
A Comparative Study of Oxygen and Hydrogen Adsorption on Strained and Alloy-Supported Pt(111) Monolayers

School of Science and Technology, Nottingham Trent University, Nottingham NG11 8NS, UK
Magnetochemistry 2021, 7(7), 101; https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7070101
Submission received: 8 June 2021
/
Revised: 1 July 2021
/
Accepted: 6 July 2021
/
Published: 9 July 2021
Abstract
:A comparative study of the unreacted and reacted uniaxially strained Pt(111) and the layered (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces has been performed using density functional theory (DFT). An in-depth study of the unreacted surfaces has been performed to evaluate the importance of geometric, magnetic and ligand effects in determining the reactivity of these different Pt surfaces. An analysis of the binding energies of oxygen and hydrogen over the high-symmetry binding positions of all surfaces has been performed. The study has shown that O and H tend to bind more strongly to the (111)-Pt/Ni/Pt3Ni surface and less strongly to the (111)-Pt/Ni/PtNi3 surface compared to binding on the equivalently strained Pt(111) surfaces. Changes in the surface magnetisation of the surfaces overlaying the ferromagnetic alloys during adsorption are discussed, as well as the behaviour of the d-band centre across all surfaces, to evaluate the potential mechanisms for these differences in binding. An accompanying comparison of the accessible density functionals has been included to estimate the error in the computational binding energies.
1. Introduction
Understanding the mechanisms of the use of the oxygen reduction reaction (ORR) and the hydrogen evolution reaction (HER) is key to the development of modern energy storage and transport materials. The technology inherent in these materials contributes significantly to sustainable chemical production, whose energy footprint is an imperative concern when designing catalysts and networks of catalysts.
There are several different ways that catalytic surfaces can be trained towards a particular reaction sequence or energy budget. Lattice strain control [1] has been conceptually available for several decades [2] and generally involves applying biaxial or, less commonly, uniaxial strain within the surface plane of the catalyst. There are a number of possible consequences of this action, and their relative importance depends sensitively on a number of factors, including the stoichiometry and structure of the catalyst under investigation, the working conditions of the catalysts, the way that the strain is applied and the type of investigation being performed. In the latter point, the main distinction made in the current work is between investigations which are performed experimentally and those that are performed theoretically, or computationally. In these latter investigations, systems are reduced in their complexity, such as the trial systems commonly presented in density functional theory (DFT) investigations. Other computational methods can model larger systems and investigate larger scale phenomena, e.g., reaction processes around extended surface clusters, but the computational tools used, such as empirical force fields, require considerable refinement and training to produce simulations which are physically meaningful.
The current manuscript is a purely computational work. However, it is worthwhile to review some of the more important experimental phenomena observed to reinforce the importance of some of the investigations that will be performed in the current work and how applying strain can enable some of these phenomena. Fundamentally, there are three classes of phenomena: ensemble, ligand and geometric. Ensemble effects occur when dissimilar species that form small groups (ensembles) acting individually demonstrate distinct mechanistic characteristics. These groups can form on a surface by, for example, relaxing the surface tension and applying heat: this reduces the energy barriers to diffusion and provides individual atoms with sufficient energy to move between binding positions. Ligand effects are associated with charge transfer between atoms, and geometric effects are associated with the relative position of atoms. These latter effects are the primary control in the current work, as the strain applied in the current simulations will move the surface atoms either closer together or farther apart. They can also manifest in a way similar to the ensemble effects earlier: if, for example, a surface phase transition is encountered under strain, then the symmetry and surface structure can change significantly.
Though these effects are often presented in the literature separately, they often inter-mix. Ligand and ensemble effects can often accompany changes in surface strain. For a clean surface of a pure material, this phenomenon is well understood: the number of bonds that atoms in the surface layer form are different from the those that atoms in deeper layers will form, and consequently an internal strain arises. This phenomenon can be extended to the cases of thin film growth where an overlayer is deposited on a substrate of a different material. In this case, in addition to the differences in valence between the atoms in the overlayer compared to their valence in bulk form, a lattice mismatch may also exist between the deposited layer and the substrate, internally straining the deposited layer. These phenomena have been described in terms of single crystal systems which are of significant interest to computational researchers—particularly those specializing in density functional theory and ab initio methods—as they are computationally accessible. However, several related phenomena exist which are of more interest to experimental teams or practitioners of force-field techniques. These phenomena exist across nanoparticles, which because of their large surface area, are particularly practical catalysts. Reducing the size of the nanoparticles will generally reduce the size of the lattice constants of these particles [3] as a way of relieving changes in the surface energy. This phenomenon becomes more complex in the case of bimetallic and alloy systems where, as well as changes in the surface strain and energy, thermal processing of the nanoparticles—a common feature in modern catalysts, which are often run at elevated temperatures—will cause the formation of pure metal overlayers. This is the case in the current investigations into PtxNi1-x and is central to the investigations in the current work.
The application of mechanical strain to a surface can also effect changes to the reactivity of the surface and its effectiveness as a catalyst. For example, applying a uniaxial compression to Pt(100) [4] causes a reduction in the energy difference between the initial and transition state energies, breaking the scaling catalysis laws [5] and improving the effectiveness of the surface as a catalyst. In addition [4], several auxiliary promotion methods, including the application of magnetic and electric fields, have been observed experimentally for similar systems.
Recent experimental studies [6] have used aberration-corrected high-resolution transmission electron microscopy on Pt−Fe alloy core-shell nanoparticles with Pt-rich surfaces to directly evidence the presence of lattice strain and its effect on oxygen reduction reactivity. More generally, the experimental technique of seed-mediated coreduction (SMCR) has recently been developed [7] and allows PtM (where M = Ni, Co, Cu or Fe) shells to be deposited on intermetallic seeds. The performance of these nanocatalysts has been significantly better than the equivalent Pt reference, with PtCu and PtNi shells notably demonstrating a 230% and 270% activity increase in activity towards the oxygen reduction reaction (ORR). Pt-skinned CoPtx [8] has shown similar effectiveness towards the hydrogen evolution reaction (HER). Equally, the application of external strain to Pt(111) has been shown to improve the rate of formate yield during formic acid oxidation [9]. Improvements in activity are not simply confined to materials with Pt incorporated into a surface layer. The manipulation of Ni3Fe thin films via externally applied strains [10] has been shown to control the activity of the electrocatalytic oxygen evolution reaction (OER). This study is significant in the current context, as both Ni and Fe are ferromagnetic compared to the nonferromagnetic Pt; the importance of this ferromagnetism and its relative importance to the externally applied strain have not been fully investigated in the original work [10] or in subsequent publications but may prove significant.
Based on the current published database of catalyst performance, some systematic design protocols have been proposed. Examples of the design protocols have been based on the chemical composition, atomic coordination, and strains of the top surface layers of nanocatalysts [11]. Other examples use strain engineering and localized lattice strain [12], which focuses development on the d-band model of surface reactivity and the known high reactivity of defects compared to defect-free surfaces and facets. The importance of defects and subsurface vacancies has been shown in fundamental studies of Pt-based catalysts [13]. Further protocols have focused on ensemble effects in Pt overlayer [14] using a kinetic-mapping technique.
Whilst these broader characterizations have demonstrated some success, there is still the need to develop a fundamental understanding of Pt-based catalysts—for example, in the empirical modelling of the pure metal under strain [15]—as a characterization and potential systematic design tool. This approach has been applied, in part, in recent studies which evaluated both the strain and the ligand effects [16] by systematically geometrically constraining Pt-bearing nanoparticles but allowing adsorbates to fully relax on them. These studies identified Pt–Au–M nanoparticles (M = Cr, Mn, Co, Cu, Zn) as promising electrocatalysts for the ORR, due to their inherently weak Pt–OH binding with respect to a pure Pt nanoparticle. This fundamental approach highlights the importance of single crystal methods, e.g., [17], even amongst the literature which is increasingly dominated by nanoparticle and multiple-facet studies, and as an essential part of the important bridging studies [18,19] between the more idealized single crystal methodology and the less computationally accessible nanoparticle simulations. One recent example of single crystal studies has been a broad survey [20] of low index transition metal surfaces and their potential reactivity towards the HER. This survey concluded that under applied strain, the hydrogen atoms across a range of surfaces could migrate between preferred binding sites with a concurrent change in local valence, binding energy, and potentially significant changes to their reactivity.
The current work will focus on density functional theory investigations of single crystal systems. This approach has very recently proven successful in determining the stability of Pt skins overlying a range of intermetallic layers [21] and in the activity of Pt/PtV(111) towards the ORR [22]. The current investigations will focus on the multilayered (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtN3i using a full spin-polarized approach. Recent studies [23] of the magnetic character of the ordered bulk L10 and L12 phases of Pt3Ni, PtNi and PtNi3 using both scalar and vector relativistic density functional theory (DFT) have shown that the effect of applying compressive or tensile strain to each alloy is to decrease or increase the magnetism of the alloy, respectively. The PtxNi1-x bulk alloys considered in that work [23] are part of a series of experimentally accessible alloys, which include PtxM1-x (M = Fe,Co), and therefore, each contain a permanently magnetised (Ni, Fe or Co) component. Comparative studies of these materials are therefore beneficial as they can highlight trends within this class of bimetallics. The response of the magnetic moment of the L10 and L12 ordered phases of Pt3M, PtM and PtM3 (M = Fe,Co) to compressive and tensile strain [24] has shown that the magnetic moment of the Pt3M and PtM phases varies linearly with applied strain. The response of the PtM3 alloys shows significant transition in the rate of change of magnetic moment at approximately zero strain. The magnetic moment changes of PtxFe1-x and PtxCo1-x during strain arise from intraorbital charge transfer between d orbitals. These background studies demonstrate some of the characteristics of the ordering of the substrate layers considered in the current work.
The computational investigations in the current work have been prompted by the experimental observation [25] that the Pt3Ni(111) activity towards the ORR is 10-fold the activity seen on Pt(111), and similar observations have been made on Pt3M surfaces [26]. Due to the technological importance of both the HER and ORR systematic investigations, a systematic series of DFT investigations have been completed for the strained H/Pt(111) [27], O/Pt(111) and OH/Pt(111) [28] systems. These investigations have shown that for the H patterned surfaces, and for smaller unit cells of the oxygenated surfaces, applying biaxial strain in the surface (111) plane changes the lowest energy binding position of the adsorbate. This result is of fundamental importance, particularly with adsorbates that have directional bonds with the substrate, such as oxygen, because changing the valence of these adsorbates with the surface will significantly change the available valence charge of that adsorbate to other reacting molecules.
Single crystal experimental studies of the Pt3Ni(100), (110) and (111) [29] have demonstrated that some refinement of the single overlayer model routinely used in computational investigations in this field is necessary. After thermal processing of nanoparticle Pt3Ni, a conventional core-shell structure develops composed of pure metal overlayers and an alloy. However, in the case of the PtxNi1-x, the Pt surface layer has been shown to lie on a purely Ni second (selvedge) layer. The current work will therefore investigate the effect of this magnetic selvedge layer. The current work will also investigate the effects of unit cell size during the oxygenation of Pt(111) under strain and will further provide a benchmark equation of state and surface energy data for the clean Pt(111) under strain. These investigations will quantitatively evaluate the density functional used and seek to establish a broad computational error bar in the binding energies and bond lengths, determined using both local density approximation (LDA) and the generalised gradient approximation (GGA).
2. Computational Methods
The investigations performed in this work used the Quantum Espresso density functional theory (DFT) package [30]. Spin-polarized simulations were performed using both the local density approximation (LDA) and the generalized gradient approximation (GGA). Hybrid functionals, which would nominally have required an order of magnitude more computational time, were not considered in the current work.
The reason that the two sets of calculations were performed was to estimate the computational error in the current set of investigations, which will assist experimental teams who may wish to compare their experiment results with the results presented in the current work. The reason that these two approximations were used was because the current work focusses on the bond lengths and the cohesive, surface and adsorption energies. The LDA is conventionally found to overestimate cohesive energies and underestimate bond lengths. The GGA tends to overcompensate for the LDA overbinding and consequently overestimates the bond lengths. Comparative studies of the two functionals can then at least approximately provide an upper and lower estimate of the expected values of these quantities. Furthermore, the two functionals are computationally accessible for the size of systems that are being investigated in the current work.
The Perdew–Zunger (PZ) LDA and the Perdew–Burke–Ernzerhof (PBE) GGA exchange-correlation functionals were used to generate the norm-conserving pseudo-potentials [31] used in the current simulations. A wave-function kinetic energy cut-off of 75 Ry and a charge density/potential cut-off of 300 Ry were used throughout these investigations, and a Brillouin zone sampling of (10 × 10 × 10) and (6 × 6 × 1) was used for the bulk and surface investigations, respectively. A first-order Methfessel–Paxton smearing of 0.02 Ry is used throughout this work [32].
The cohesive energy of the bulk Pt crystal is defined in Equation (1):
where is the energy of the primitive bulk crystal, which contains 1 Pt atom/unit cell. is the energy of a single Pt atom in the gas phase, which was calculated by setting the atom in an orthogonal box with dimensions 10 × 10 × 10 Å3. (1 × 1 × 1) unit cells were used to perform the bulk Pt and bulk alloy investigations.
The surface energy of the clean Pt(111) slab was calculated using Equation (2):
where A is the total surface area of a slab containing atoms with a total energy . (1 × 1) surface unit cells were used to perform the unreacted (clean) Pt(111) investigations.
The energies E of any density of states which have been considered in this work are normalized to the Fermi level EF, i.e., EF = 0 eV. The centre and width of each of the density of states are defined in terms of their first and second moment, respectively [33].
Strain σ was defined in Equation (5) in terms of general lattice constants L and L0, where L0 is the lattice constant L at equilibrium.
We applied the strains biaxially throughout the current work, so the surface lattice constant along the directions was changed by the same fractional amount when strain was applied to surface FCC (111) simulations. All atoms in each strained slab were allowed to fully relax, except for the central atomic layers. Each Pt(111) simulation had 6 Pt layers, whilst each alloy simulation had 7 layers of Pt3Ni or PtNi3 alloy with further Ni and Pt layers—detailed later in the text—on each side of the slab. Subsequent slabs were separated by a vacuum with a width of approximately ten interplanar distances. For bulk simulations, the strains were applied identically and simultaneously along the [100], [010] and [001] directions. In the bulk case, however, no subsequent relaxation was considered.
To simulate the adsorption of atoms, the slabs were patterned symmetrically by attaching adsorbate atoms—either oxygen or hydrogen—in the same binding position on either side of a symmetric slab. The adsorption energy of was then defined as
where is the Kohn–Sham energy of the adsorbate patterned slab, is the energy of the unpatterned slab and is the energy of the adsorbate. The factors of 2 and ½ account for the patterning by adsorbates on both faces of the slab. Symmetric slabs were used throughout the current—with one minor exception, which is discussed in the text. This means that all quantitative outcomes were calculated using slabs which were either entirely unreacted or which were reacted identically adsorbate on either face. (3 × 3) and (2 × 2) surface unit cells were used to simulate the reacted Pt(111), and the Pt/Ni/Pt3Ni-(111) and Pt/Ni/PtNi3-(111) surfaces, respectively.
3. Results and Discussion
This section is structured in the following way: first, the bulk Pt and unreacted Pt(111) surface are investigated comparatively using the LDA and GGA exchange correlation functionals before investigating the reactivity of the Pt(111) surface towards oxygen and hydrogen. A similar methodology is then performed to investigate the more complex Pt/Ni/Pt3Ni-(111) and Pt/Ni/PtNi3-(111) surfaces before investigating their reactivity towards oxygen and hydrogen. The effects of adsorption on these surfaces are discussed with some particular focus on the magnetisation of the various surface, the origins of this magnetisation and the effects of adsorption on the magnetisation.
3.1. Unreacted Bulk Pt and Pt(111)
The orthogonal unit cell of the bulk FCC Pt crystal is shown in Figure 1.
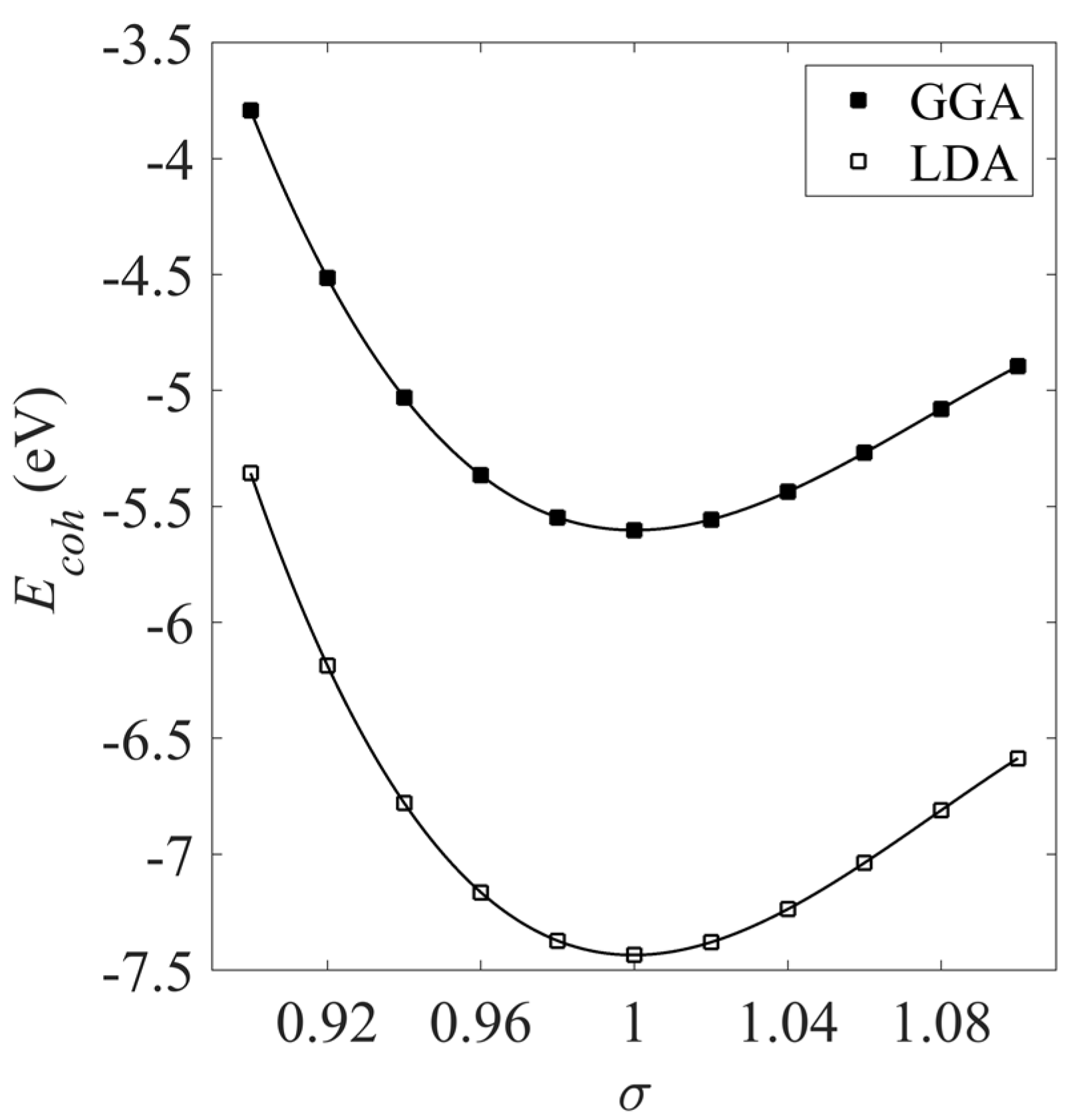
To determine the equilibrium lattice parameter a, the cohesive energy Ecoh of the unit cell was calculated at different strains σ. The resulting equations of state are shown in Figure 2 and were calculated using the LDA and GGA approximations. The equilibrium lattice spacing occurs at the minimum of these curves.
Table 1 summarizes the bulk lattice parameters, equilibrium cohesive energies and magnetic moments for both the bulk Pt system (shown in Figure 1) and for the bulk Ni system, which has the same FCC structure as Pt. It can be seen from Table 1 that the LDA tends to underestimate the bond lengths compared to the experimental values. To compensate for this, the GGA estimates a lower cohesive energy compared to the LDA cohesive energies, and this can be seen in Figure 1. This lower estimation of the energy predicts longer equilibrium bond lengths for the GGA in Table 1.
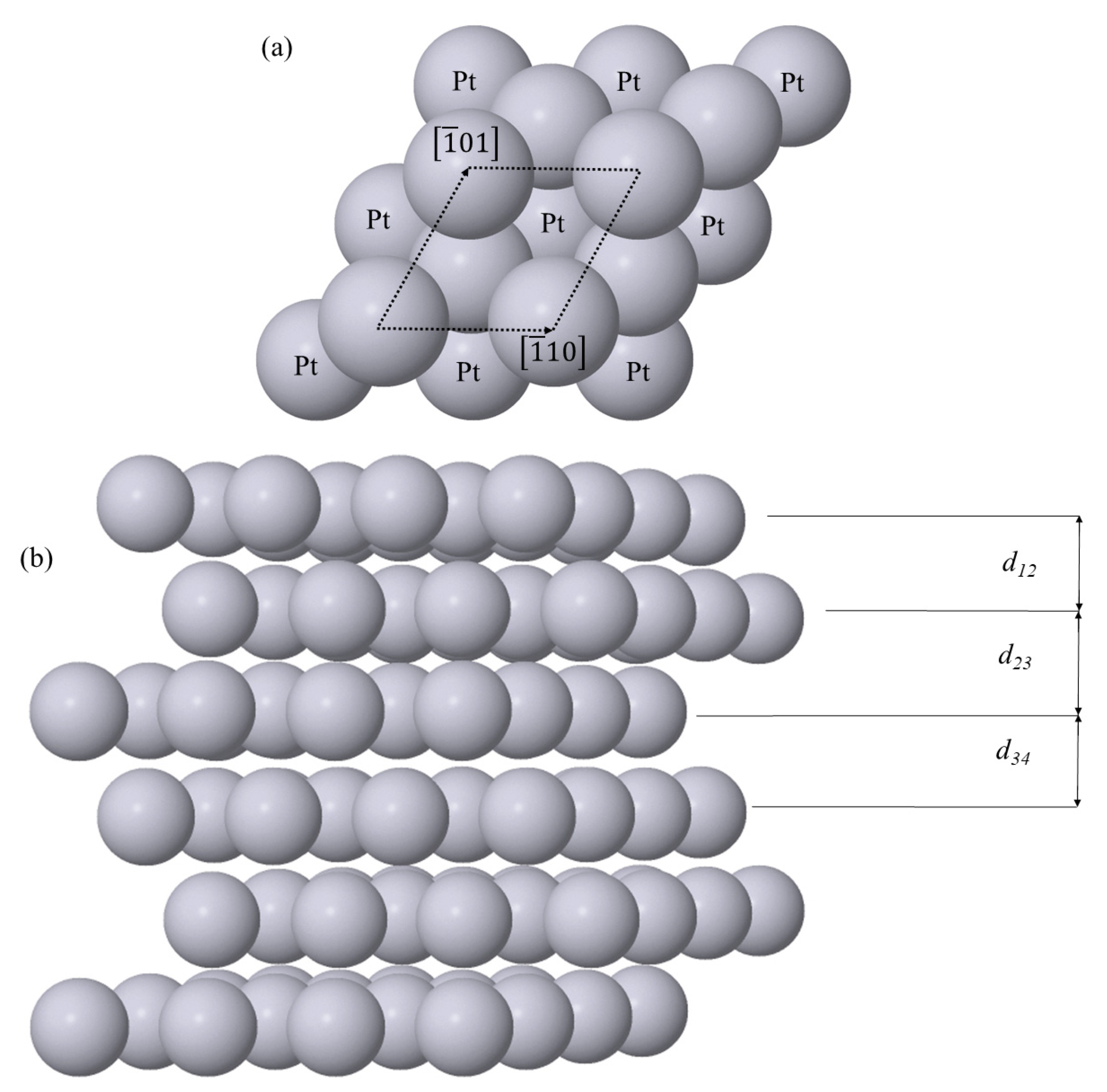
Figure 3 shows the (a) Pt(111) surface and (b) the same slab in cross-sectional view. When strain is applied to the surface, the lattice constant along both the orthogonal directions was changed by the same fractional amount. Once this strain was applied, the atoms within the slab could relax freely, except for the central two layers which were constrained.
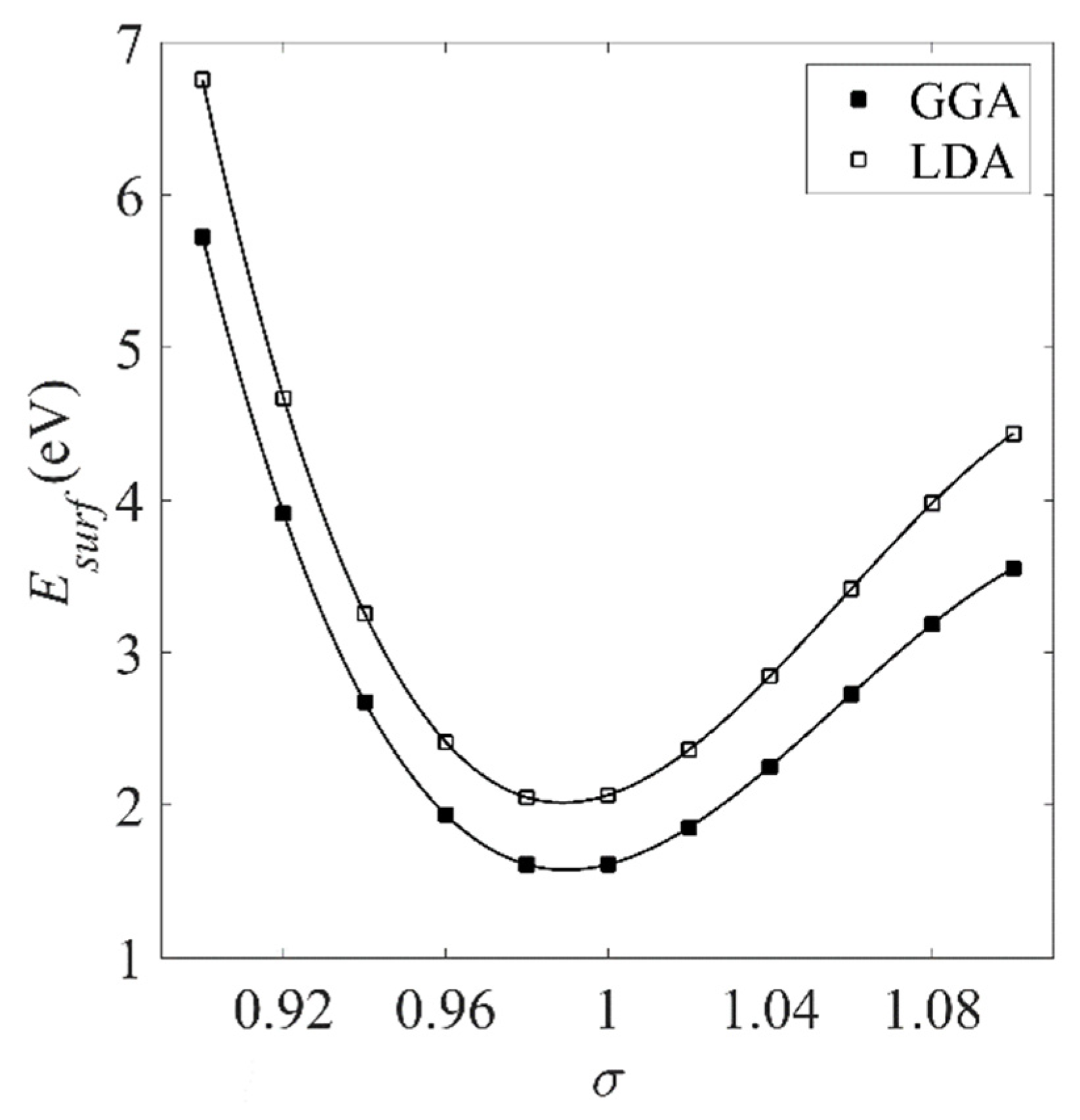
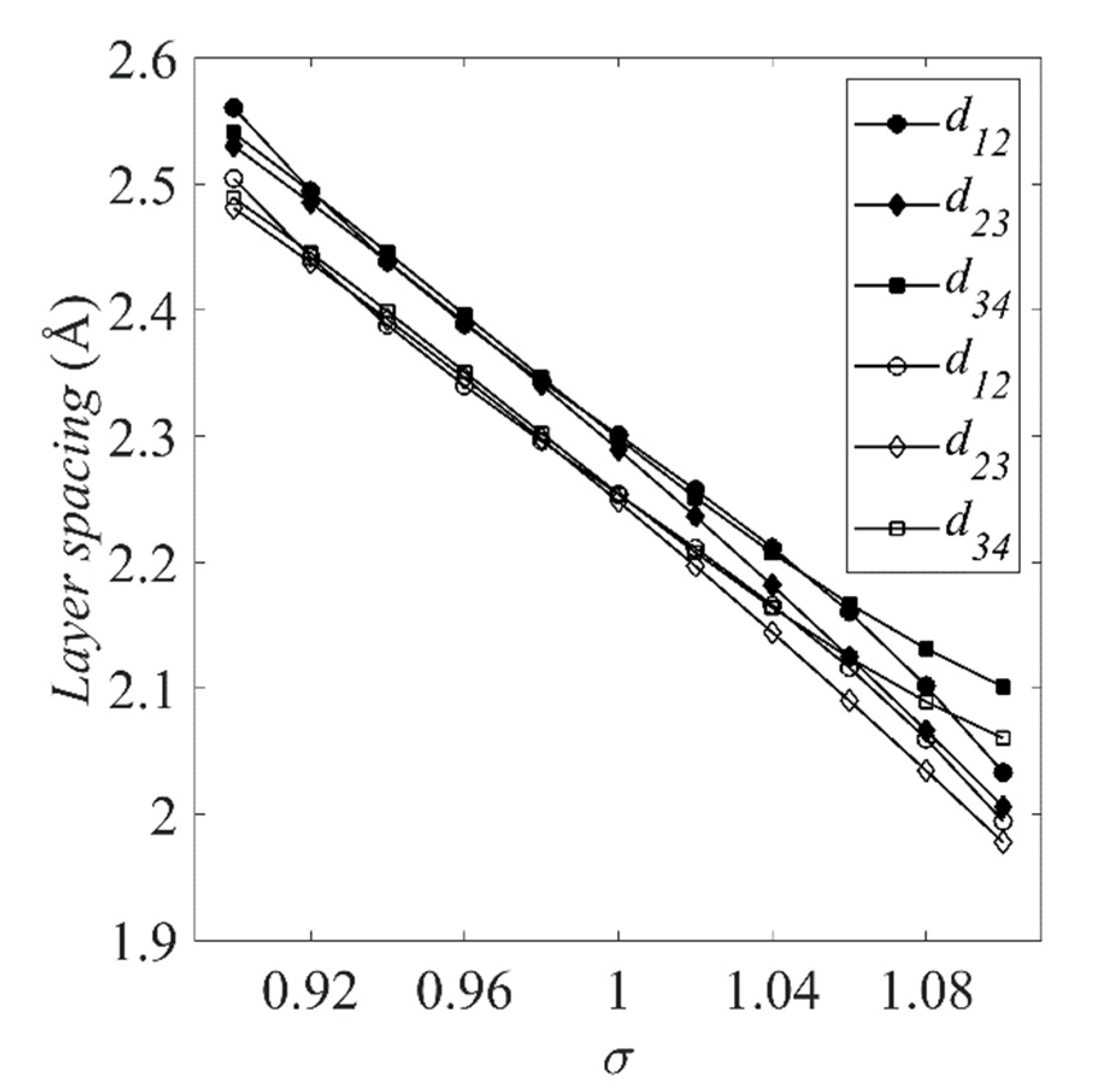
The resulting surface energies and layer spacings are shown in Figure 4 and Figure 5, respectively. Esurf is overestimated by the LDA in the same way that the LDA cohesive energies were overestimated in Figure 2, with the GGA reducing the overestimation in both cases.
Figure 5 shows the layer spacings for the Pt(111) slab defined in Figure 3. For σ < 1.02, the GGA estimates larger layer spacings.
The results and analysis in this section have so far focused on the energetic and the structural character of the bulk and unreacted pure metal surfaces. In order to develop the description of these systems, the projected density of states is now considered. Following Equations (3) and (4) in the ‘Computational Methods’ section, the key metrics which will be used in the current work are the centre Ec and width w of the density of states. Figure 6 shows explicitly how these quantities develop for bulk Pt. The centre of both the d and the sp bands approaches the Fermi level as the strain σ increases, i.e., as the atoms in the bulk become increasingly separated. Accompanying this shift in Ec, the width w narrows. These two phenomena can be interpreted as the atoms approaching a more gas-like state as σ increases and are indicative that over the range of strains considered for the bulk Pt phase, a structural or electronic phase transition might not ordinarily be expected.
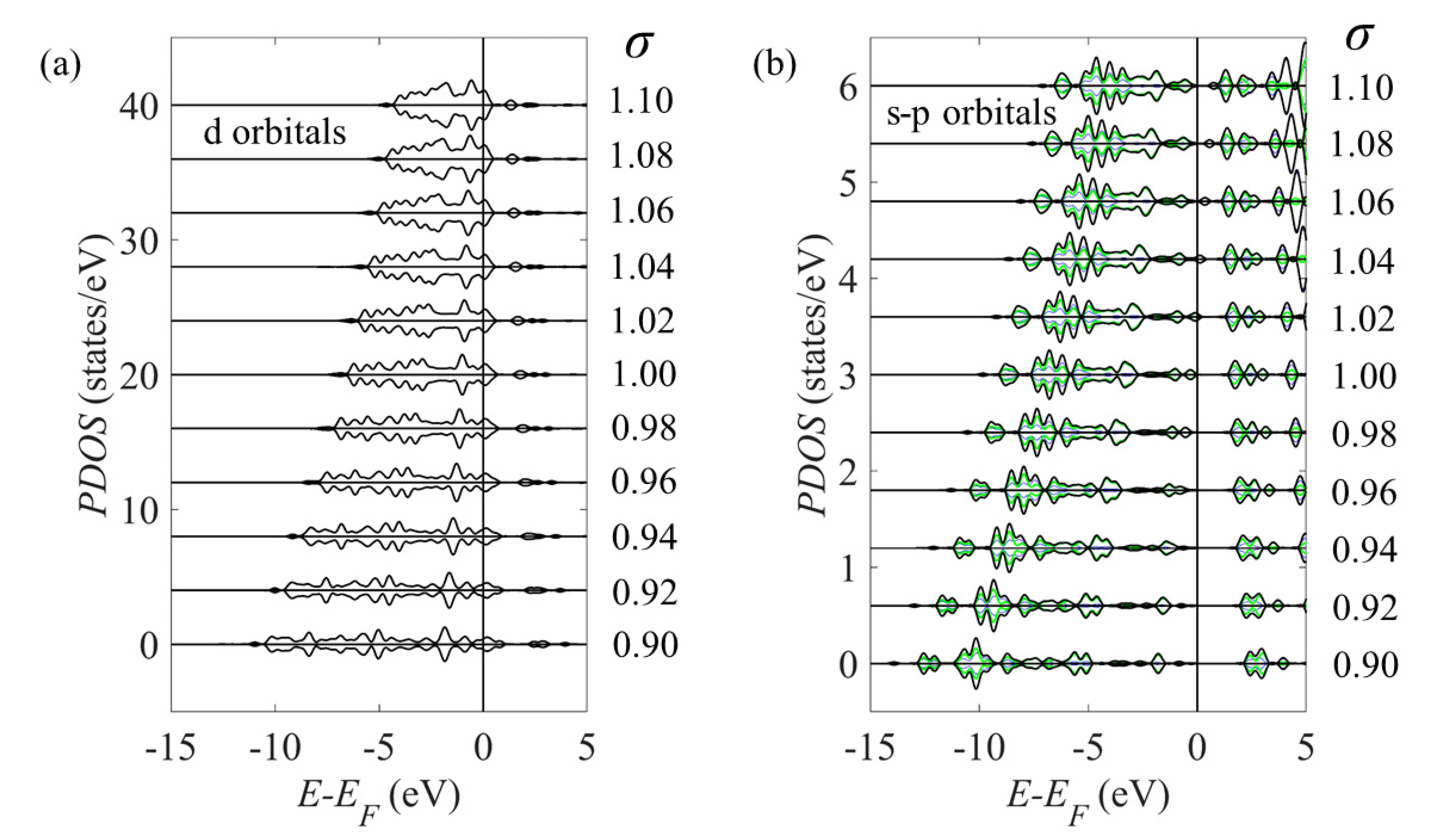
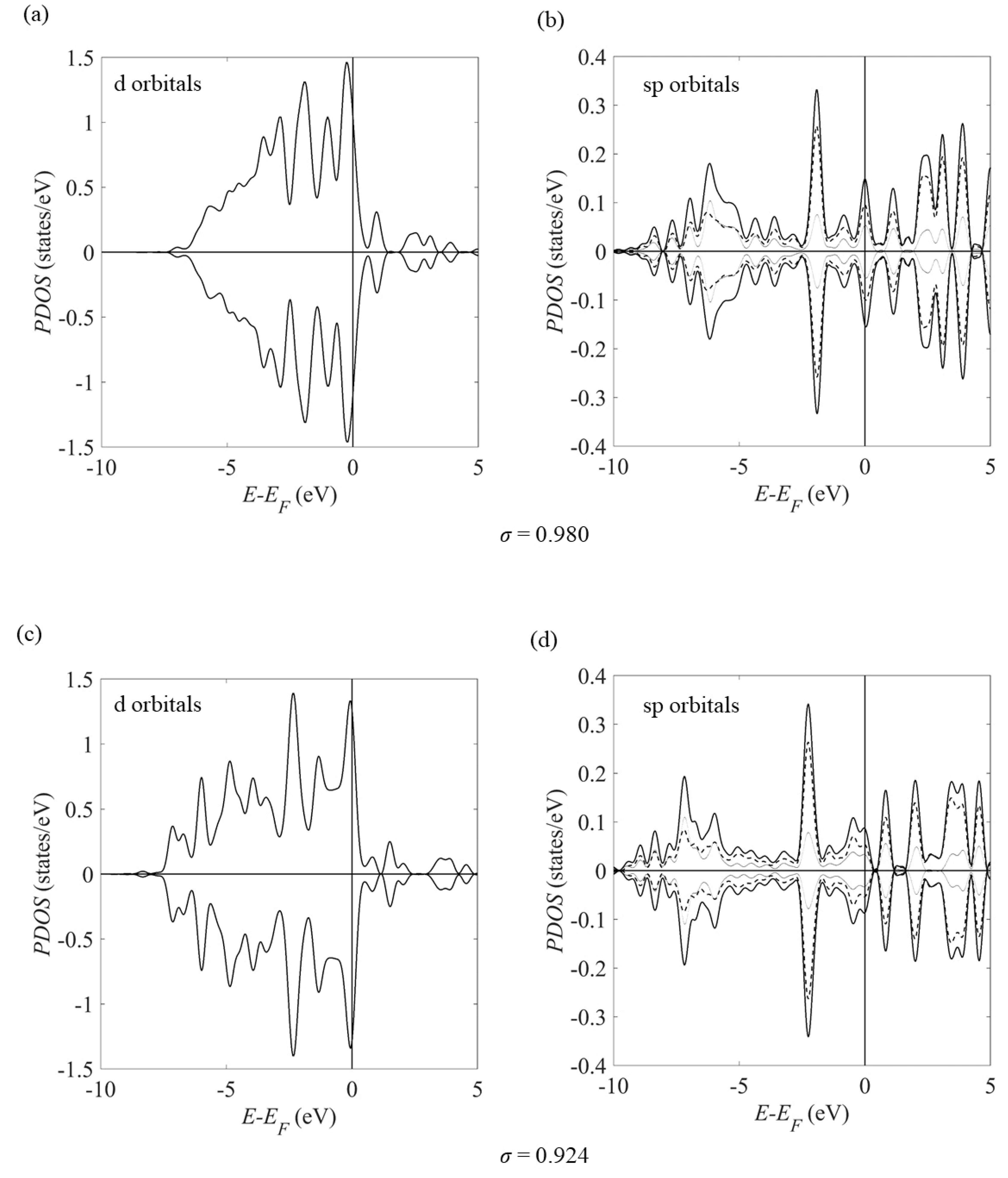
Figure 7 shows the surface projected density of states (PDOS) in a strained Pt(111) slab at strains which correspond to the surface lattice constants of (111)-Pt3Ni and PtNi3 and shows little evidence that the surface Pt atoms become magnetised simply due to strain alone.
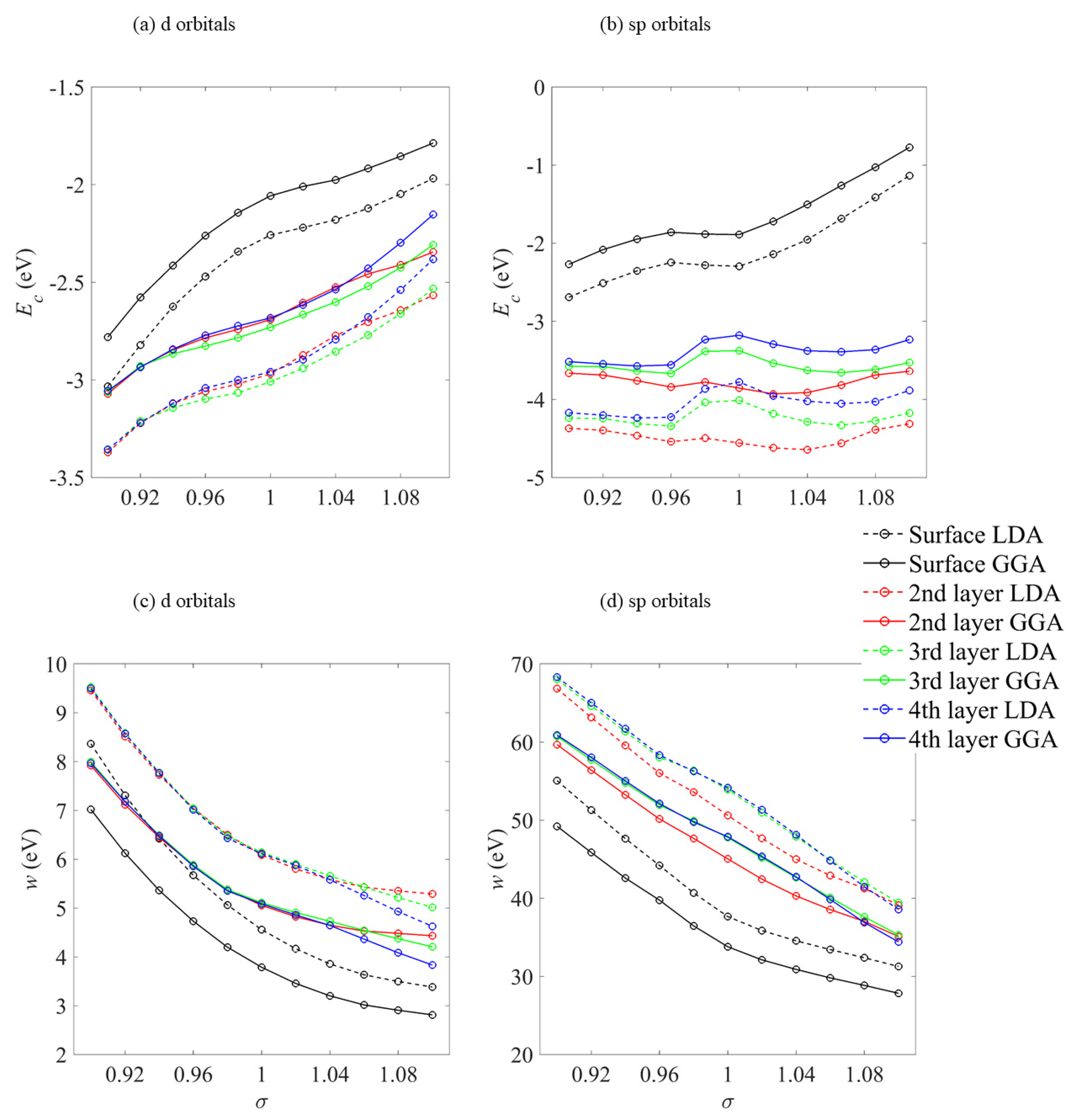
The layer-wise variation in the centre Ec and width w of the density of states under strain is summarized in Figure 8 which shows that the surface Pt Ec approach the Fermi level as strain becomes increasingly tensile and the distance between the atoms becomes greater. This effect is largely independent of the functional. For the deeper level atoms, the effect is only apparent for the more directional d bonds. The sp band is less directional, containing the diffuse s electrons which are less sensitive to the changes in the lattice constant. This is evidenced in the small gradient of the second and deeper layer atoms in Figure 8b.
3.2. Reacted Pt(111)
In the previous section, the energetic, structural, magnetic and electronic character of the Pt(111) surface under strain was presented. In the current section, the consequences of this character will be demonstrated in the response of the surface to oxygen and hydrogen.
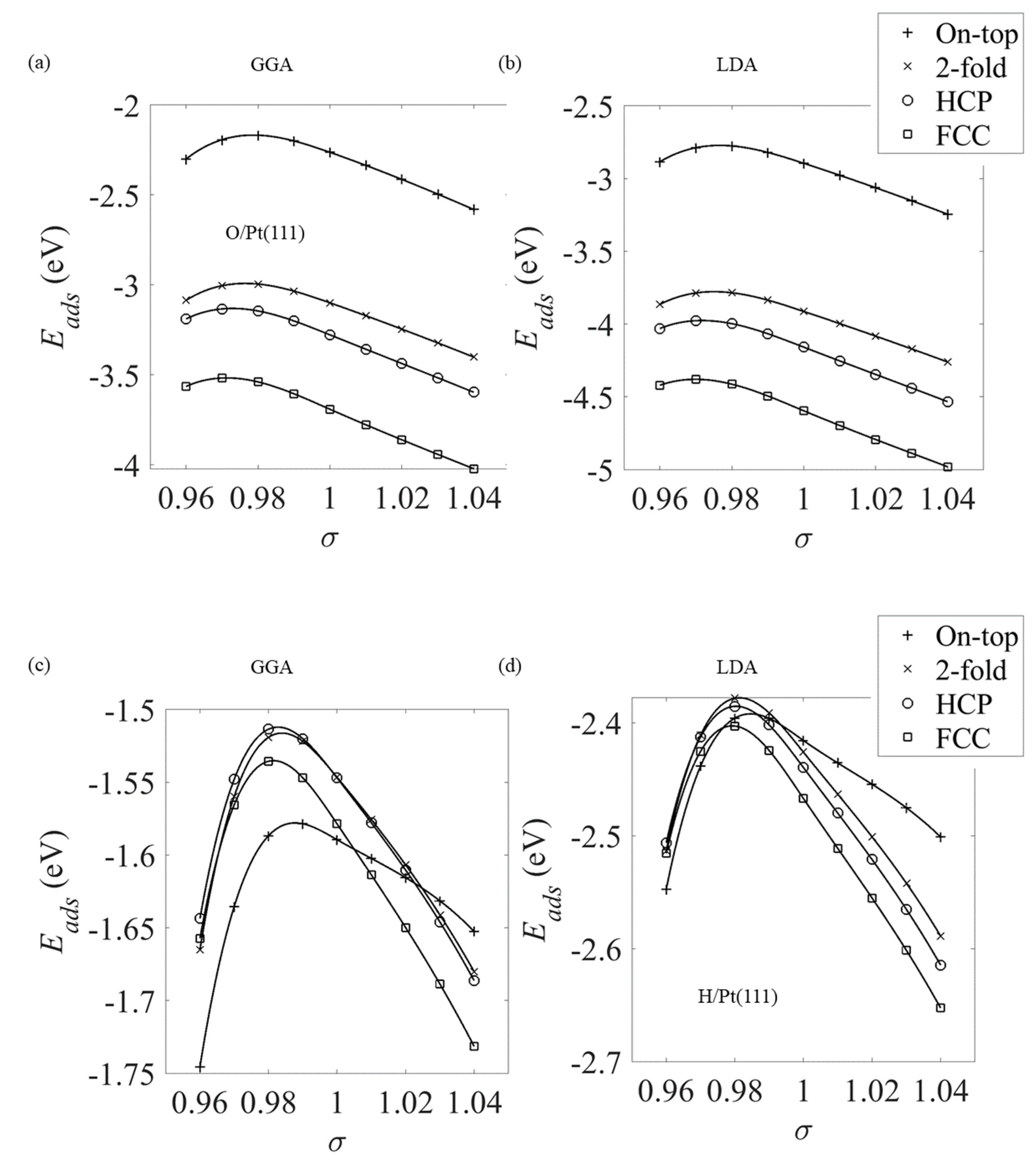
Figure 9 shows the high-symmetry binding positions on the Pt(111) surface together with the (3 × 3) unit cell used in the current simulations. Figure 10 shows the adsorption energy under strain σ for (3 × 3)-O/Pt(111) and (3 × 3)-H/Pt(111). The oxygen atoms are seen to preferentially occupy the FCC sites for all strains. This observation is different from previous simulations [28], which showed that for compressive strains (σ < 0.97), the oxygen atoms preferentially occupy a bridge site. However, these earlier studies used a smaller (2 × 2) surface unit cell and observed a reconstruction of the surface, particularly for the related case of hydroxyl adsorption. This reconstruction does lead to a lower energy binding position—the values of Eads in the earlier work are noted to be of the order of 0.5 eV lower than those in the current work. Consequentially, oxygen adsorption on strained Pt(111) should be considered in low coverage (current work) and high coverage regimes. The interactions in the high coverage regime are not fully characterized, but the current evidence does suggest that lateral O-Pt interactions are significant.
To provide a direct quantitative comparison between oxygen and hydrogen adsorption on the strained Pt(111) surface considered in the current section and adsorption on the alloy-based surfaces considered later in this work, Table 2 shows the adsorption energy Eads for the unstrained (σ = 1) and compressively strained surfaces. The results in this table are consistent with those seen earlier in the current work—for example, in Figure 2 and Figure 4—where the LDA is seen to overbind and consequently predict larger Eads. The results in Table 2, more importantly, can be used to discuss the error in this energy, which arises from the choice of functional. Generally, oxygen is more tightly bound to the Pt(111) surface independent of the strain state.
3.3. Unreacted PtxNi1-x and Pt/Ni/PtxNi1-x
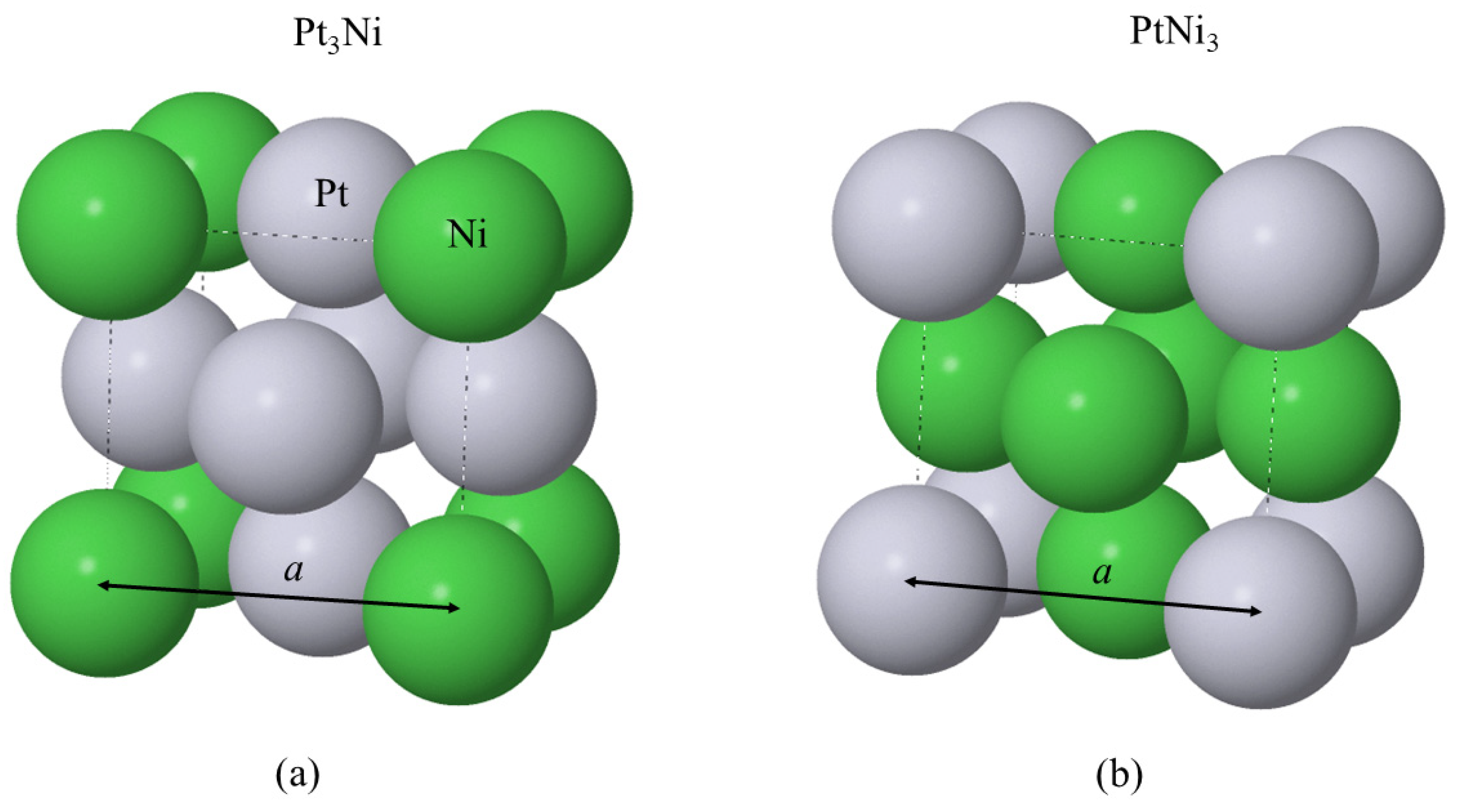
Thus far, the results in the current work have focused on pure Pt in either bulk or slab form. This section now investigates the unreacted Pt3Ni and PtNi3 alloys in both bulk and slab form. Figure 11 shows the bulk unit cells of these alloys.
Table 3 shows the equilibrium lattice parameters, cohesive energies and magnetic moments for both unit cells. The Pt atoms can be seen to have developed a permanent magnetic moment in the presence of Ni, which is particularly noticeable for the PtNi3 alloy.
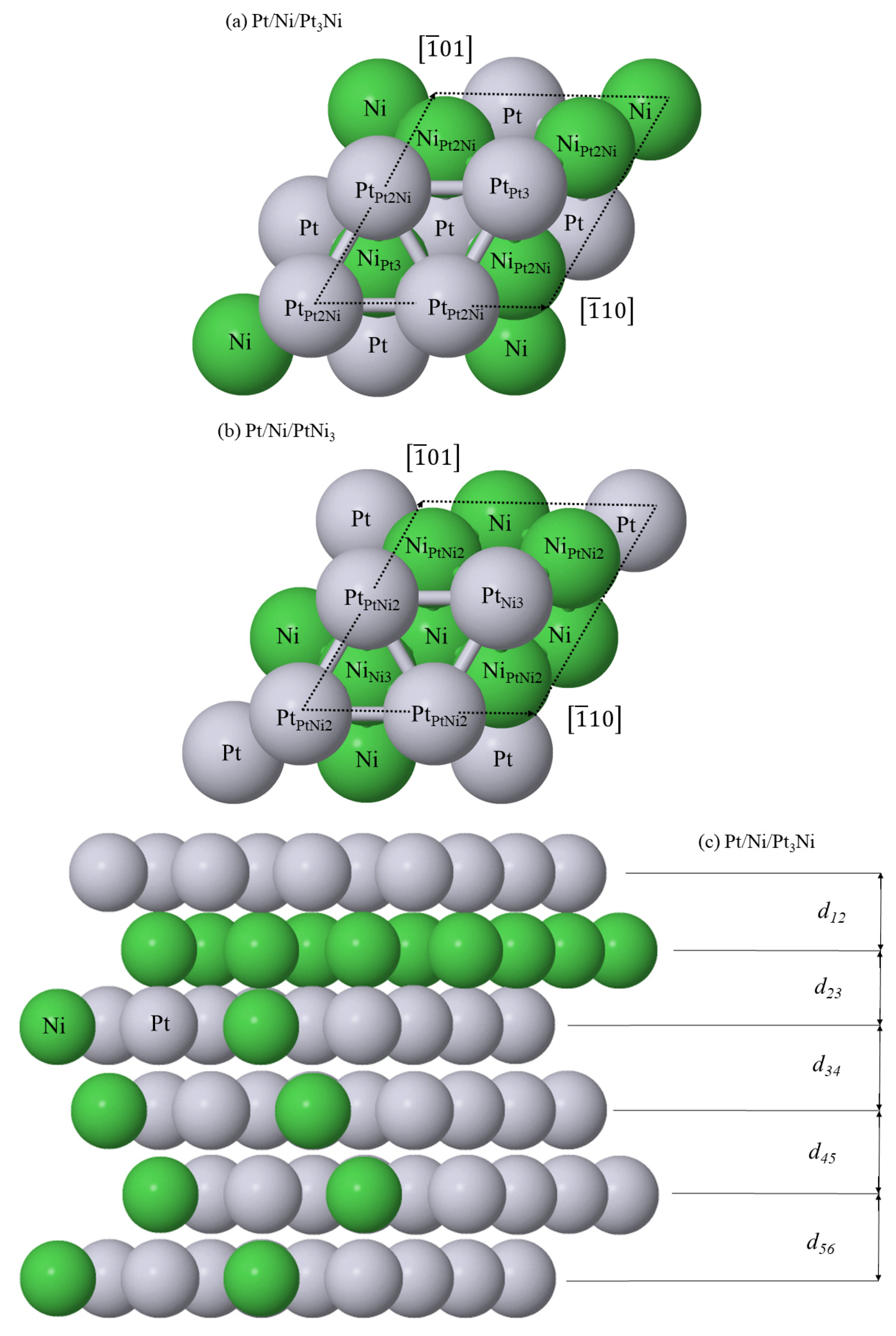
Using the bulk lattice parameters presented in Table 3, the surfaces of unreacted (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 were fully relaxed. Based on the experimental studies of these systems [29], a Ni selvedge layer is set beneath the surface Pt layer. However, because of the complexity of the bulk alloy phase, the surface and selvedge layers are comprised of non-equivalent atoms. This is show in Figure 12a,b, where a subscript notation is adopted to identify the different types of surface and second layer Pt and Ni atom.
Table 4 summarizes the interlayer spacing for the alloy systems. The variation in height between the different surface Pt and second layer Ni species is averaged across each layer.
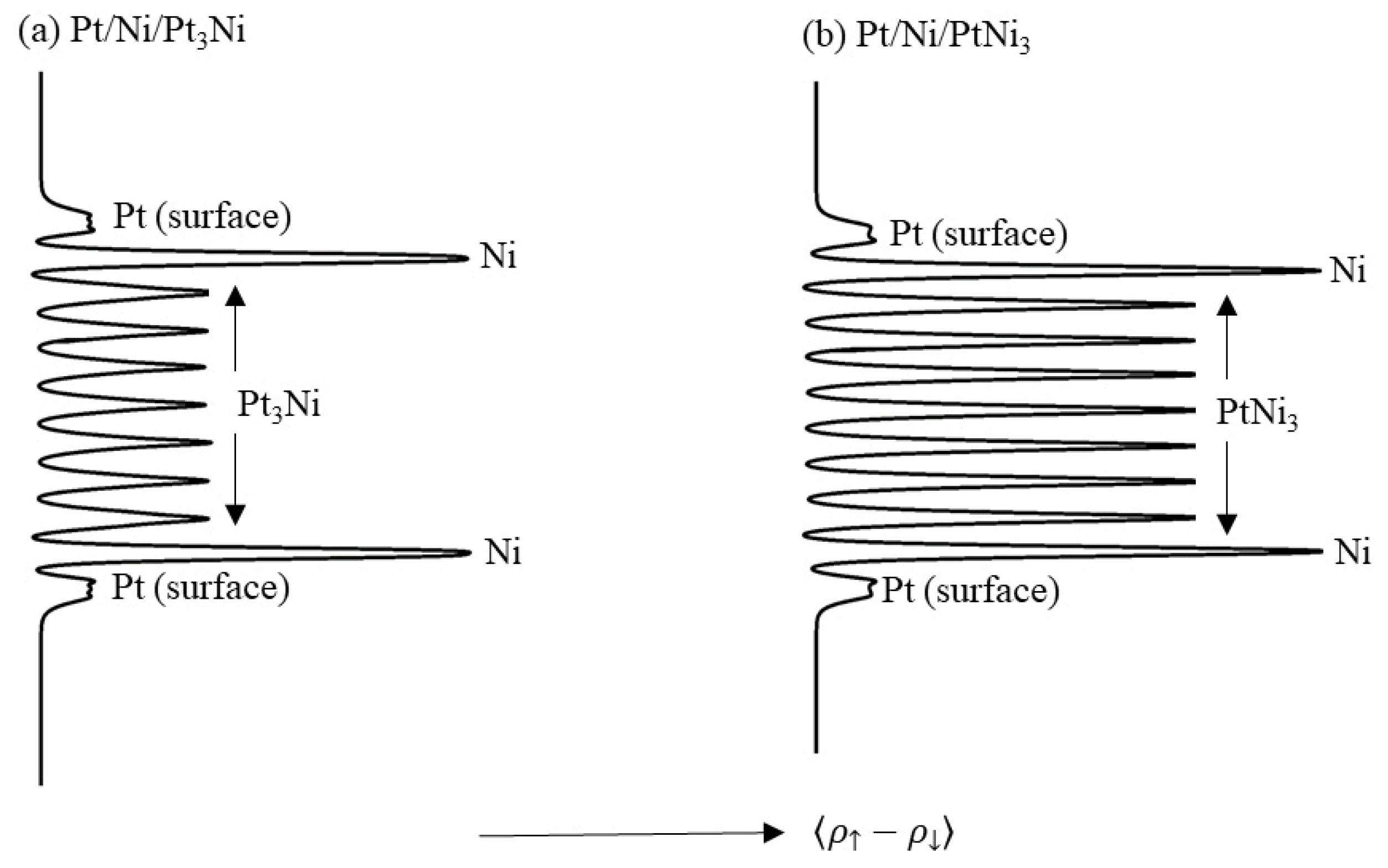
Figure 13 shows the planar-averaged spin density for the clean (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 slabs. Each slab is composed of 7 layers of alloy bounded by Ni selvedge and Pt surface layers, and the averaging is taken parallel to the (111) plane.
Quantitatively, the Pt surface layer has an average spin polarization of 0.006 Å−1 for both slabs shown in Figure 13. The Ni second layer has an average spin polarization of 0.051 Å−1 and 0.061 Å−1 for the Pt3Ni and PtNi3 alloys, respectively. The values for the Ni atoms are consistent with the ferromagnetic moment that the Ni atoms carry. The induced magnetisation in the surface Pt layers is significant, particularly when it is compared to the negligible moment carried by the surface atoms in a pure Pt(111) slab, which was discussed earlier in this manuscript. The consequences of this induced moment in the surface Pt layer for the adsorption of oxygen and hydrogen and the effects of adsorption on this moment will be discussed in the next section.
3.4. Reacted Pt/Ni/PtxNi1-x
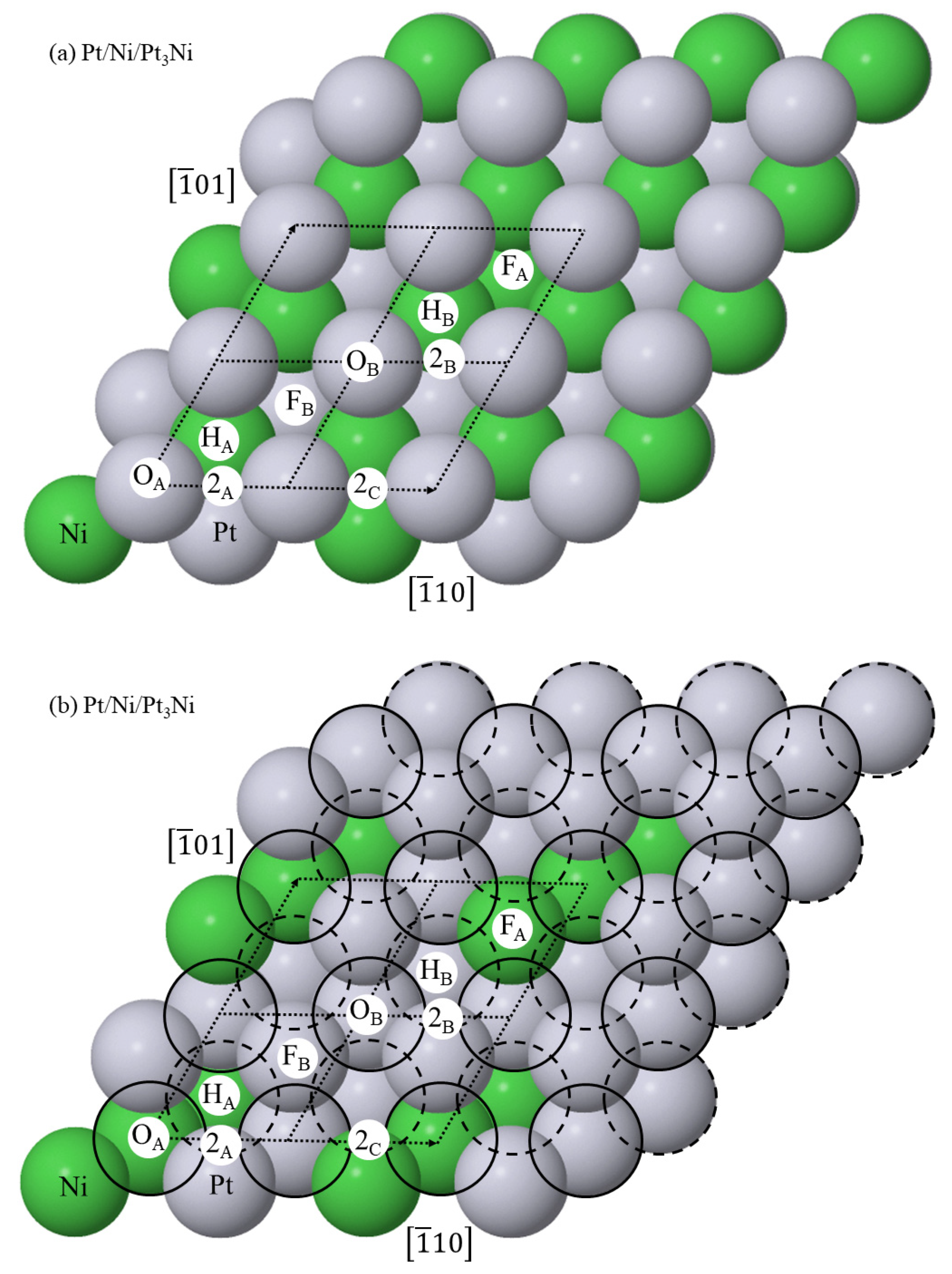
Figure 14 shows the high-symmetry binding positions on the (111)-Pt/Ni/Pt3Ni surface. The binding positions on the (111)-Pt/Ni/PtNi3 surface are identically arranged, though the Pt (Ni) atoms are replaced with Ni (Pt) atoms in the alloy layers.
Table 5 shows the adsorption energies Eads at the high symmetry sites for oxygen and hydrogen. These energies can be compared directly with the adsorption energies for the strained Pt(111) surface presented in Table 2. For oxygen adsorption, the adsorption energies are between 0.176 and 1.081 eV greater on the (111)-Pt/Ni/Pt3Ni surface than on the strained Pt(111) surface, across both functionals. This shows that a ligand effect exists on the (111)-Pt/Ni/Pt3Ni surface which causes the oxygen atoms to bind more strongly to that surface than to the strained Pt(111) surface. Conversely, on the (111)-Pt/Ni/PtNi3 surface, the adsorption energies are 0.863 to 1.874 eV lower than on the equivalently strained Pt(111) surface. The ligand effect does have some depth sensitivity. It does not depend on the nearest neighbour interactions between the adsorbate and the surface Pt atoms and does extend even beyond the second nearest-neighbour interactions involving the Ni atoms in the second (selvedge) layer. For hydrogen adsorption, a similar set of energy differences can be determined. The hydrogen atoms bind between 0.320 and 0.636 eV more strongly to the (111)-Pt/Ni/Pt3Ni surface than to the strained Pt(111) surface, and between 0.897 and 1.437 eV less strongly to the (111)-Pt/Ni/PtNi3 surface than to the strained Pt(111) surface.
Table 6 compares the nearest-neighbour O-Pt and H-Pt bond lengths obtained using both the LDA and the GGA functionals. The LDA predicts shorter bond lengths than the GGA in almost all cases. This is indicative of a functional which is overestimating the bonding and therefore predicting shorter bond lengths than might be observed experimentally. Conversely the GGA tends to underestimate the binding and therefore predicts generally longer bond lengths. For these reasons, experimentalists might anticipate measuring a bond length between the LDA and GGA values presented in this table.
The next step in characterizing the oxygen and hydrogen adsorption is to look in more detail at the density of states of the adsorbate and substrate for the alloy-bearing systems, in the same way that these quantities were analysed for the Pt(111) systems earlier in this work. Table 7 shows the first and second moments of the surface Pt atoms for these systems. As the surface Pt atoms in the strained Pt(111) are not magnetised, both the centre and width of the spin up and down components are identical, so only one value is quoted for each for Pt(111). For both alloys, the width of the spin polarized d-band orbital is larger than the equivalent width for the strained Pt(111). However, the behaviour of the centre of the d-band is less clearly defined. For the Pt/Ni/Pt3Ni, the Ec;Pt are close to the values for both the LDA and GGA simulations. This result is surprising within the conventional d-band model, as it was noted earlier in this section that comparisons of the adsorption energy between the Pt/Ni/Pt3Ni and strained Pt(111) varied significantly with the density functional. For Pt/Ni/PtNi3, the situations appear more consistent with the d-band model, as there is some variation in the Ec;Pt and the and for both the LDA and GGA simulations, with the Ec;Pt changing alignment with the functional, which is more consistent with the d-band model.
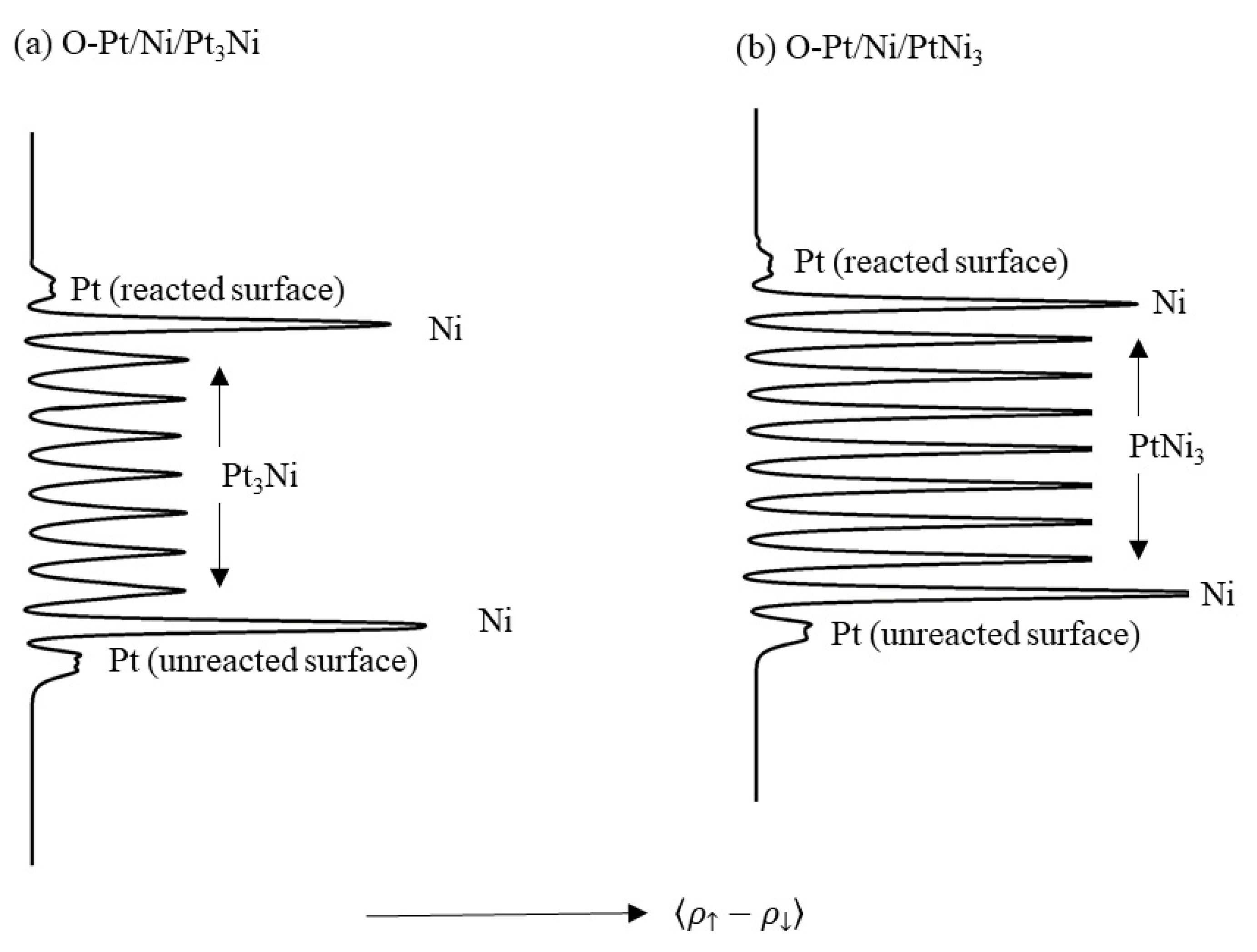
A further observation is that the magnetisation of the surface Pt atoms is generally reduced by both oxygen and hydrogen adsorption. This is shown qualitatively in Figure 15, where the surface average spin moment of the Pt atoms in the reacted surface is less than that on the clean surface.
The observation that the magnetisation of the surface Pt atoms is reduced by both oxygen and hydrogen adsorption is presented quantitatively in Table 8. Comparing the reacted and unreacted surfaces, the adsorption generally reduces the surface Pt atoms moment by up to 75%. This is not the case for on-top adsorption, however, where the magnetic moment is seen to increase.
4. Conclusions
The reacted (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces have been investigated using density functional theory (DFT). These investigations have focused on the binding energy, bond lengths, magnetisation and electronic density of states of adsorbates and the surface Pt atoms during the incorporation of oxygen and hydrogen into the high symmetry ad-sites.
The current work has shown that the surface Pt atoms in the unreacted (111)-Pt/Ni/PtxNi1-x slabs are permanently magnetised. In comparison with appropriately strained clean Pt(111), this magnetisation has been seen to be due to a ligand effect between the surface Pt and the selvedge Ni atoms, instead of being seen as a geometric effect due to the strain.
The adsorption of oxygen and hydrogen into the high symmetry sites quenches this magnetisation. Comparing the adsorption energies of the reacted (111)-Pt/Ni/PtxNi1-x slabs and the reacted, strained Pt(111) surface, it has been shown that this quenching of the magnetisation significantly increases the binding energy of the adsorbate. This effect has been shown to be predicted with both the LDA and the GGA.
A survey of the behaviour of the clean Pt(111) and bulk Pt systems has been included in the current work. This survey has quantified the shifting in the d- and sp-band centres and changes to the width of these bands under strain and has explained this behaviour using a simple bulk-to-gas phase model. The correlation between the adsorption energy of H/Pt(111) and O/Pt(111) under strain and the position of the centre of the d-band has been qualitatively discussed.
Funding
This work was supported by the author’s membership to the UK’s HEC Materials Chemistry Consortium, which is funded by the EPSRC (EP/L000202), and used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.P.; More, K.; Yu, C.; Liu, Z.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts. Nat. Chem. 2010, 2, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Mavrikakis, M.; Hammer, B.; Nørskov, J.K. Effect of Strain on the Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819–2822. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, P.; Pan, Y.-T.F.; Warren, S.; Yin, X.; Yang, H. Surface lattice-engineered bimetallic nanoparticles and their catalytic properties. Chem. Soc. Rev. 2012, 41, 8066–8074. [Google Scholar] [CrossRef] [PubMed]
- Hülsey, M.J.; Lim, C.W.; Yan, N. Promoting heterogeneous catalysis beyond catalyst design. Chem. Sci. 2020, 11, 1456–1468. [Google Scholar] [CrossRef] [Green Version]
- Khorshidi, A.; Violet, J.; Hashemi, J.; Peterson, A.A. How strain can break the scaling relations of catalysis. Nat. Catal. 2018, 1, 263–268. [Google Scholar] [CrossRef]
- Gan, L.; Yu, R.; Luo, J.; Cheng, Z.; Zhu, J. Lattice Strain Distributions in Individual Dealloyed Pt–Fe Catalyst Nanoparticles. J. Phys. Chem. Lett. 2012, 3, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Gamler, J.T.L.; Ashberry, H.M.; Sang, X.; Unocic, R.R.; Skrabalak, S.E. Building Random Alloy Surfaces from Intermetallic Seeds: A General Route to Strain-Engineered Electrocatalysts with High Durability. ACS Appl. Nano Mater. 2019, 2, 4538–4546. [Google Scholar] [CrossRef]
- Lin, L.; Sun, Z.; Yao, H.; Yuan, M.; Yang, H.; Li, H.; Zhang, Q.; Wang, D.; Gu, L.; Sun, G.; et al. Tuning Surface Lattice Strain toward a Pt–Skin CoPtx Truncated Octahedron for Hydrogen Evolution Reaction. J. Phys. Chem. C 2019, 123, 29722–29728. [Google Scholar] [CrossRef]
- Niu, Z.; Wan, Y.; Li, X.; Zhang, M.; Liu, B.; Chen, Z.; Lu, G.; Yan, K. In-situ regulation of formic acid oxidation via elastic strains. J. Catal. 2020, 389, 631–635. [Google Scholar] [CrossRef]
- Wang, A.; Zhao, Z.; Hu, D.; Niu, J.; Zhang, M.; Yan, K.; Lu, G. Tuning the oxygen evolution reaction on a nickel–iron alloy via active straining. Nanoscale 2019, 11, 426–430. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, Z.; Duan, X.; Huang, Y. Nanoscale Structure Design for High-Performance Pt-Based ORR Catalysts. Adv. Mater. 2019, 31, e1802234. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Guo, S. Strain engineering of metal-based nanomaterials for energy electrocatalysis. Chem. Soc. Rev. 2019, 48, 3265–3278. [Google Scholar] [CrossRef] [PubMed]
- Callejas-Tovar, R.; Balbuena, P.B. Effect of Subsurface Vacancies on Oxygen Reduction Reaction Activity of Pt-Based Alloys. J. Phys. Chem. C 2012, 116, 14414–14422. [Google Scholar] [CrossRef]
- Jagannath, M.S.P.; Divi, S.; Chatterjee, A. Kinetic Map for Destabilization of Pt-Skin Au Nanoparticles via Atomic Scale Rearrangements. J. Phys. Chem. C 2018, 122, 26214–26225. [Google Scholar] [CrossRef]
- Shuttleworth, I. Non-linear modelling of the effects of strain on transition metal surfaces. Chem. Phys. Lett. 2016, 666, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Jennings, P.C.; Lysgaard, S.; Hansen, H.A.; Vegge, T. Decoupling strain and ligand effects in ternary nanoparticles for improved ORR electrocatalysis. Phys. Chem. Chem. Phys. 2016, 18, 24737–24745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J. Physical and chemical properties of bimetallic surfaces. Surf. Sci. Rep. 1996, 24, 223–287. [Google Scholar] [CrossRef]
- Yu, W.; Porosoff, M.; Chen, J.G. Review of Pt-Based Bimetallic Catalysis: From Model Surfaces to Supported Catalysts. Chem. Rev. 2012, 112, 5780–5817. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, H. Platinum-Based Oxygen Reduction Electrocatalysts. Acc. Chem. Res. 2013, 46, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, I. Strain engineering of H/transition metal systems. Surf. Sci. 2017, 661, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.K.; Shin, K.; Henkelman, G. Stability of Pt Skin Intermetallic Core Catalysts and Adsorption Properties for the Oxygen Reduction Reaction. J. Phys. Chem. C 2021, 125, 3527–3534. [Google Scholar] [CrossRef]
- Haile, A.S.; Yohannes, W.; Mekonnen, Y.S. Oxygen reduction reaction on Pt-skin Pt3V(111) fuel cell cathode: A density functional theory study. RSC Adv. 2020, 10, 27346–27356. [Google Scholar] [CrossRef]
- Shuttleworth, I. The effects of strain on the ordered phases of NixPt1-x (x = 0.25, 0.5, and 0.75). Chem. Phys. Lett. 2017, 689, 41–47. [Google Scholar] [CrossRef]
- Shuttleworth, I. Magnetism in the strained ordered phases of Pt x Fe 1-x and Pt x Co 1-x (x = 0.25, 0.5, and 0.75). J. Phys. Chem. Solids 2018, 114, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Stamenkovic, V.R.; Fowler, B.; Mun, B.S.; Wang, G.; Ross, P.N.; Lucas, C.A.; Marković, N.M. Improved Oxygen Reduction Activity on Pt3Ni(111) via Increased Surface Site Availability. Science 2007, 315, 493–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamenkovic, V.; Mun, B.S.; Mayrhofer, K.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Nørskov, J.K. Changing the Activity of Electrocatalysts for Oxygen Reduction by Tuning the Surface Electronic Structure. Angew. Chem. Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef]
- Shuttleworth, I. Controlled FCC/on-top binding of H/Pt(111) using surface stress. Appl. Surf. Sci. 2016, 378, 286–292. [Google Scholar] [CrossRef]
- Shuttleworth, I.G. Binding Site Transitions Across Strained Oxygenated and Hydroxylated Pt(111). ChemistryOpen 2018, 7, 356–369. [Google Scholar] [CrossRef]
- Mun, B.S.; Watanabè, M.; Rossi, M.; Stamenković, V.; Markovic, N.M.; Ross, P.N. The Study of Surface Segregation, Structure, and Valence Band Density of States of Pt3Ni(100),(110), and (111) Crystals. Surf. Rev. Lett. 2006, 13, 697–702. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Corso, A.D. Pseudopotentials periodic table: From H to Pu. Comput. Mater. Sci. 2014, 95, 337–350. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vojvodic, A.; Norskov, J.K.; Abildpedersen, F. Electronic Structure Effects in Transition Metal Surface Chemistry. Top. Catal. 2014, 57, 25–32. [Google Scholar] [CrossRef]
- Pisanty, A.; Amador-Bedolla, C.; Ruiz, Y.; de la Vega, M. Band structures of Ni3Pt and NiPt3. Eur. Phys. J. B 1990, 80, 237–239. [Google Scholar] [CrossRef]
Figure 1.
Orthogonal unit cell of the face-centred cubic (FCC) bulk Pt. The lattice parameter a is indicated by the solid arrow. The dashed lines are the edges of the orthogonal unit cell.
Figure 1.
Orthogonal unit cell of the face-centred cubic (FCC) bulk Pt. The lattice parameter a is indicated by the solid arrow. The dashed lines are the edges of the orthogonal unit cell.

Figure 2.
Cohesive energy Ecoh of the bulk Pt crystal estimated using both the LDA and GGA estimates of the exchange-correlation.
Figure 2.
Cohesive energy Ecoh of the bulk Pt crystal estimated using both the LDA and GGA estimates of the exchange-correlation.

Figure 3.
Structure of the (a) surface and (b) cross-section of Pt(111). In the latter, d12, d23 and d34 are the surface-second layer, second-third layer and third-fourth layer spacings, respectively.
Figure 3.
Structure of the (a) surface and (b) cross-section of Pt(111). In the latter, d12, d23 and d34 are the surface-second layer, second-third layer and third-fourth layer spacings, respectively.

Figure 4.
Surface energy Esurf of unreacted Pt(111) estimated using both the LDA and GGA exchange-correlation functionals.
Figure 4.
Surface energy Esurf of unreacted Pt(111) estimated using both the LDA and GGA exchange-correlation functionals.

Figure 5.
Layer spacings of the clean Pt(111) slab shown in Figure 3b. The filled (solid) symbols correspond to results obtained using the GGA; the unfilled (clear) symbols correspond to those obtained using the LDA.
Figure 5.
Layer spacings of the clean Pt(111) slab shown in Figure 3b. The filled (solid) symbols correspond to results obtained using the GGA; the unfilled (clear) symbols correspond to those obtained using the LDA.

Figure 6.
Projected density of states (PDOS) of the (a) d and (b) s-p orbitals for strained bulk Pt. The thin blue and green lines in (b) are the s and p PDOS, respectively, and the thick black line is the sum of the s and p PDOS. Each panel shows the spin up and down components as positive and negative curves, respectively. Subsequent curves are offset vertically for clarity and correspond to different amounts of strain σ.
Figure 6.
Projected density of states (PDOS) of the (a) d and (b) s-p orbitals for strained bulk Pt. The thin blue and green lines in (b) are the s and p PDOS, respectively, and the thick black line is the sum of the s and p PDOS. Each panel shows the spin up and down components as positive and negative curves, respectively. Subsequent curves are offset vertically for clarity and correspond to different amounts of strain σ.

Figure 7.
Projected density of states (PDOS) of the surface Pt atoms in a strained Pt(111) slab. The strains σ correspond to the strain in the Pt surface on a (a,b) Pt/Ni/Pt3Ni and (c,d) Pt/Ni/PtNi3 alloy. The sp orbitals panels (b,d) show the s, p and the sum of the s and p PDOS as thin dotted, dashed and thick solid curves, respectively. Each graph shows the spin up and down components as positive and negative curves, respectively.
Figure 7.
Projected density of states (PDOS) of the surface Pt atoms in a strained Pt(111) slab. The strains σ correspond to the strain in the Pt surface on a (a,b) Pt/Ni/Pt3Ni and (c,d) Pt/Ni/PtNi3 alloy. The sp orbitals panels (b,d) show the s, p and the sum of the s and p PDOS as thin dotted, dashed and thick solid curves, respectively. Each graph shows the spin up and down components as positive and negative curves, respectively.

Figure 8.
Layer-resolved band centre Ec and width w for the d and sp bands on an 7-layer Pt(111) slab. The dashed (solid) lines correspond to estimates made using the LDA (GGA) exchange-correlation functional.
Figure 8.
Layer-resolved band centre Ec and width w for the d and sp bands on an 7-layer Pt(111) slab. The dashed (solid) lines correspond to estimates made using the LDA (GGA) exchange-correlation functional.

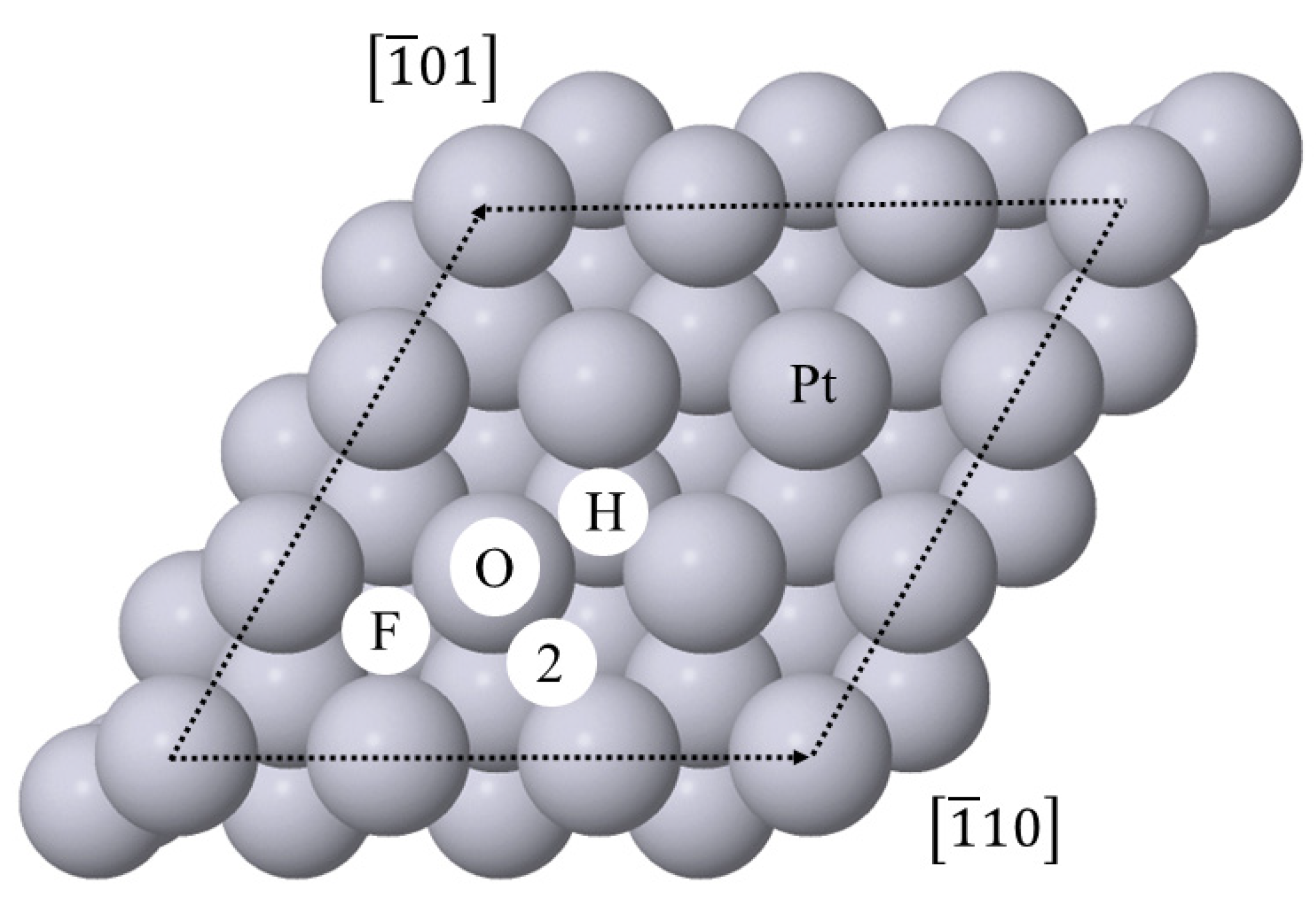
Figure 9.
The ‘O’ on-top, ‘F’ FCC, ‘H’ HCP and ‘2’ two-fold bridge high-symmetry binding positions on the Pt(111) surface. The dashed lines show the (3 × 3) surface unit cell used in the current simulations.
Figure 9.
The ‘O’ on-top, ‘F’ FCC, ‘H’ HCP and ‘2’ two-fold bridge high-symmetry binding positions on the Pt(111) surface. The dashed lines show the (3 × 3) surface unit cell used in the current simulations.

Figure 10.
The binding energy of (a,b) oxygen and (c,d) hydrogen to the on-top, FCC, HCP and two-fold bridge high-symmetry binding positions on the Pt(111) surface as a function of strain σ. The curves were obtained using the (a,c) GGA and (b,d) LDA estimates of the exchange-correlation.
Figure 10.
The binding energy of (a,b) oxygen and (c,d) hydrogen to the on-top, FCC, HCP and two-fold bridge high-symmetry binding positions on the Pt(111) surface as a function of strain σ. The curves were obtained using the (a,c) GGA and (b,d) LDA estimates of the exchange-correlation.

Figure 11.
Orthogonal unit cell of the bulk (a) Pt3Ni and (b) PtNi3 (L12) structures. The lattice parameter a for both alloys is shown by the solid arrows. Dashed lines are the edges of the orthogonal unit cell. The Pt (Ni) atoms are shown as grey (green) spheres.
Figure 11.
Orthogonal unit cell of the bulk (a) Pt3Ni and (b) PtNi3 (L12) structures. The lattice parameter a for both alloys is shown by the solid arrows. Dashed lines are the edges of the orthogonal unit cell. The Pt (Ni) atoms are shown as grey (green) spheres.

Figure 12.
Structure of the surfaces of unreacted (a) (111)-Pt/Ni/Pt3Ni and (b) (111)-Pt/Ni/PtNi3 and (c) the cross-section of unreacted (111)-Pt/Ni/Pt3Ni. In the latter, d12, d23, d34, d45 and d56 are interlayer spacings. In (a,b), the third layer Pt and Ni atoms are labelled by their unannotated chemical symbol, whereas the surface Pt and second layer Ni atoms carry a subscript which is the stoichiometry of their nearest third layer neighbours. This stoichiometry gives rise to non-equivalent on-top, FCC and HCP surface binding positions and three non-equivalent 2-fold bridge sites.
Figure 12.
Structure of the surfaces of unreacted (a) (111)-Pt/Ni/Pt3Ni and (b) (111)-Pt/Ni/PtNi3 and (c) the cross-section of unreacted (111)-Pt/Ni/Pt3Ni. In the latter, d12, d23, d34, d45 and d56 are interlayer spacings. In (a,b), the third layer Pt and Ni atoms are labelled by their unannotated chemical symbol, whereas the surface Pt and second layer Ni atoms carry a subscript which is the stoichiometry of their nearest third layer neighbours. This stoichiometry gives rise to non-equivalent on-top, FCC and HCP surface binding positions and three non-equivalent 2-fold bridge sites.

Figure 13.
Average spin density of the surfaces of unreacted (a) (111)-Pt/Ni/Pt3Ni and (b) (111)-Pt/Ni/PtNi3. The averaging was performed parallel to the (111) plane. The labels show the regions of bulk Pt3Ni and PtNi3, selvedge Ni and surface Pt in symmetric slabs, which each have 11 atomic layers in total.
Figure 13.
Average spin density of the surfaces of unreacted (a) (111)-Pt/Ni/Pt3Ni and (b) (111)-Pt/Ni/PtNi3. The averaging was performed parallel to the (111) plane. The labels show the regions of bulk Pt3Ni and PtNi3, selvedge Ni and surface Pt in symmetric slabs, which each have 11 atomic layers in total.

Figure 14.
(a) The ‘OA’ and ‘OB’ on-top, ‘FA’ and ‘FB’ FCC, ‘HA’ and ‘HB’ HCP and ‘2A’, ‘2B’ and ‘2C’ two-fold bridge high-symmetry binding positions on the reacted Pt/Ni/Pt3Ni-(111) surface. In (b), the surface and second layers—the pure Pt and Ni layers—are replaced with transparent circles bounded by solid and dashed lines, respectively. The straight dotted lines show the (2 × 2) surface unit cell used in the current simulations. The Pt/Ni/PtNi3-(111) is generated by changing every Pt (Ni) atom in layer 3 and deeper for a Ni (Pt) atom.
Figure 14.
(a) The ‘OA’ and ‘OB’ on-top, ‘FA’ and ‘FB’ FCC, ‘HA’ and ‘HB’ HCP and ‘2A’, ‘2B’ and ‘2C’ two-fold bridge high-symmetry binding positions on the reacted Pt/Ni/Pt3Ni-(111) surface. In (b), the surface and second layers—the pure Pt and Ni layers—are replaced with transparent circles bounded by solid and dashed lines, respectively. The straight dotted lines show the (2 × 2) surface unit cell used in the current simulations. The Pt/Ni/PtNi3-(111) is generated by changing every Pt (Ni) atom in layer 3 and deeper for a Ni (Pt) atom.

Figure 15.
Average spin density of the surfaces of (a) O-Pt/Ni/Pt3Ni(111) and (b) O-Pt/Ni/PtNi3(111). The oxygen atoms are bound in the FCC ‘A’ (‘FA’) sites defined in Figure 14 in the upper ‘reacted’ surfaces, whilst the lower surface is unreacted. This asymmetry is simply intended to highlight the change in the magnetic character of the respective surface Pt layers during adsorption, and quantitative results from these simulations were not subsequently used in this work. The averaging was performed parallel to the (111) plane. The labels show the regions of bulk Pt3Ni and PtNi3, selvedge Ni and surface Pt.
Figure 15.
Average spin density of the surfaces of (a) O-Pt/Ni/Pt3Ni(111) and (b) O-Pt/Ni/PtNi3(111). The oxygen atoms are bound in the FCC ‘A’ (‘FA’) sites defined in Figure 14 in the upper ‘reacted’ surfaces, whilst the lower surface is unreacted. This asymmetry is simply intended to highlight the change in the magnetic character of the respective surface Pt layers during adsorption, and quantitative results from these simulations were not subsequently used in this work. The averaging was performed parallel to the (111) plane. The labels show the regions of bulk Pt3Ni and PtNi3, selvedge Ni and surface Pt.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The bulk lattice parameters a, cohesive energies Ecoh and magnetic moments μPt and μNi for the bulk Ni and Pt metals. Cohesive energies are at the minima in the equations of state shown in Figure 2. The ‘Theory/Experiment’ column shows the exchange-correlation functional used (LDA/GGA) in the case of theoretical estimates of each of the physical parameter or shows the experimental value (‘Exptl’) where available.
Table 1.
The bulk lattice parameters a, cohesive energies Ecoh and magnetic moments μPt and μNi for the bulk Ni and Pt metals. Cohesive energies are at the minima in the equations of state shown in Figure 2. The ‘Theory/Experiment’ column shows the exchange-correlation functional used (LDA/GGA) in the case of theoretical estimates of each of the physical parameter or shows the experimental value (‘Exptl’) where available.
| Metal | Theory/Experiment | a (Å) | Ecoh (eV) | μPt (μB) | μNi (μB) |
|---|---|---|---|---|---|
| Pt | LDA | 3.890 | −7.141 | 0.000 | - |
| GGA | 3.962 | −5.586 | 0.000 | - | |
| Exptl | 3.912 | - | |||
| Ni | LDA | 3.427 | −6.125 | - | 0.694 |
| GGA | 3.518 | −4.927 | - | 0.699 | |
| Exptl | 3.508 | - |
Table 2.
Oxygen and hydrogen adsorption energies on strained Pt(111). The rightmost column sub-headings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. The strains used are those estimated within a Pt surface layer formed on a Pt3Ni alloy (0.980/0.978) using the GGA and LDA approximations, respectively, and similarly (0.924/0.918) for a PtNi3 alloy. All energies are in eV.
Table 2.
Oxygen and hydrogen adsorption energies on strained Pt(111). The rightmost column sub-headings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. The strains used are those estimated within a Pt surface layer formed on a Pt3Ni alloy (0.980/0.978) using the GGA and LDA approximations, respectively, and similarly (0.924/0.918) for a PtNi3 alloy. All energies are in eV.
| Binding Position | Strain σ | O/Pt(111) | H/Pt(111) | ||
|---|---|---|---|---|---|
| LDA | GGA | LDA | GGA | ||
| On-top | 1.000 | −2.895 | −2.263 | −2.416 | −1.589 |
| 0.980 | - | −2.168 | - | −1.586 | |
| 0.978 | −2.773 | - | −2.400 | - | |
| 0.924 | - | −3.760 | - | −2.976 | |
| 0.918 | −4.842 | - | −4.283 | - | |
| FCC | 1.000 | −3.913 | −3.100 | −2.426 | −1.547 |
| 0.980 | - | −2.996 | - | −1.519 | |
| 0.978 | −3.780 | - | −2.380 | - | |
| 0.924 | - | −4.313 | - | −2.906 | |
| 0.918 | −5.622 | - | −4.2664 | - | |
| HCP | 1.000 | −4.159 | −3.278 | −2.440 | −1.547 |
| 0.980 | - | −3.146 | - | −1.513 | |
| 0.978 | −3.989 | - | −2.387 | - | |
| 0.924 | - | −4.286 | - | −2.807 | |
| 0.918 | −5.666 | - | −4.161 | - | |
| 2-fold | 1.000 | −4.596 | −3.692 | −2.467 | −1.579 |
| 0.980 | - | −3.541 | - | −1.535 | |
| 0.978 | −4.402 | - | −2.403 | - | |
| 0.924 | - | −4.666 | - | −2.818 | |
| 0.918 | −6.027 | - | −4.188 | - | |
Table 3.
The bulk lattice parameters a, cohesive energies Ecoh and magnetic moments μPt and μNi for the Pt3Ni and PtNi3. The ‘Theory/Experiment’ column shows the exchange-correlation functional used (LDA/GGA) in the case of theoretical estimates of each of the physical parameters or shows the experimental value (‘Exptl’) where available.
Table 3.
The bulk lattice parameters a, cohesive energies Ecoh and magnetic moments μPt and μNi for the Pt3Ni and PtNi3. The ‘Theory/Experiment’ column shows the exchange-correlation functional used (LDA/GGA) in the case of theoretical estimates of each of the physical parameters or shows the experimental value (‘Exptl’) where available.
| Alloy | Theory/ Experiment | a (Å) | Ecoh (eV) | μPt (μB) | μNi (μB) |
|---|---|---|---|---|---|
| Pt3Ni (L12) | LDA | 3.805 | −27.357 | 0.103 | 0.517 |
| GGA | 3.884 | −21.595 | 0.173 | 0.721 | |
| Exptl [34] | 3.836 | ||||
| PtNi3 (L12) | LDA | 3.570 | −25.533 | 0.290 | 0.584 |
| GGA | 3.662 | −20.470 | 0.373 | 0.806 | |
| Exptl [34] | 3.645 |
Table 4.
The interlayer spacing d12, d23, d34, d45 and d56 defined in Figure 12 of the unreacted (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. The ‘Functional’ column shows the exchange-correlation functional used (LDA/GGA). All lengths are given in units of Å.
Table 4.
The interlayer spacing d12, d23, d34, d45 and d56 defined in Figure 12 of the unreacted (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. The ‘Functional’ column shows the exchange-correlation functional used (LDA/GGA). All lengths are given in units of Å.
| Alloy | Functional | d12 | d23 | d34 | d45 | d56 |
|---|---|---|---|---|---|---|
| Pt3Ni (L12) | LDA | 2.042 | 1.977 | 2.226 | 2.217 | 2.207 |
| GGA | 2.090 | 2.034 | 2.285 | 2.269 | 2.259 | |
| PtNi3 (L12) | LDA | 2.155 | 1.984 | 2.084 | 2.081 | 2.073 |
| GGA | 2.193 | 2.035 | 2.141 | 2.137 | 2.128 |
Table 5.
Oxygen and hydrogen adsorption energies Eads on the high symmetry sites of the (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. The binding positions are defined in Figure 14. The column subheadings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. The values shown in bold are the lowest energy for a particular type of binding position. All energies are in eV.
Table 5.
Oxygen and hydrogen adsorption energies Eads on the high symmetry sites of the (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. The binding positions are defined in Figure 14. The column subheadings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. The values shown in bold are the lowest energy for a particular type of binding position. All energies are in eV.
| Binding Position | O/(111)-Pt/Ni/Pt3Ni | O/(111)-Pt/Ni/PtNi3 | |||
|---|---|---|---|---|---|
| LDA | GGA | LDA | GGA | ||
| On-top | A | −3.496 | −2.782 | −3.382 | −2.672 |
| B | −3.411 | −2.701 | −3.390 | −2.668 | |
| FCC | A | −4.861 | −3.805 | −4.389 | −3.422 |
| B | −4.887 | −3.825 | −4.424 | −3.450 | |
| HCP | A | −4.625 | −3.589 | −4.203 | −3.239 |
| B | −4.558 | −3.526 | −4.201 | −3.241 | |
| 2-fold | A | −4.332 | −3.366 | −4.102 | −3.178 |
| B | −4.578 | −3.328 | −4.153 | −3.203 | |
| C | −4.337 | −3.356 | −4.067 | −3.138 | |
| Binding Position | H/(111)-Pt/Ni/Pt3Ni | H/(111)-Pt/Ni/PtNi3 | |||
| LDA | GGA | LDA | GGA | ||
| On-top | A | −2.833 | −1.879 | −2.861 | −1.919 |
| B | −2.867 | −1.906 | −2.833 | −1.881 | |
| FCC | A | −2.998 | −1.992 | −2.813 | −1.895 |
| B | −3.016 | −2.010 | −2.829 | −1.892 | |
| HCP | A | −2.988 | −1.979 | −2.835 | −1.910 |
| B | −2.997 | −1.989 | −2.848 | −1.908 | |
| 2-fold | A | −2.940 | −1.940 | −2.859 | −1.911 |
| B | −2.952 | −1.953 | −2.875 | −1.908 | |
| C | −2.947 | −1.947 | −2.846 | −1.895 | |
Table 6.
Nearest-neighbour oxygen-Pt and hydrogen-Pt bond lengths on the high symmetry sites of the (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. All the nearest-neighbour Pt atoms lie in the surface Pt layer. The binding positions are defined in Figure 14. The column subheadings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. All bond lengths are in Å.
Table 6.
Nearest-neighbour oxygen-Pt and hydrogen-Pt bond lengths on the high symmetry sites of the (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. All the nearest-neighbour Pt atoms lie in the surface Pt layer. The binding positions are defined in Figure 14. The column subheadings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. All bond lengths are in Å.
| Binding Position | O/(111)-Pt/Ni/Pt3Ni | O/(111)-Pt/Ni/PtNi3 | |||
|---|---|---|---|---|---|
| LDA | GGA | LDA | GGA | ||
| On-top | A | 1.83 | 1.86 | 1.78 | 1.80 |
| B | 1.84 | 1.86 | 1.80 | 1.80 | |
| FCC | A | 2.03 | 2.07 | 2.04 | 2.07 |
| B | 2.04 | 2.07 | 2.05 | 2.09 | |
| HCP | A | 2.04 | 2.07 | 2.05 | 2.09 |
| B | 2.04 | 2.07 | 2.06 | 2.10 | |
| 2-fold | A | 1.98 | 2.01 | 1.98 | 2.01 |
| B | 2.02 | 2.02 | 1.97 | 2.02 | |
| C | 1.98 | 2.02 | 1.98 | 2.02 | |
| Binding Position | H/(111)-Pt/Ni/Pt3Ni | H/(111)-Pt/Ni/PtNi3 | |||
| LDA | GGA | LDA | GGA | ||
| On-top | A | 1.57 | 1.58 | 1.56 | 1.56 |
| B | 1.57 | 1.57 | 1.56 | 1.56 | |
| FCC | A | 1.83 | 1.85 | 1.79 | 1.81 |
| B | 1.85 | 1.85 | 1.88 | 1.87 | |
| HCP | A | 1.85 | 1.86 | 1.81 | 1.86 |
| B | 1.86 | 1.87 | 1.83 | 1.86 | |
| 2-fold | A | 1.75 | 1.76 | 1.73 | 1.74 |
| B | 1.77 | 1.77 | 1.77 | 1.78 | |
| C | 1.76 | 1.77 | 1.76 | 1.77 | |
Table 7.
The spin-resolved first and second moments of the d-band of the surface Pt atoms in the Pt3Ni and PtNi3 alloy-bearing systems and in the surface Pt atoms in strained Pt(111). The up and down arrows denote the spin up and down components of the surface Pt atoms in the alloy systems, and ‘Pt’ denotes the surface Pt atoms in strained Pt(111). The number in brackets after the Ec;Pt and is the strain σ at which the quantity was determined. The ‘Functional’ column shows the exchange-correlation functional used (LDA/GGA). All the band centers are relative to the Fermi energy; all the energies and widths are in eV.
Table 7.
The spin-resolved first and second moments of the d-band of the surface Pt atoms in the Pt3Ni and PtNi3 alloy-bearing systems and in the surface Pt atoms in strained Pt(111). The up and down arrows denote the spin up and down components of the surface Pt atoms in the alloy systems, and ‘Pt’ denotes the surface Pt atoms in strained Pt(111). The number in brackets after the Ec;Pt and is the strain σ at which the quantity was determined. The ‘Functional’ column shows the exchange-correlation functional used (LDA/GGA). All the band centers are relative to the Fermi energy; all the energies and widths are in eV.
| Alloy | Functional | Ec;Pt | |||||
|---|---|---|---|---|---|---|---|
| Pt/Ni/Pt3Ni (L12) | LDA | −2.439 | −2.330 | −2.343 (0.9781) | 8.046 | 8.155 | 5.211 (0.9781) |
| GGA | −2.313 | −2.161 | −2.144 (0.9803) | 6.803 | 6.987 | 4.199 (0.9803) | |
| Pt/Ni/PtNi3 (L12) | LDA | −2.837 | −2.724 | −2.846 (0.9177) | 10.597 | 10.721 | 4.510 (0.9177) |
| GGA | −2.567 | −2.418 | −2.380 (0.9243) | 8.787 | 8.978 | 3.670 (0.92428) |
Table 8.
Magnetisation of the surface Pt atoms on the oxygen and hydrogen reacted and clean (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. For the reacted surfaces, the Pt atoms are the nearest neighbours to either the O or the H adsorbate. The binding positions are defined in Figure 14, and the column subheadings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. The numbers in brackets shown are the magnetic moments of the adsorbate. All magnetic moments in these tables are in units of μB.
Table 8.
Magnetisation of the surface Pt atoms on the oxygen and hydrogen reacted and clean (111)-Pt/Ni/Pt3Ni and (111)-Pt/Ni/PtNi3 surfaces. For the reacted surfaces, the Pt atoms are the nearest neighbours to either the O or the H adsorbate. The binding positions are defined in Figure 14, and the column subheadings (‘LDA’/‘GGA’) indicate the exchange-correlation functional used. The numbers in brackets shown are the magnetic moments of the adsorbate. All magnetic moments in these tables are in units of μB.
| Binding Position | O/(111)-Pt/Ni/Pt3Ni | O/(111)-Pt/Ni/PtNi3 | |||
|---|---|---|---|---|---|
| LDA | GGA | LDA | GGA | ||
| On-top | A | 0.263 (0.575) | 0.303 (0.641) | 0.310 (0.543) | 0.422 (0.649) |
| B | 0.257 (0.565) | 0.321 (0.654) | 0.251 (0.499) | 0.403 (0.632) | |
| FCC | A | 0.049 (0.007) | 0.068 (0.003) | 0.028 (−0.008) | 0.039 (−0.001) |
| B | 0.038 (−0.010) | 0.047 (−0.020) | 0.032 (0.004) | 0.042 (0.008) | |
| HCP | A | 0.040 (−0.014) | 0.074 (−0.014) | 0.030 (−0.015) | 0.056 (0.004) |
| B | 0.042 (−0.008) | 0.073 (−0.002) | 0.028 (−0.020) | 0.051 (−0.003) | |
| 2-fold | A | 0.052 (0.045) | 0.095 (0.090) | 0.026 (−0.004) | 0.032 (0.013) |
| B | 0.037 (−0.004) | 0.072 (0.040) | 0.024 (0.007) | 0.035 (0.025) | |
| C | 0.133 (0.041) | 0.186 (0.078) | 0.069 (−0.007) | 0.102 (0.016) | |
| Unreacted | - | 0.180 | 0.216 | 0.188 | 0.211 |
| Binding Position | H/(111)-Pt/Ni/Pt3Ni | H/(111)-Pt/Ni/PtNi3 | |||
| LDA | GGA | LDA | GGA | ||
| On-top | A | 0.033 (−0.008) | 0.035 (−0.015) | 0.024 (−0.008) | 0.028 (−0.012) |
| B | 0.034 (−0.010) | 0.033 (−0.017) | 0.025 (−0.007) | 0.027 (−0.010) | |
| FCC | A | 0.072 (−0.001) | 0.090 (−0.002) | 0.063 (−0.001) | 0.064 (−0.002) |
| B | 0.080 (−0.002) | 0.088 (−0.003) | 0.051 (−0.001) | 0.061 (−0.003) | |
| HCP | A | 0.074 (−0.002) | 0.093 (−0.003) | 0.077 (−0.001) | 0.071 (−0.002) |
| B | 0.081 (−0.002) | 0.088 (−0.003) | 0.054 (−0.001) | 0.066 (−0.002) | |
| 2-fold | A | 0.059 (−0.003) | 0.073 (−0.005) | 0.046 (−0.002) | 0.051 (−0.004) |
| B | 0.065 (−0.003) | 0.070 (−0.005) | 0.045 (−0.002) | 0.050 (−0.004) | |
| C | 0.106 (−0.002) | 0.116 (−0.004) | 0.074 (−0.002) | 0.089 (−0.004) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shuttleworth, I. A Comparative Study of Oxygen and Hydrogen Adsorption on Strained and Alloy-Supported Pt(111) Monolayers. Magnetochemistry 2021, 7, 101. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7070101
AMA Style
Shuttleworth I. A Comparative Study of Oxygen and Hydrogen Adsorption on Strained and Alloy-Supported Pt(111) Monolayers. Magnetochemistry. 2021; 7(7):101. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7070101
Chicago/Turabian StyleShuttleworth, Ian. 2021. "A Comparative Study of Oxygen and Hydrogen Adsorption on Strained and Alloy-Supported Pt(111) Monolayers" Magnetochemistry 7, no. 7: 101. https://0-doi-org.brum.beds.ac.uk/10.3390/magnetochemistry7070101
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.