Second Tier Molecular Genetic Testing in Newborn Screening for Pompe Disease: Landscape and Challenges

 and
and
Abstract
:1. Introduction
2. Current Approach to Second Tier and Follow-Up Testing
3. The Variant Spectrum of Pompe Disease
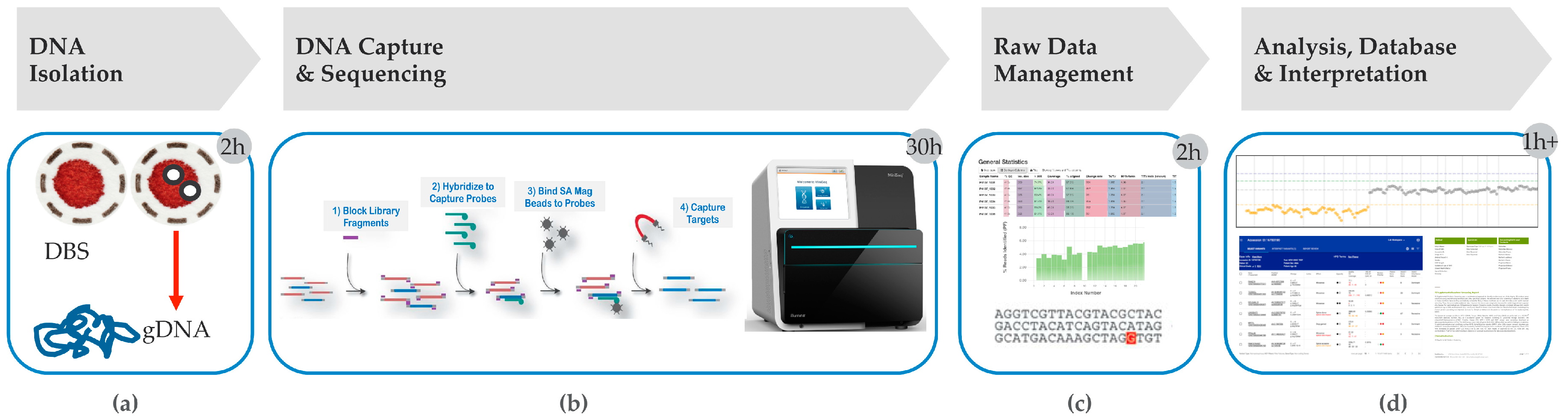
4. Hybrid Capture tNGS as a Second Tier Method
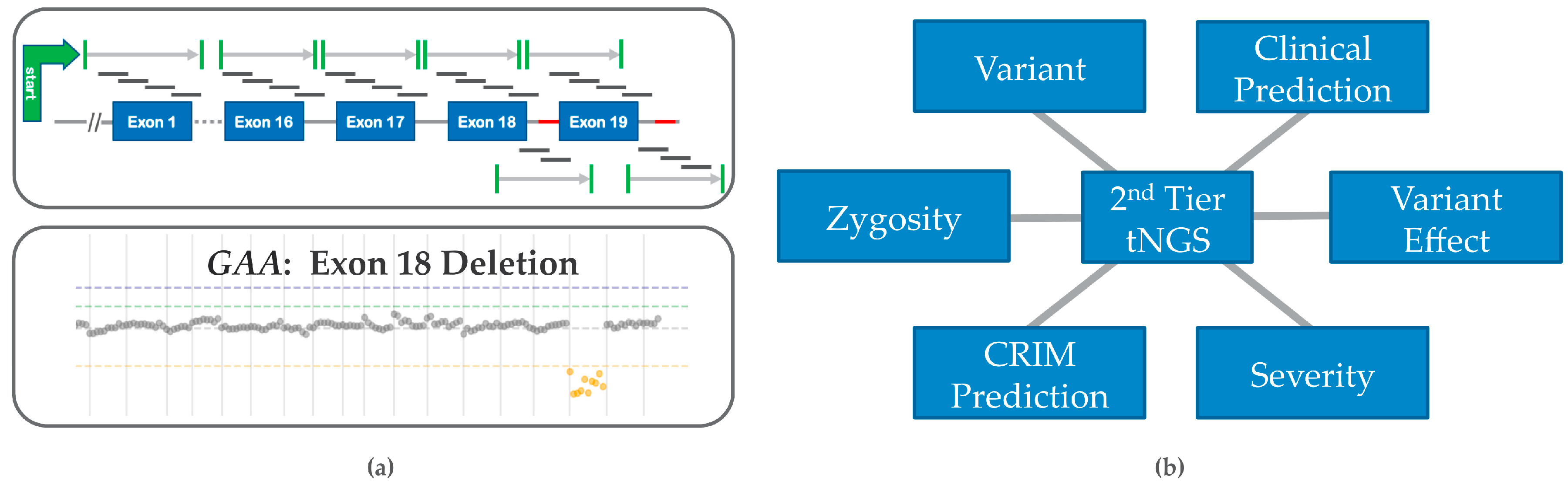
5. Ideal PD Second Tier tNGS Testing Features
5.1. Timeliness
5.2. Prediction of Genotype-Phenotype Effects
5.3. Prediction of CRIM Status
5.4. Copy Number Variation (CNV) and False Negative Risk
5.5. False Positives Resolved by Second Tier tNGS Testing
5.6. Other Considerations
6. Genome Scale Data and Its Impact on PD Screening
7. Variant ‘Cut off’ for PD
8. Follow-Up Infrastructure for Families, Screen Positive Infants and Carrier Status
9. Current and Future Utilization of tNGS Testing
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Gelb, M.; Lukacs, Z.; Ranieri, E.; Schielen, P. Newborn Screening for Lysosomal Storage Disorders: Methodologies for Measurement of Enzymatic Activities in Dried Blood Spots. Int. J. Neonatal Screen. 2018, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.-H.; Hwu, W.-L.; Lee, N.-C. Newborn Screening: Taiwanese Experience. Ann. Transl. Med. 2019, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-F.; Liu, H.-C.; Hsu, T.-R.; Tsai, F.-C.; Chiang, S.-F.; Chiang, C.-C.; Ho, H.-C.; Lai, C.-J.; Yang, T.-F.; Chuang, S.-Y.; et al. A Large-Scale Nationwide Newborn Screening Program for Pompe Disease in Taiwan: Towards Effective Diagnosis and Treatment. Am. J. Med Genet. Part A 2013, 164, 54–61. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York Pilot Newborn Screening Program for Lysosomal Storage Diseases: Report of the First 65,000 Infants. Genet. Med. 2018, 21, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting Newborn Screening Markers: The Incidental Discovery of a Second-Tier Test for Pompe Disease. Genet. Med. 2017, 20, 840–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, B.K.; Kronn, D.F.; Hwu, W.-L.; Kishnani, P.S. The Initial Evaluation of Patients after Positive Newborn Screening: Recommended Algorithms Leading to a Confirmed Diagnosis of Pompe Disease. Pediatrics 2017, 140, S14–S23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saich, R.; Brown, R.; Collicoat, M.; Jenner, C.; Primmer, J.; Clancy, B.; Holland, T.; Krinks, S. Is Newborn Screening the Ultimate Strategy to Reduce Diagnostic Delays in Pompe Disease? The Parent and Patient Perspective. Int. J. Neonatal Screen. 2020, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Reuser, A.J.J.; Ploeg, A.T.; Chien, Y.H.; Llerena, J.; Abbott, M.A.; Clemens, P.R.; Kimonis, V.E.; Leslie, N.; Maruti, S.S.; Sanson, B.J.; et al. On Behalf of the Pompe Registry Sit. GAA Variants and Phenotypes among 1079 Patients with Pompe Disease: Data from the Pompe Registry. Hum. Mutat. 2019, 40, 2146–2164. [Google Scholar] [CrossRef] [Green Version]
- Owens, P.; Wong, M.; Bhattacharya, K.; Ellaway, C. Infantile-Onset Pompe Disease: A Case Series Highlighting Early Clinical Features, Spectrum of Disease Severity and Treatment Response. J. Paediatr. Child Health 2018, 54, 1255–1261. [Google Scholar] [CrossRef]
- Desai, A.K.; Li, C.; Rosenberg, A.S.; Kishnani, P.S. Immunological Challenges and Approaches to Immunomodulation in Pompe Disease: A Literature Review. Ann. Transl. Med. 2019, 7, 285. [Google Scholar] [CrossRef]
- Groot, A.D.; Kazi, Z.; Martin, R.; Terry, F.; Desai, A.; Martin, W.; Kishnani, P. HLA- and Genotype-Based Risk Assessment Model to Identify Infantile Onset Pompe Disease Patients at High-Risk of Developing Significant Anti-Drug Antibodies (ADA). Clin. Immunol. 2019, 200, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-F.; Chu, T.-H.; Huang, L.-Y.; Liao, H.-C.; Soong, W.-J.; Niu, D.-M. AB028. Very Early Treatment for Infantile-Onset Pompe Disease Contributes to Better Outcomes: 10-Year Experience in One Institute. Ann. Transl. Med. 2017, 169, 174–180. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.-H.; Lee, N.-C.; Chen, C.-A.; Tsai, F.-J.; Tsai, W.-H.; Shieh, J.-Y.; Huang, H.-J.; Hsu, W.-C.; Tsai, T.-H.; Hwu, W.-L. Long-Term Prognosis of Patients with Infantile-Onset Pompe Disease Diagnosed by Newborn Screening and Treated since Birth. J. Pediatr. 2015, 166, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Bali, D.; Goldstein, J.; Banugaria, S.; Dai, J.; Mackey, J.; Rehder, C.; Kishnani, P. Predicting Cross Reactive Immunological Material (CRIM) Status in Pompe Disease Using GAA Mutations: Lessons Learned from 10 Years of Clinical Laboratory Testing Experience. In American Journal of Medical Genetics Part C: Seminars in Medical Genetics; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Peruzzo, P.; Pavan, E.; Dardis, A. Molecular Genetics of Pompe Disease: A Comprehensive Overview. Ann. Transl. Med. 2019, 7, 278. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Sokolsky, T.; Wyman, S.K.; Reese, M.G.; Puffenberger, E.; Strauss, K.; Morton, H.; Parad, R.B.; Naylor, E.W. Development of DNA Confirmatory and High-Risk Diagnostic Testing for Newborns Using Targeted Next-Generation DNA Sequencing. Genet. Med. 2014, 17, 337–347. [Google Scholar] [CrossRef]
- Millington, D.; Bali, D. Current State of the Art of Newborn Screening for Lysosomal Storage Disorders. Int. J. Neonatal Screen. 2018, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- Kishnani, P.S.; Goldenberg, P.C.; Dearmey, S.L.; Heller, J.; Benjamin, D.; Young, S.; Bali, D.; Smith, S.A.; Li, J.S.; Mandel, H.; et al. Cross-Reactive Immunologic Material Status Affects Treatment Outcomes in Pompe Disease Infants. Mol. Genet. Metab. 2010, 99, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Haskell, G.; Kazi, Z.; Zhu, X.; Dearmey, S.M.; Goldstein, J.L.; Bali, D.; Rehder, C.; Cirulli, E.T.; Kishnani, P.S. Sensitivity of Whole Exome Sequencing in Detecting Infantile- and Late-Onset Pompe Disease. Mol. Genet. Metab. 2017, 122, 189–197. [Google Scholar] [CrossRef]
- Holm, I.A.; Agrawal, P.B.; Ceyhan-Birsoy, O.; Christensen, K.D.; Fayer, S.; Frankel, L.A.; Genetti, C.A.; Krier, J.B.; Lamay, R.C.; Levy, H.L.; et al. The BabySeq Project: Implementing Genomic Sequencing in Newborns. BMC Pediatr. 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Ceyhan-Birsoy, O.; Machini, K.; Lebo, M.S.; Yu, T.W.; Agrawal, P.B.; Parad, R.B.; Holm, I.A.; Mcguire, A.; Green, R.C.; Beggs, A.H.; et al. A Curated Gene List for Reporting Results of Newborn Genomic Sequencing. Genet. Med. 2017, 19, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Whiffin, N.; Minikel, E.; Walsh, R.; O’Donnell-Luria, A.H.; Karczewski, K.; Ing, A.Y.; Barton, P.J.R.; Funke, B.; Cook, S.A.; Macarthur, D.; et al. Using High-Resolution Variant Frequencies to Empower Clinical Genome Interpretation. Genet. Med. 2017, 19, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botkin, J.R.; Belmont, J.W.; Berg, J.S.; Berkman, B.E.; Bombard, Y.; Holm, I.A.; Levy, H.P.; Ormond, K.E.; Saal, H.M.; Spinner, N.B.; et al. Points to Consider: Ethical, Legal, and Psychosocial Implications of Genetic Testing in Children and Adolescents. Am. J. Hum. Genet. 2015, 97, 501. [Google Scholar] [CrossRef] [Green Version]
- Report on the Genetic Testing of Children 2010; British Society for Human Genetics: Birmingham, UK, 2010.
- Ethical and Policy Issues in Genetic Testing and Screening of Children. Pediatrics 2013, 131, 620–622. [CrossRef] [PubMed] [Green Version]
- Tang, H.; Feuchtbaum, L.; Sciortino, S.; Matteson, J.; Mathur, D.; Bishop, T.; Olney, R.S. The First Year Experience of Newborn Screening for Pompe Disease in California. Int. J. Neonatal Screen. 2020, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hitchins, L.; Hickey, R.; Christensen, K.M.; Groeppner, D.; et al. Newborn Screening for Pompe Disease in Illinois: Experience with 684,290 Infants. Int. J. Neonatal Screen. 2020, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Schultz, N.; Oakson, K.; Jones, D.; Rindler, M.; Hart, K.; Rohrwasser, A. Targeted Second-Tier Confirmatory Next Generation Sequencing Newborn Screening Pipeline. In Poster Abstracts 2019; Newborn Screening & Genetic Testing Symposium: Chicago, IL, USA, 7–10 April 2019. [Google Scholar]
- Milko, L.V.; Odaniel, J.M.; Decristo, D.M.; Crowley, S.B.; Foreman, A.K.M.; Wallace, K.E.; Mollison, L.F.; Strande, N.T.; Girnary, Z.S.; Boshe, L.J.; et al. An Age-Based Framework for Evaluating Genome-Scale Sequencing Results in Newborn Screening. J. Pediatr. 2019, 209, 68–76. [Google Scholar] [CrossRef]
- Bergsma, A.J.; In’t Groen, S.L.; van den Dorpel, J.J.; van den Hout, H.J.; van der Beek, N.A.; Schoser, B.; Toscano, A.; Musumeci, O.; Bembi, B.; Dardis, A.; et al. A Genetic Modifier of Symptom Onset in Pompe Disease. EBioMedicine 2019, 43, 553–561. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Prevalence | Max. Allelic Contribution | Penetrance | MCAF (Whiffin et al. [22]) | Max. MAF in ClinVar | Final Cut Off |
|---|---|---|---|---|---|
| 0.000025 | 0.05 | 1 | 0.00025 | 0.00358 | 0.00025 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, L.D.; Bainbridge, M.N.; Parad, R.B.; Bhattacharjee, A. Second Tier Molecular Genetic Testing in Newborn Screening for Pompe Disease: Landscape and Challenges. Int. J. Neonatal Screen. 2020, 6, 32. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6020032
Smith LD, Bainbridge MN, Parad RB, Bhattacharjee A. Second Tier Molecular Genetic Testing in Newborn Screening for Pompe Disease: Landscape and Challenges. International Journal of Neonatal Screening. 2020; 6(2):32. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6020032
Chicago/Turabian StyleSmith, Laurie D., Matthew N. Bainbridge, Richard B. Parad, and Arindam Bhattacharjee. 2020. "Second Tier Molecular Genetic Testing in Newborn Screening for Pompe Disease: Landscape and Challenges" International Journal of Neonatal Screening 6, no. 2: 32. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns6020032