Reducing False-Positive Results in Newborn Screening Using Machine Learning

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Summary
2.2. NBS Metabolic Data Analysis Using Random Forest
2.3. Validation of the Random Forest Model
2.4. Web-Based RF Tool and Statistical Analysis
3. Results
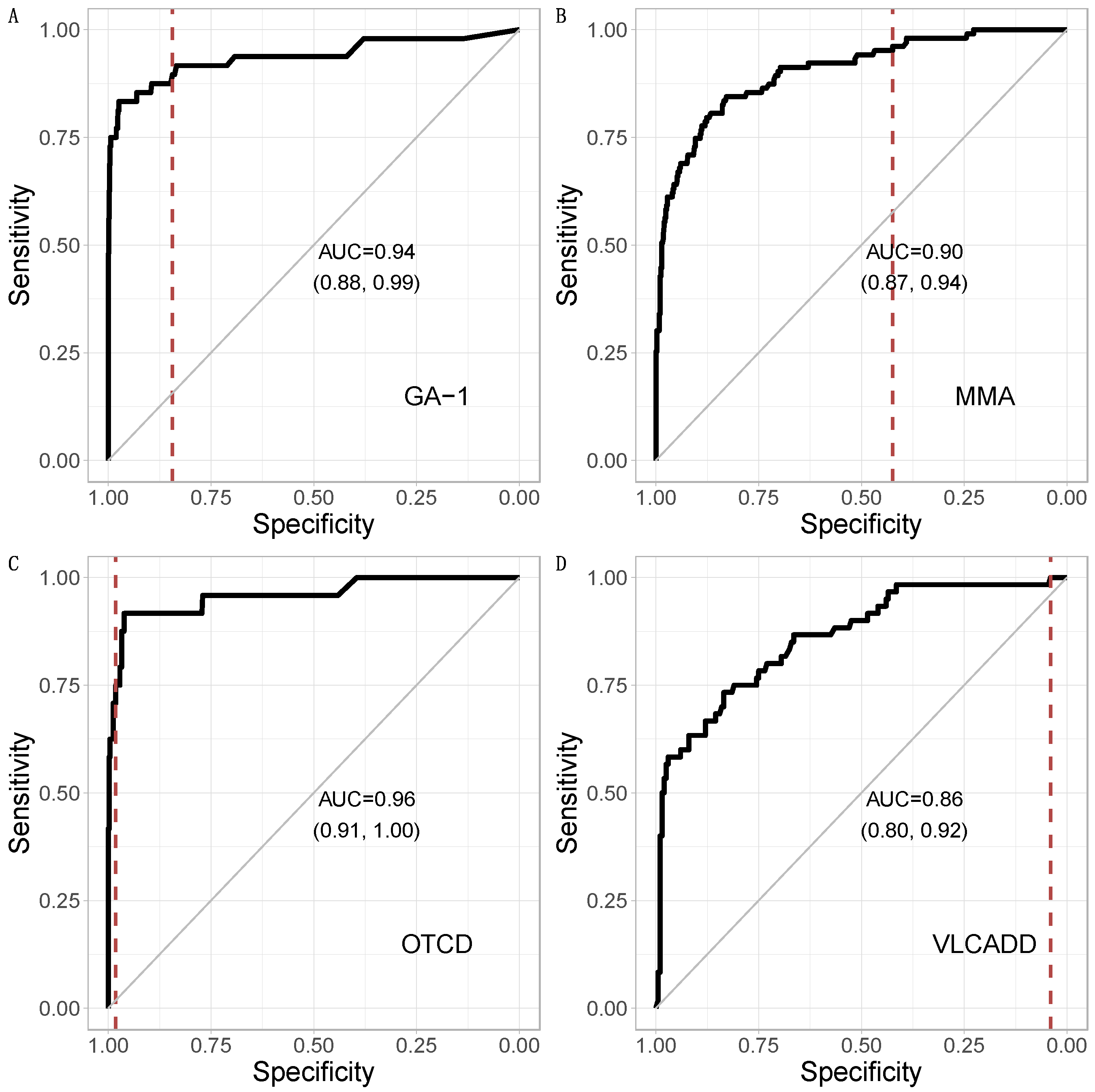
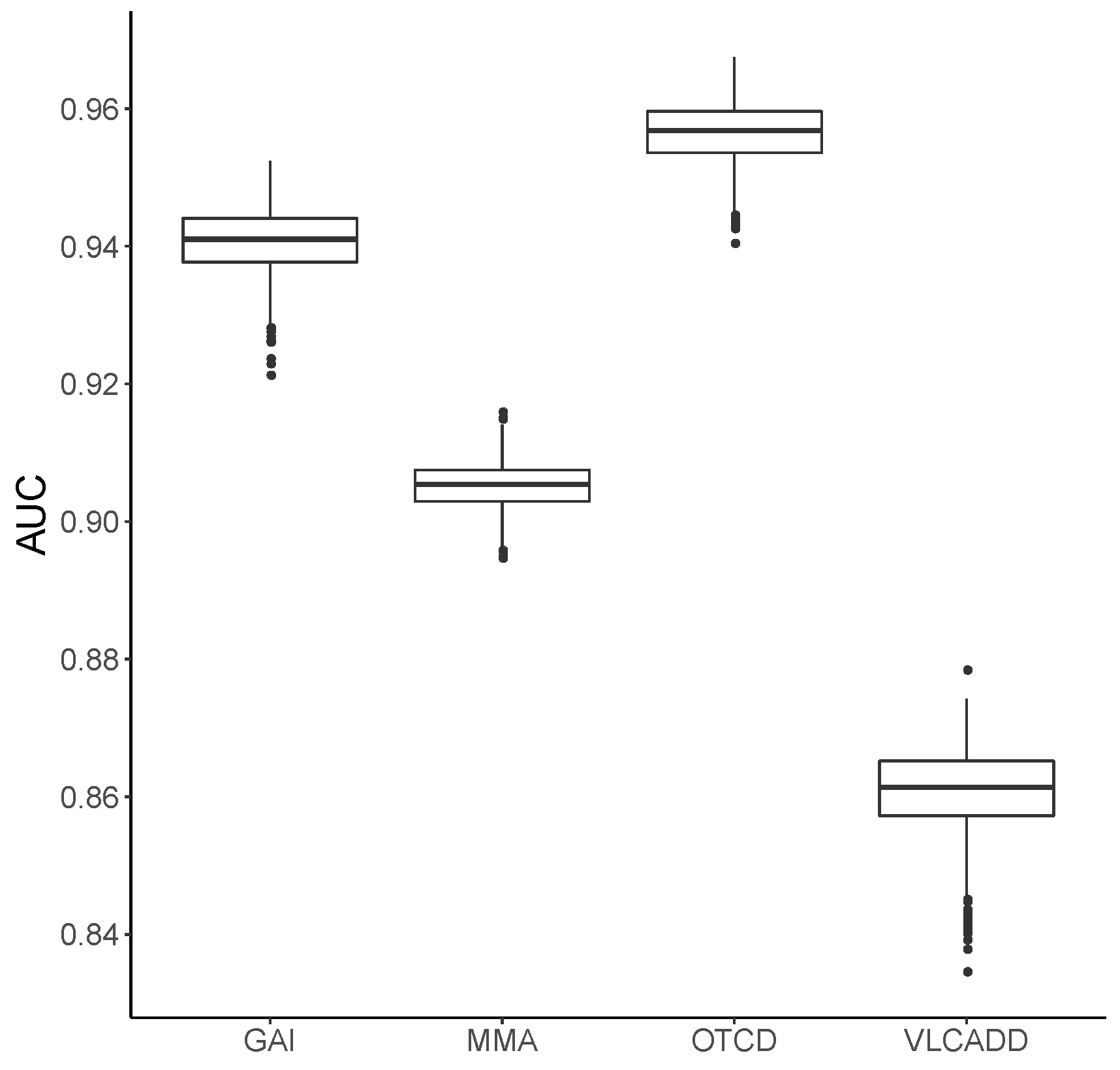
3.1. Metabolic Pattern Analysis Using Random Forest
3.2. Ranking of Metabolic Analytes
3.3. Comparison of CLIR and Random Forest
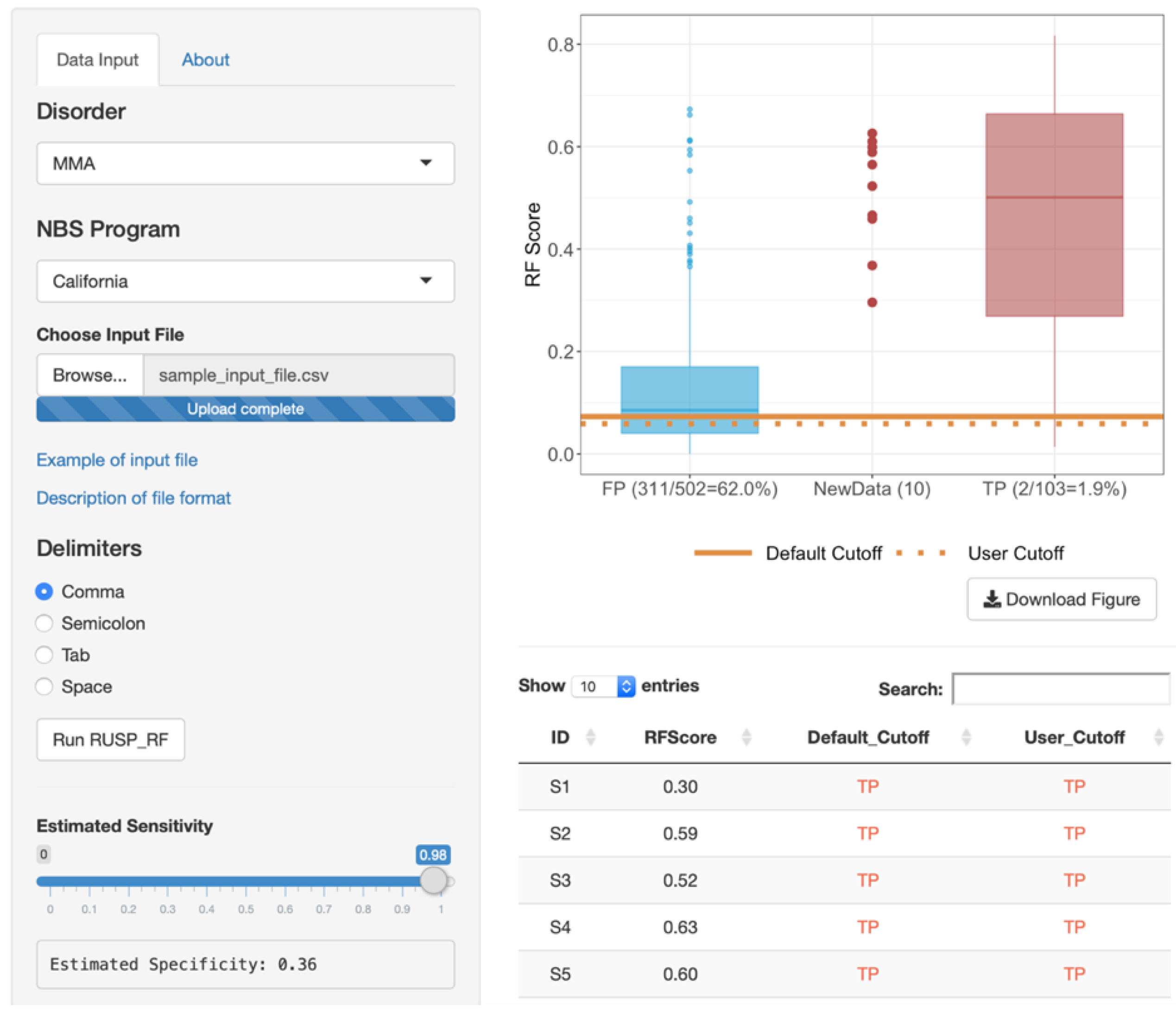
3.4. Web-Based RF Tool
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwon, C.; Farrell, P.M. The magnitude and challenge of false-positive newborn screening test results. Arch. Pediatr. Adolesc. Med. 2000, 154, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, G.; Currier, R.; McHugh, D.M.S.; Gavrilov, D.; Magera, M.J.; Matern, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Smith, E.H.; et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet. Med. 2012, 14, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting newborn screening markers: The incidental discovery of a second-tier test for Pompe disease. Genet. Med. 2018, 20, 840–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.; Currier, R.J.; Tang, T.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef] [Green Version]
- Morkrid, L.; Rowe, A.D.; Elgstoen, K.B.; Olesen, J.H.; Ruijter, G.; Hall, P.L.; Tortorelli, S.; Schulze, A.; Kyriakopoulou, L.; Wamelink, M.M.; et al. Continuous age- and sex-adjusted reference intervals of urinary markers for cerebral creatine deficiency syndromes: A novel approach to the definition of reference intervals. Clin. Chem. 2015, 61, 760–768. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner, C.; Bohm, C.; Baumgartner, D.; Marini, G.; Weinberger, K.; Olgemöller, B.; Liebl, B.; Roscher, A.A. Supervised machine learning techniques for the classification of metabolic disorders in newborns. Bioinformatics 2004, 20, 2985–2996. [Google Scholar] [CrossRef]
- Chen, W.H.; Hsieh, S.L.; Hsu, K.P.; Chen, H.P.; Su, X.Y.; Tseng, Y.J.; Chien, Y.H.; Hwu, W.L.; Lai, F. Web-based newborn screening system for metabolic diseases: Machine learning versus clinicians. J. Med. Internet Res. 2013, 15, e98. [Google Scholar] [CrossRef]
- Ho, T.K. Random decision forests. In Paper Presented at: Proceedings of the 3rd International Conference on Document Analysis and Recognition, Montreal, QC, Canada, 14–16 August 1995. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ishwaran, H. Random forests for genomic data analysis. Genomics 2012, 99, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Abbott, T.; Fishman, D.; McMurray, W.; Mor, G.; Stone, K.; Ward, D.; Williams, K.; Zhao, H. Comparison of statistical methods for classification of ovarian cancer using mass spectrometry data. Bioinformatics 2003, 19, 1636–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, C.; Navarro, L.C.; de Oliveira, D.N.; Guerreiro, T.M.; Lima, E.O.; Delafiori, J.; Dabaja, M.Z.; Ribeiro, M.D.S.; de Menezes, M.; Rodrigues, R.G.M.; et al. A Machine Learning Application Based in Random Forest for Integrating Mass Spectrometry-Based Metabolomic Data: A Simple Screening Method for Patients With Zika Virus. Front. Bioeng Biotechnol. 2018, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, B.T.; Joseloff, E.; Goetz, D.; Ingram, B.; Heltshe, S.L.; Leung, D.H.; Ramsey, B.W.; McCoy, K.; Borowitz, D. Urinary metabolomics reveals unique metabolic signatures in infants with cystic fibrosis. J. Cyst. Fibros. 2019, 18, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.; Shen, P.; Gandotra, N.; Le, A.; Fung, E.; Jelliffe-Pawlowski, L.; Davis, R.W.; Enns, G.M.; Zhao, H.; Cowan, T.M.; et al. Combining newborn metabolic and DNA analysis for second-tier testing of methylmalonic acidemia. Genet. Med. 2019, 21, 896–903. [Google Scholar] [CrossRef] [Green Version]
- American College of Medical Genetics Newborn Screening Expert Group. Newborn screening: Toward a uniform screening panel and system—Executive summary. Pediatrics 2006, 117, S296–S307. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, T.M.; Perez, P.S.; Baranauskas., J.A. How Many Trees in a Random Forest? In Machine Learning and Data Mining in Pattern Recognition; Perner, P., Ed.; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Varma, S.; Simon, R. Bias in error estimation when using cross-validation for model selection. BMC Bioinform. 2006, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Nicodemus, K.K. Letter to the editor: On the stability and ranking of predictors from random forest variable importance measures. Brief. Bioinform. 2011, 12, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Shiny: Web Application Framework for R. Available online: https://shiny.rstudio.com (accessed on 1 August 2019).
- R: A Language and Environment for Statistical Computing. Available online: https://www.r-project.org (accessed on 1 August 2019).
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R. News. 2002, 2, 18–22. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Zytkovicz, T.H.; Fitzgerald, E.F.; Marsden, D.; Larson, C.A.; Shih, V.E.; Johnson, D.M.; Strauss, A.W.; Comeau, A.M.; Eaton, R.B.; Grady, G.F. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: A two-year summary from the New England Newborn Screening Program. Clin. Chem. 2001, 47, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Weisfeld-Adams, J.D.; Morrissey, M.A.; Kirmse, B.M.; Salveson, B.R.; Wasserstein, M.P.; McGuire, P.J.; Sunny, S.; Cohen-Pfeffer, J.L.; Yu, C.; Caggana, M.; et al. Newborn screening and early biochemical follow-up in combined methylmalonic aciduria and homocystinuria, cblC type, and utility of methionine as a secondary screening analyte. Mol. Genet. Metab. 2010, 99, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldo, P.; Whitley, R.J.; Rhead, W.J.; Hannon, W.H. Evidence-Based Rationale for Expanded Newborn Screening. N. Engl. J. Med. 2009, 348, 2304–2312. [Google Scholar]
- McClead, R.E.J.; Rozen, R.; Fox, J.; Rosenberg, L.; Menke, J.; Bickers, R.; Morrow, G., 3rd. Clinical application of DNA analysis in a family with OTC deficiency. Am. J. Med. Genet. 1986, 25, 513–518. [Google Scholar] [CrossRef]
- McHugh, D.; Cameron, C.A.; Abdenur, J.E.; Abdulrahman, M.; Adair, O.; Al Nuaimi, S.A.; Ahlman, H.; Allen, J.J.; Antonozzi, I.; Archer, S.; et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: A worldwide collaborative project. Genet. Med. 2011, 13, 230–254. [Google Scholar] [CrossRef]
- Di Donato, S.; Rimoldi, M.; Garavaglia, B.; Uziel, G. Propionylcarnitine excretion in propionic and methylmalonic acidurias: A cause of carnitine deficiency. Clin. Chim. Acta 1984, 139, 13–21. [Google Scholar] [CrossRef]
- Bisanzi, S.; Morrone, A.; Donati, M.A.; Pasquini, E.; Spada, M.; Strisciuglio, P.; Parenti, G.; Parini, R.; Papadia, F.; Zammarchi, E. Genetic analysis in nine unrelated Italian patients affected by OTC deficiency: Detection of novel mutations in the OTC gene. Mol. Genet. Metab. 2002, 76, 137–144. [Google Scholar] [CrossRef]
- De Sain-van der Velden, M.G.; Rinaldo, P.; Elvers, B.; Henderson, M.; Walter, J.H.; Prinsen, B.H.; Verhoeven-Duif, N.M.; de Koning, T.J.; van Hasselt, P. The Proline/Citrulline Ratio as a Biomarker for OAT Deficiency in Early Infancy. JIMD Rep. 2012, 6, 95–99. [Google Scholar]
- Hennermann, J.B.; Roloff, S.; Gellermann, J.; Gruters, A.; Klein, J. False-positive newborn screening mimicking glutaric aciduria type I in infants with renal insufficiency. J. Inherited Metab. Dis. 2009, 32, S355–S359. [Google Scholar] [CrossRef]
- Diekman, E.; de Sain-van der Velden, M.; Waterham, H.; Kluijtmans, L.; Schielen, P.; van Veen, E.B.; Ferdinandusse, S.; Wijburg, F.; Visser, G. The Newborn Screening Paradox: Sensitivity vs. Overdiagnosis in VLCAD Deficiency. JIMD Rep. 2016, 27, 101–106. [Google Scholar]
- Kolker, S.; Boy, S.P.; Heringer, J.; Müller, E.; Maier, E.M.; Ensenauer, R.; Mühlhausen, C.; Schlune, A.; Greenberg, C.R.; Koeller, D.M.; et al. Complementary dietary treatment using lysine-free, arginine-fortified amino acid supplements in glutaric aciduria type I—A decade of experience. Mol. Genet. Metab. 2012, 107, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Atkins, A.E.; Tarini, B.A.; Phillips, E.K.; Calhoun, A. Misclassification of VLCAD carriers due to variable confirmatory testing after a positive NBS result. J. Community Genet. 2019, 10, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Merritt, J.L., 2nd; Vedal, S.; Abdenur, J.E.; Au, S.M.; Barshop, B.A.; Feuchtbaum, L.; Harding, C.O.; Hermerath, C.; Lorey, F.; Sesser, D.E.; et al. Infants suspected to have very-long chain acyl-CoA dehydrogenase deficiency from newborn screening. Mol. Genet. Metab. 2014, 111, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Vernooij-van Langen, A.M.; Loeber, J.G.; Elvers, B.; Triepels, R.H.; Roefs, J.; Gille, J.J.; Reijntjens, S.; Dompeling, E.; Dankert-Roelse, J.E. The influence of sex, gestational age, birth weight, blood transfusion, and timing of the heel prick on the pancreatitis-associated protein concentration in newborn screening for cystic fibrosis. J. Inherited Metab. Dis. 2013, 36, 147–154. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Confirmed Positive | First-Tier NBS | Second-Tier Analysis (RF, This Study) | ||
|---|---|---|---|---|---|
| False Positives | PPV | False Positives * | PPV | ||
| GA-1 | 48 | 1344 | 3.10% | 150 | 22.30% |
| MMA | 103 | 502 | 16.40% | 276 | 26.40% |
| OTCD | 24 | 496 | 3.50% | 11 | 62.10% |
| VLCADD | 60 | 200 | 23.10% | 196 | 23.40% |
| GA-1 | MMA | OTCD | VLCADD | Control * | |
|---|---|---|---|---|---|
| Gestational Age, week | |||||
| <37 | 340 (24.4%) | 175 (28.9%) | 181 (34.8%) | 42 (16.2%) | 5490 (5.5%) |
| 37–41 | 1005 (72.2%) | 412 (68.1%) | 325 (62.5%) | 206 (79.2%) | 93,603 (94.0%) |
| >41 | 47 (3.4%) | 18 (3.0%) | 14 (2.7%) | 12 (4.6%) | 444 (0.4%) |
| Birth Weight, g | |||||
| <2500 | 279 (20.0%) | 173 (28.6%) | 130 (25.0%) | 26 (10.0%) | 4045 (4.1%) |
| 2500–4000 | 1025 (73.6%) | 381 (63.0%) | 354 (68.1%) | 223 (85.8%) | 87,268 (87.7%) |
| >4000 | 88 (6.3%) | 51 (8.4%) | 36 (6.9%) | 11 (4.2%) | 8224 (8.3%) |
| Sex | |||||
| Male | 845 (60.7%) | 321 (53.1%) | 325 (62.5%) | 165 (63.5%) | 51,352 (51.6%) |
| Female | 542 (38.9%) | 281 (46.4%) | 194 (37.3%) | 93 (35.8%) | 47,882 (48.1%) |
| Unknown | 5 (0.4%) | 3 (0.5%) | 1 (0.2%) | 2 (0.8%) | 303 (0.3%) |
| Race/Ethnicity | |||||
| Asian | 136 (9.8%) | 63 (10.4%) | 33 (6.3%) | 40 (15.4%) | 14275 (14.3%) |
| Black | 212 (15.2%) | 25 (4.1%) | 50 (9.6%) | 15 (5.8%) | 6630 (6.7%) |
| Hispanic | 444 (31.9%) | 407 (67.3%) | 224 (43.1%) | 94 (36.2%) | 49,400 (49.6%) |
| White | 554 (39.8%) | 92 (15.2%) | 197 (37.9%) | 102 (39.2%) | 26341 (26.5%) |
| Other/Unknown | 46 (3.3%) | 18 (3.0%) | 16 (3.1%) | 9 (3.5%) | 2891 (2.9%) |
| Age at Blood Collection, hour | |||||
| <12 | 246 (17.7%) | 142 (23.5%) | 45 (8.7%) | 47 (18.1%) | 21,564 (21.7%) |
| 12–24 | 877 (63.0%) | 319 (52.7%) | 259 (49.8%) | 183 (70.4%) | 71,396 (71.7%) |
| >24 | 269 (19.3%) | 144 (23.8%) | 216 (41.5%) | 30 (11.5%) | 6577 (6.6%) |
| Total Parenteral Nutrition | |||||
| No | 1178 (84.6%) | 393 (65.0%) | 453 (87.1%) | 248 (95.4%) | 97,269 (97.7%) |
| Yes | 146 (10.5%) | 187 (30.9%) | 57 (11.0%) | 3 (1.2%) | 998 (1.0%) |
| Unknown | 68 (4.9%) | 25 (4.1%) | 10 (1.9%) | 9 (3.5%) | 1270 (1.3%) |
| MDA Ranking | GA-1 | MMA | OTCD | VLCADD |
|---|---|---|---|---|
| 1 | C5DC a | Methionine b [28] | Methionine b [29,30] | C14:1 a |
| 2 | C3DC | Free Carnitine [31] | Proline b [29,32,33] | C14 a |
| 3 | C8 b [28] | C2 a | Alanine [29] | C3DC |
| 4 | C10 [34] | C3 a | Glycine [29] | C2 b [35] |
| 5 | Ornithine [36] | C4 | Citrulline a | C5DC |
| Predicted by Algorithm | NBS Results (Truth) | ||
|---|---|---|---|
| TP | FP | ||
| CLIR | TP | 44 | 67 |
| FP | 4 | 911 | |
| Random Forest | TP | 43 | 53 |
| FP | 5 | 925 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, G.; Tang, Y.; Cowan, T.M.; Enns, G.M.; Zhao, H.; Scharfe, C. Reducing False-Positive Results in Newborn Screening Using Machine Learning. Int. J. Neonatal Screen. 2020, 6, 16. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6010016
Peng G, Tang Y, Cowan TM, Enns GM, Zhao H, Scharfe C. Reducing False-Positive Results in Newborn Screening Using Machine Learning. International Journal of Neonatal Screening. 2020; 6(1):16. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6010016
Chicago/Turabian StylePeng, Gang, Yishuo Tang, Tina M. Cowan, Gregory M. Enns, Hongyu Zhao, and Curt Scharfe. 2020. "Reducing False-Positive Results in Newborn Screening Using Machine Learning" International Journal of Neonatal Screening 6, no. 1: 16. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6010016