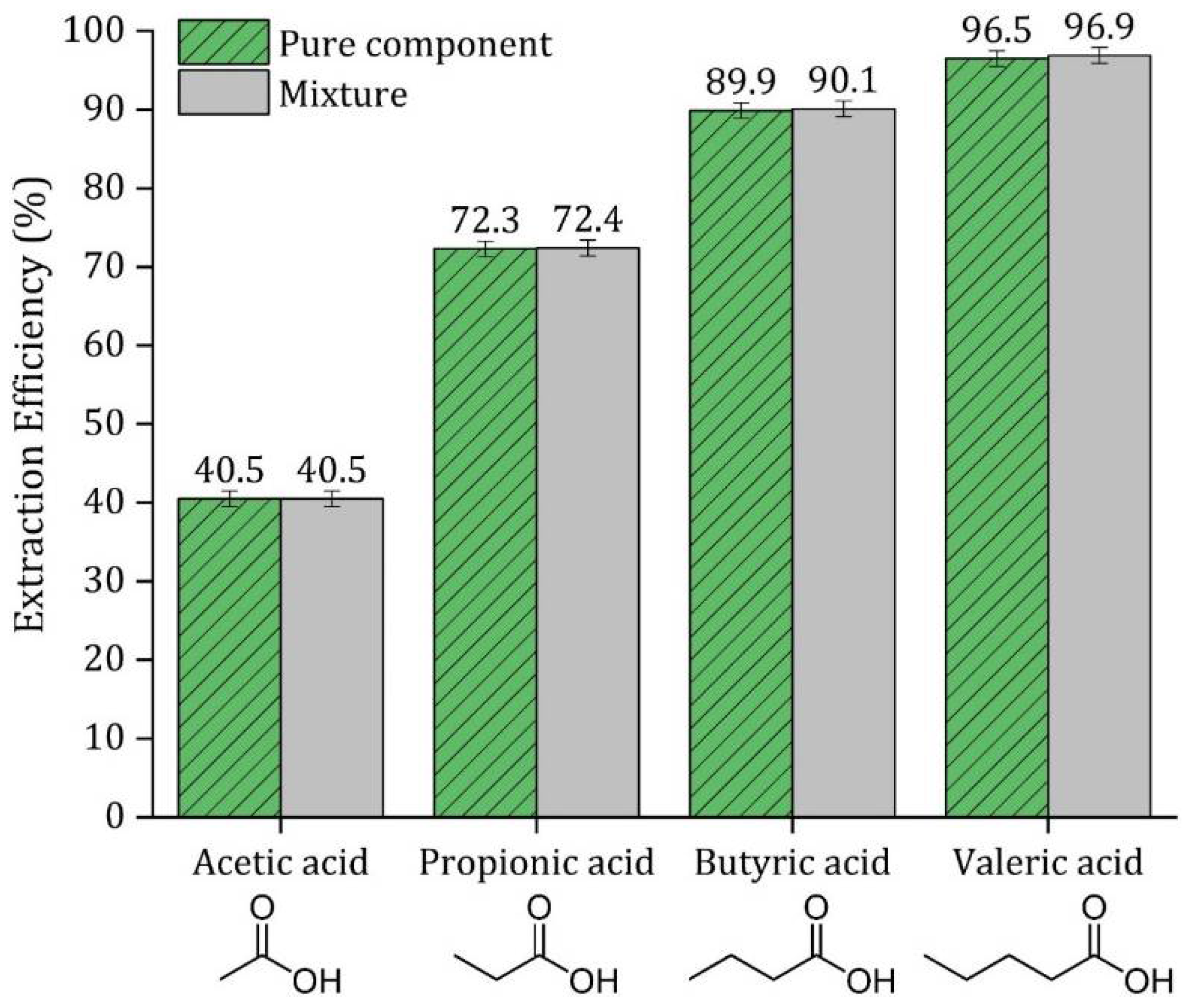
3.9. Extraction Efficiency of a Mixture of VFA
3.10. Literature Comparison
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, W.S.; Chua, A.S.M.; Yeoh, H.K.; Ngoh, G.C. A review of the production and applications of waste-derived volatile fatty acids. Chem. Eng. J. 2014, 235, 83–99. [Google Scholar] [CrossRef]
- Straathof, A.J.J. Transformation of biomass into commodity chemicals using enzymes or cells. Chem. Rev. 2014, 114, 1871–1908. [Google Scholar] [CrossRef] [PubMed]
- Grootscholten, T.I.M.; Strik, D.P.B.T.B.; Steinbusch, K.J.J.; Buisman, C.J.N.; Hamelers, H.V.M. Two-stage medium chain fatty acid (MCFA) production from municipal solid waste and ethanol. Appl. Energy 2014, 116, 223–229. [Google Scholar] [CrossRef]
- Pratt, S.; Liew, D.; Batstone, D.J.; Werker, A.G.; Morgan-Sagastume, F.; Lant, P.A. Inhibition by fatty acids during fermentation of pre-treated waste activated sludge. J. Biotechnol. 2012, 159, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Royce, L.A.; Liu, P.; Stebbins, M.J.; Hanson, B.C.; Jarboe, L.R. The damaging effects of short chain fatty acids on Escherichia coli membranes. Appl. Microbiol. Biotechnol. 2013, 97, 8317–8327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arslan, D.; Steinbusch, K.J.J.; Diels, L.; Hamelers, H.V.M.; Strik, D.P.B.T.B.; Buisman, C.J.N.; De Wever, H. Selective short-chain carboxylates production: A review of control mechanisms to direct mixed culture fermentations. Crit. Rev. Environ. Sci. Technol. 2016, 46, 592–634. [Google Scholar] [CrossRef]
- Dionisi, D.; Silva, I.M.O. Production of ethanol, organic acids and hydrogen: An opportunity for mixed culture biotechnology? Rev. Environ. Sci. Biotechnol. 2016, 15, 213–242. [Google Scholar] [CrossRef]
- Monso, E.; Monso, E. Acetic acid. Eur. Chem. News 2001, 74, 20. [Google Scholar] [CrossRef]
- Kannengiesser, J.; Sakaguchi-Söder, K.; Mrukwia, T.; Jager, J.; Schebek, L. Extraction of medium chain fatty acids from organic municipal waste and subsequent production of bio-based fuels. Waste Manag. 2016, 47, 78–83. [Google Scholar] [CrossRef]
- Reyhanitash, E.; Zaalberg, B.; Kersten, S.R.A.; Schuur, B. Extraction of volatile fatty acids from fermented wastewater. Sep. Purif. Technol. 2016, 161, 61–68. [Google Scholar] [CrossRef]
- Alkaya, E.; Kaptan, S.; Ozkan, L.; Uludag-Demirer, S.; Demirer, G.N. Recovery of acids from anaerobic acidification broth by liquid-liquid extraction. Chemosphere 2009, 77, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Rocha, M.A.A.; Raeissi, S.; Hage, P.; Weggemans, W.M.A.; van Spronsen, J.; Peters, C.J.; Kroon, M.C. Recovery of volatile fatty acids from water using medium-chain fatty acids and a cosolvent. Chem. Eng. Sci. 2017, 165, 74–80. [Google Scholar] [CrossRef]
- Rodríguez-Llorente, D.; Bengoa, A.; Pascual-Muñoz, G.; Navarro, P.; Águeda, V.I.; Delgado, J.A.; Álvarez-Torrellas, S.; García, J.; Larriba, M. Sustainable Recovery of Volatile Fatty Acids from Aqueous Solutions Using Terpenoids and Eutectic Solvents. ACS Sustain. Chem. Eng. 2019, 7, 16786–16794. [Google Scholar] [CrossRef]
- van den Bruinhorst, A.; Raes, S.; Maesara, S.A.; Kroon, M.C.; Esteves, A.C.C.; Meuldijk, J. Hydrophobic eutectic mixtures as volatile fatty acid extractants. Sep. Purif. Technol. 2019, 216, 147–157. [Google Scholar] [CrossRef]
- Reyhanitash, E.; Kersten, S.R.A.; Schuur, B. Recovery of Volatile Fatty Acids from Fermented Wastewater by Adsorption. ACS Sustain. Chem. Eng. 2017, 5, 9176–9184. [Google Scholar] [CrossRef]
- Alghezawi, N.; Şanli, O.; Aras, L.; Asman, G. Separation of acetic acid-water mixtures through acrylonitrile grafted poly(vinyl alcohol) membranes by pervaporation. Chem. Eng. Process. Process Intensif. 2005, 44, 51–58. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, B. Liquid-Liquid Extraction (LLE). In Separation and Purification Technologies in Biorefineries; John Wiley & Sons: West Sussex, UK, 2013; pp. 61–78. ISBN 9780470977965. [Google Scholar]
- Lemaoui, T.; Benguerba, Y.; Darwish, A.S.; Hatab, F.A.; Warrag, S.E.E.; Kroon, M.C.; Alnashef, I.M. Simultaneous dearomatization, desulfurization, and denitrogenation of diesel fuels using acidic deep eutectic solvents as extractive agents: A parametric study. Sep. Purif. Technol. 2021, 256, 117861. [Google Scholar] [CrossRef]
- Sprakel, L.M.J.; Schuur, B. Solvent developments for liquid-liquid extraction of carboxylic acids in perspective. Sep. Purif. Technol. 2019, 211, 935–957. [Google Scholar] [CrossRef]
- Krzyzaniak, A.; Leeman, M.; Vossebeld, F.; Visser, T.J.; Schuur, B.; De Haan, A.B. Novel extractants for the recovery of fermentation derived lactic acid. Sep. Purif. Technol. 2013, 111, 82–89. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 9, 70–71. [Google Scholar] [CrossRef] [Green Version]
- van Osch, D.J.G.P.; Zubeir, L.F.; van den Bruinhorst, A.; Rocha, M.A.A.; Kroon, M.C. Hydrophobic deep eutectic solvents as water-immiscible extractants. Green Chem. 2015, 17, 4518–4521. [Google Scholar] [CrossRef] [Green Version]
- Lemaoui, T.; Darwish, A.S.; Attoui, A.; Hatab, F.A.; El, N.; Hammoudi, H.; Benguerba, Y.; Vega, L.F.; Alnashef, I.M. Predicting the density and viscosity of hydrophobic eutectic solvents: Towards the development of sustainable solvents. Green Chem. 2020, 22, 15–17. [Google Scholar] [CrossRef]
- Lemaoui, T.; Darwish, A.S.; Hammoudi, N.E.H.; Abu Hatab, F.; Attoui, A.; Alnashef, I.M.; Benguerba, Y. Prediction of Electrical Conductivity of Deep Eutectic Solvents Using COSMO-RS Sigma Profiles as Molecular Descriptors: A Quantitative Structure–Property Relationship Study. Ind. Eng. Chem. Res. 2020, 59, 13343–13354. [Google Scholar] [CrossRef]
- Lemaoui, T.; Abu Hatab, F.; Darwish, A.S.; Attoui, A.; Hammoudi, N.E.H.; Almustafa, G.; Benaicha, M.; Benguerba, Y.; Alnashef, I.M. Molecular-Based Guide to Predict the pH of Eutectic Solvents: Promoting an Efficient Design Approach for New Green Solvents. ACS Sustain. Chem. Eng. 2021, 9, 5783–5808. [Google Scholar] [CrossRef]
- García, G.; Aparicio, S.; Ullah, R.; Atilhan, M. Deep Eutectic Solvents: Physicochemical Properties and Gas Separation Applications. Energy Fuels 2015, 29, 2616–2644. [Google Scholar] [CrossRef]
- Francisco, M.; van den Bruinhorst, A.; Zubeir, L.F.; Peters, C.J.; Kroon, M.C. A new low transition temperature mixture (LTTM) formed by choline chloride+lactic acid: Characterization as solvent for CO2 capture. Fluid Phase Equilib. 2013, 340, 77–84. [Google Scholar] [CrossRef]
- Smith, E.L.; Abbott, A.P.; Ryder, K.S. Deep Eutectic Solvents (DESs) and Their Applications. Chem. Rev. 2014, 114, 11060–11082. [Google Scholar] [CrossRef] [Green Version]
- Benabid, S.; Haddaoui, N.; Lemaoui, T.; Darwish, A.S.; Benguerba, Y.; Alnashef, I.M. Computational modeling of polydecanediol-co-citrate using benzalkonium chloride-based hydrophobic eutectic solvents: COSMO-RS, reactivity, and compatibility insights. J. Mol. Liq. 2021, 339, 116674. [Google Scholar] [CrossRef]
- Van Osch, D.J.G.P.; Dietz, C.H.J.T.; Warrag, S.E.E.; Kroon, M.C. The Curious Case of Hydrophobic Deep Eutectic Solvents: A Story on the Discovery, Design, and Applications. ACS Sustain. Chem. Eng. 2020, 8, 10591–10612. [Google Scholar] [CrossRef]
- Chemat, F.; Vian, M.A.; Cravotto, G. Green extraction of natural products: Concept and principles. Int. J. Mol. Sci. 2012, 13, 8615–8627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almustafa, G.; Sulaiman, R.; Kumar, M.; Adeyemi, I.; Arafat, H.A.; AlNashef, I. Boron extraction from aqueous medium using novel hydrophobic deep eutectic solvents. Chem. Eng. J. 2020, 395, 125173. [Google Scholar] [CrossRef]
- Almustafa, G.; Darwish, A.S.; Lemaoui, T.; O’Conner, M.J.; Amin, S.; Arafat, H.A.; AlNashef, I. Liquification of 2,2,4-trimethyl-1,3-pentanediol into hydrophobic eutectic mixtures: A multi-criteria design for eco-efficient boron recovery. Chem. Eng. J. 2021, 426, 131342. [Google Scholar] [CrossRef]
- Darwish, A.S.; Abu Hatab, F.; Lemaoui, T.; Ibrahim, O.A.Z.; Almustafa, G.; Zhuman, B.; Warrag, S.E.E.; Hadj-Kali, M.K.; Benguerba, Y.; Alnashef, I.M. Multicomponent extraction of aromatics and heteroaromatics from diesel using acidic eutectic solvents: Experimental and COSMO-RS predictions. J. Mol. Liq. 2021, 336, 116575. [Google Scholar] [CrossRef]
- Warrag, S.E.E.; Darwish, A.S.; Adeyemi, I.A.; Hadj-Kali, M.K.; Kroon, M.C.; Alnashef, I.M. Extraction of pyridine from n-alkane mixtures using methyltriphenylphosphonium bromide-based deep eutectic solvents as extractive denitrogenation agents. Fluid Phase Equilib. 2020, 517, 112622. [Google Scholar] [CrossRef]
- Hatab, F.A.; Darwish, A.S.; Lemaoui, T.; Warrag, S.E.E.; Benguerba, Y.; Kroon, M.C.; Alnashef, I.M. Extraction of Thiophene, Pyridine, and Toluene from n-Decane Model Diesel Using Betaine-Based Natural Deep Eutectic Solvents. J. Chem. Eng. Data 2020, 65, 5443–5457. [Google Scholar] [CrossRef]
- Lalikoglu, M. Separation of butyric acid from aqueous media using menthol-based hydrophobic deep eutectic solvent and modeling by response surface methodology. Biomass Convers. Biorefinery 2021, 2021, 1–11. [Google Scholar] [CrossRef]
- van Osch, D.J.G.P. Design and Applications of Hydrophobic Deep Eutectic Solvents. Ph.D. Thesis, Technische Universiteit Eindhoven, Eindhoven, The Netherlands, 2018. [Google Scholar]
- Florindo, C.; Branco, L.C.; Marrucho, I.M. Development of hydrophobic deep eutectic solvents for extraction of pesticides from aqueous environments. Fluid Phase Equilib. 2017, 448, 135–142. [Google Scholar] [CrossRef]
- Florindo, C.; Romero, L.; Rintoul, I.; Branco, L.C.; Marrucho, I.M. From Phase Change Materials to Green Solvents: Hydrophobic Low Viscous Fatty Acid-Based Deep Eutectic Solvents. ACS Sustain. Chem. Eng. 2018, 6, 3888–3895. [Google Scholar] [CrossRef]
- Martins, M.A.R.; Crespo, E.A.; Pontes, P.V.A.; Silva, L.P.; Bülow, M.; Maximo, G.J.; Batista, E.A.C.; Held, C.; Pinho, S.P.; Coutinho, J.A.P. Tunable Hydrophobic Eutectic Solvents Based on Terpenes and Monocarboxylic Acids. ACS Sustain. Chem. Eng. 2018, 6, 8836–8846. [Google Scholar] [CrossRef]
- Ribeiro, B.D.; Florindo, C.; Iff, L.C.; Coelho, M.A.Z.; Marrucho, I.M. Menthol-based eutectic mixtures: Hydrophobic low viscosity solvents. ACS Sustain. Chem. Eng. 2015, 3, 2469–2477. [Google Scholar] [CrossRef]
- Lemaoui, T.; Hammoudi, N.E.H.; Alnashef, I.M.; Balsamo, M.; Erto, A.; Ernst, B.; Benguerba, Y. Quantitative structure properties relationship for deep eutectic solvents using Sσ-profile as molecular descriptors. J. Mol. Liq. 2020, 309, 113165. [Google Scholar] [CrossRef]
- Valderrama, J.O.; Rojas, R.E. Critical properties of ionic liquids. Revisited. Ind. Eng. Chem. Res. 2009, 48, 6890–6900. [Google Scholar] [CrossRef]
- Warrag, S.E.E.; Darwish, A.S.; Abuhatab, F.O.S.; Adeyemi, I.A.; Kroon, M.C.; Alnashef, I.M. Combined Extractive Dearomatization, Desulfurization, and Denitrogenation of Oil Fuels Using Deep Eutectic Solvents: A Parametric Study. Ind. Eng. Chem. Res. 2020, 59, 11723–11733. [Google Scholar] [CrossRef]
- Gano, Z.S.; Mjalli, F.S.; Al-Wahaibi, T.; Al-Wahaibi, Y.; Alnashef, I.M. Desulfurization of liquid fuel via extraction with imidazole-containing deep eutectic solvent. Green Process. Synth. 2017, 6, 511–521. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical | CAS Number | Purity (wt%) | Source |
|---|---|---|---|
| Acetic acid | 64-19-7 | ≥99.5 | Surechem Products |
| Propionic acid | 79-09-4 | ≥99.0 | Acros Organics |
| Butyric acid | 107-92-6 | ≥99.0 | Sigma-Aldrich |
| Valeric acid | 109-52-4 | ≥99.0 | Sigma-Aldrich |
| Octanoic acid | 124-07-2 | ≥99.0 | Sigma-Aldrich |
| Decanoic acid | 334-48-5 | ≥98.0 | Sigma-Aldrich |
| Lauric acid | 143-07-7 | ≥98.0 | Sigma-Aldrich |
| DL-Menthol | 89-78-1 | ≥99.0 | Sigma-Aldrich |
| Tetraoctylammonium bromide | 14866-33-2 | ≥98.0 | Merck |
| Specification | |
|---|---|
| Equipment model | Agilent 1260 Infinity |
| Column | Agilent Hi-Plex H, 7.7 × 300 mm, 8 µm |
| Detector | UV/Vis, 210 nm |
| Pump | Isocratic |
| Injection volume | 5 µL |
| Column temperature | 55 °C |
| Eluent | 5 mM H2SO4 |
| Flowrate | 0.6 mL/min |
| Component 1 | Component 2 | Molar Ratio |
|---|---|---|
| Tetraoctylammonium bromide (TOABr) | Octanoic acid (OctAc) | 2:1 |
| Tetraoctylammonium bromide (TOABr) | Decanoic acid (DecAc) | 2:1 |
| Tetraoctylammonium bromide (TOABr) | Lauric acid (LaAc) | 2:1 |
| DL-Menthol (Men) | Octanoic acid (OctAc) | 2:1 |
| DL-Menthol (Men) | Decanoic acid (DecAc) | 2:1 |
| DL-Menthol (Men) | Lauric acid (LaAc) | 2:1 |
| Constituent 1 | |
| Name | DL-Menthol |
| Abbreviation | Men |
| Structure |  |
| Mole percentage | 60.0 mol% |
| Weight percentage | 53.9 wt% |
| Constituent 2 | |
| Name | Lauric Acid |
| Abbreviation | LaAc |
| Structure |  |
| Mole percentage | 40.0 mol% |
| Weight percentage | 46.1 wt% |
| Physical Property | Value |
|---|---|
| Density, (g·cm−3) | 0.888 ± 0.002 |
| Viscosity, (mPa) | 24.98 ± 0.45 |
| Degradation temperature, (K) | 485.7 |
| Melting point, Tm (K) | 296.17 [41] |
| Water Content, (ppmwt) | 893 ± 101 |
| Water Content, (ppmmol) | 8556 ± 391 |
| Tc,HDES (K) | Pc,HDES (bar) | Vc,HDES (mL·mol−1) |
|---|---|---|
| 767.5 | 21.79 | 649.3 |
| Solute (1) in Solvent (2) | Solubility |
|---|---|
| {Acetic Acid (1) in Water (2)} | Fully miscible with no turbidity |
| {Propionic Acid (1) in Water (2)} | Fully miscible but slightly turbid |
| {Butyric Acid (1) in Water (2)} | Fully miscible but highly turbid |
| {Valeric Acid (1) in Water (2)} | w1 = 4.01 ± 0.01 wt% x1 = 0.73 ± 0.01 mol% |
| {Water (1) in Men:LaAc (2)} HDES saturation with water | w1 = 2.10 ± 0.01 wt% x1 = 17.16 ± 0.10 mol% |
| {Men:LaAc (1) in Water (2)} Migration of the HDES constituents to the water-phase | Not soluble, observed using FT-IR, and ΔTOC = 29.4 ± 1.9 ppm |
| {Acetic Acid (1) in Men:LaA (2)} | Fully miscible with no turbidity |
| {Propionic Acid (1) in Men:LaAc (2)} | Fully miscible with no turbidity |
| {Butyric Acid (1) in Men:LaAc (2)} | Fully miscible with no turbidity |
| {Valeric Acid (1) in Men:LaAc (2)} | Fully miscible with no turbidity |
| Initial Concentration (wt%) | 0.25% | 0.50% | 1% | 3% | 5% | |||||
| pH | 3.1 | 2.9 | 2.8 | 2.5 | 2.4 | |||||
| [HA]|[A−] (M) a | 0.041 | 0.001 | 0.084 | 0.001 | 0.168 | 0.002 | 0.506 | 0.003 | 0.844 | 0.004 |
| Final Concentration (wt%) | 0.15% | 0.30% | 0.63% | 1.80% | 3% | |||||
| pH | 3.2 | 3.0 | 2.9 | 2.6 | 2.5 | |||||
| [HA]|[A−] (M) a | 0.025 | 0.001 | 0.05 | 0.001 | 0.105 | 0.001 | 0.303 | 0.002 | 0.506 | 0.003 |
| Solvent | Acetic Acid | Propionic Acid | Buyuric Acid | Valeric Acid | Ref |
|---|---|---|---|---|---|
| Trioctylamine | 18.6 | 45.9 | 73.5 | – | [22] |
| Geraniol | 55.9 | 74.9 | 85.6 | 92.8 | [13] |
| Eugenol | 40.9 | 63.2 | 70.4 | 91.8 | [13] |
| Citral | 45.7 | 64.2 | 70.0 | 76.0 | [13] |
| Hexanoic acid | 27.3 | 66.2 | 85.3 | – | [12] |
| Octanoic acid | 22.0 | 57.3 | 80.1 | – | [12] |
| Decanoic acid:methyltrioctylammonium chloride (2:1) | 38.0 | 70.5 | 89.8 | – | [22] |
| Decanoic acid:tetraheptylammonium chloride (2:1) | 32.0 | 76.5 | 91.5 | – | [22] |
| Decanoic acid:tetraoctylammonium chloride (2:1) | 25.0 | 52.7 | 81.3 | – | [22] |
| Decanoic acid:methyltrioctylammonium bromide (2:1) | 29.7 | 63.4 | 83.1 | – | [22] |
| Decanoic acid:tetraoctylammonium bromide (2:1) | 30.6 | 65.9 | 87.4 | – | [22] |
| Menthol: octanoic acid (1:1) | 15.7 | 60.4 | 82.0 | 91.3 | [13] |
| Thymol: octanoic acid (1:2) | 35.5 | 73.1 | 82.1 | 89.0 | [13] |
| Menthol: lauric acid (2:1) | 27.2 | – | – | – | This work |
| Trioctylphosphine oxide (20 wt%) in kerosene | ≈60 a | ≈70 a | ≈85 a | ≈95 a | [11] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darwish, A.S.; Warrag, S.E.E.; Lemaoui, T.; Alseiari, M.K.; Hatab, F.A.; Rafay, R.; Alnashef, I.; Rodríguez, J.; Alamoodi, N. Green Extraction of Volatile Fatty Acids from Fermented Wastewater Using Hydrophobic Deep Eutectic Solvents. Fermentation 2021, 7, 226. https://0-doi-org.brum.beds.ac.uk/10.3390/fermentation7040226
Darwish AS, Warrag SEE, Lemaoui T, Alseiari MK, Hatab FA, Rafay R, Alnashef I, Rodríguez J, Alamoodi N. Green Extraction of Volatile Fatty Acids from Fermented Wastewater Using Hydrophobic Deep Eutectic Solvents. Fermentation. 2021; 7(4):226. https://0-doi-org.brum.beds.ac.uk/10.3390/fermentation7040226
Chicago/Turabian StyleDarwish, Ahmad S., Samah E. E. Warrag, Tarek Lemaoui, Maha K. Alseiari, Farah Abu Hatab, Ramis Rafay, Inas Alnashef, Jorge Rodríguez, and Nahla Alamoodi. 2021. "Green Extraction of Volatile Fatty Acids from Fermented Wastewater Using Hydrophobic Deep Eutectic Solvents" Fermentation 7, no. 4: 226. https://0-doi-org.brum.beds.ac.uk/10.3390/fermentation7040226