
Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
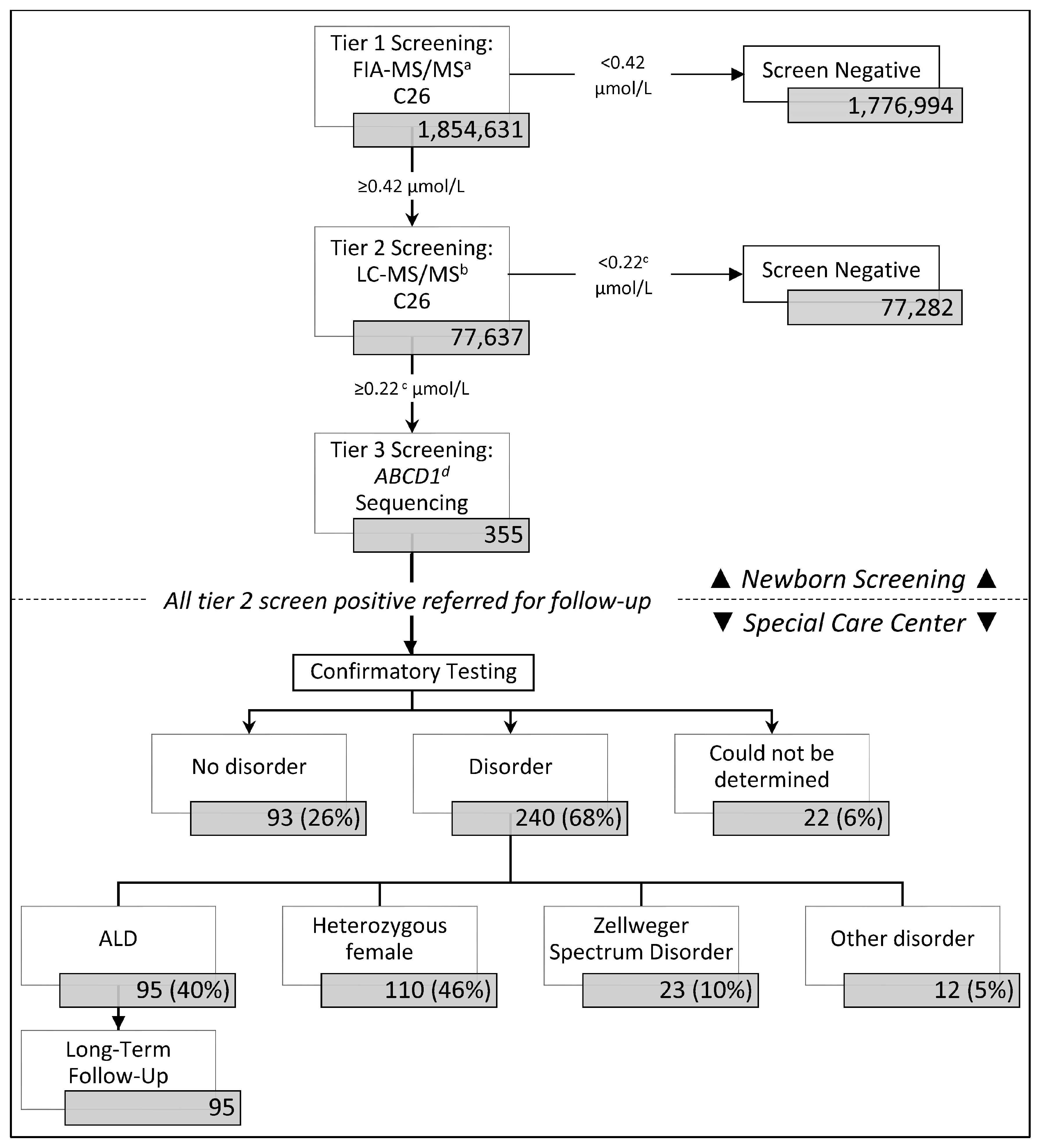
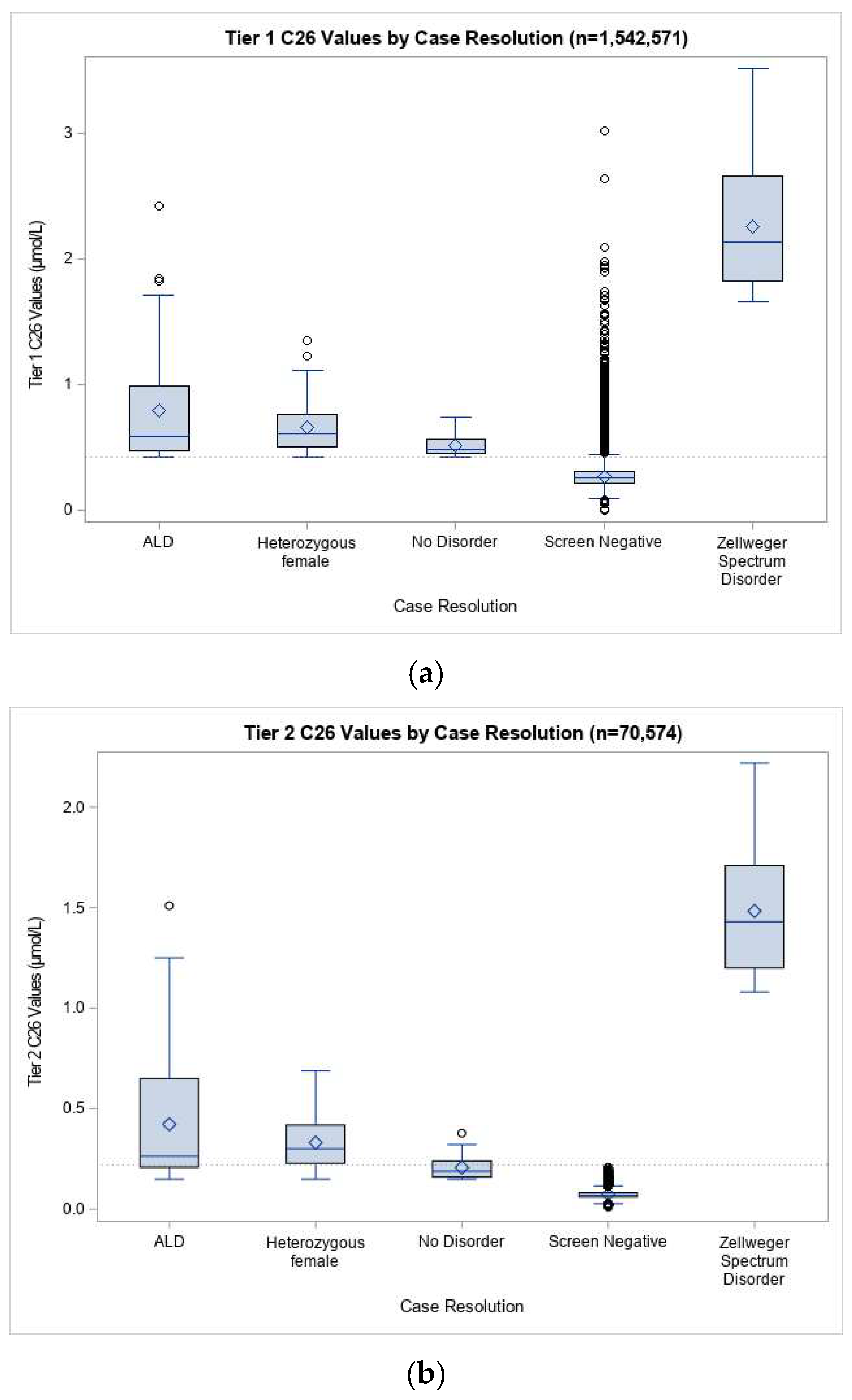
3. Results
3.1. ALD Newborn Screening Outcomes in California
3.2. ABCD1 Sequencing and Short-Term Follow-Up
3.3. ALD Prevalence and Screening Performance in California
3.4. Long-Term Follow-Up (LTFU) Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bezman, L.; Moser, A.B.; Raymond, G.V.; Rinaldo, P.; Watkins, P.A.; Smith, K.D.; Kass, N.E.; Moser, H.W. Adrenoleukodystrophy: Incidence, new mutation rate, and results of extended family screening. Ann. Neurol. 2001, 49, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Moser, A.B.; Jones, R.O.; Hubbard, W.C.; Tortorelli, S.; Orsini, J.J.; Caggana, M.; Vogel, B.H.; Raymond, G.V. Newborn Screening for X-Linked Adrenoleukodystrophy. Int. J. Neonatal Screen. 2016, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, H.W.; Mahmood, A.; Raymond, G.V. X-linked adrenoleukodystrophy. Nat. Clin. Pract. Neurol. 2007, 3, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.R.; Theda, C.; Fatemi, A.; Moser, A.B. X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int. J. Dev. Neurosci. 2020, 80, 52–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B.T. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Wiesinger, C.; Eichler, F.S.; Berger, J. The genetic landscape of X-linked adrenoleukodystrophy: Inheritance, mutations, modifier genes, and diagnosis. Appl. Clin. Genet. 2015, 8, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, S.; Pujol, A.; Waterham, H.R.; van Geel, B.M.; Boehm, C.D.; Raymond, G.V.; Cutting, G.R.; Wanders, R.J.; Moser, H.W. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: Role in diagnosis and clinical correlations. Hum. Mutat. 2001, 18, 499–515. [Google Scholar] [CrossRef]
- Van Geel, B.M.; Bezman, L.; Loes, D.J.; Moser, H.W.; Raymond, G.V. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann. Neurol. 2001, 49, 186–194. [Google Scholar] [CrossRef]
- Jangouk, P.; Zackowski, K.M.; Naidu, S.; Raymond, G.V. Adrenoleukodystrophy in female heterozygotes: Underrecognized and undertreated. Mol. Genet. Metab. 2012, 105, 180–185. [Google Scholar] [CrossRef]
- Engelen, M.; Barbier, M.; Dijkstra, I.M.; Schur, R.; de Bie, R.M.; Verhamme, C.; Dijkgraaf, M.G.; Aubourg, P.A.; Wanders, R.J.; van Geel, B.M.; et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain J. Neurol. 2014, 137, 693–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, A.; Raymond, G.V.; Dubey, P.; Peters, C.; Moser, H.W. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: A comparison study. Lancet Neurol. 2007, 6, 687–692. [Google Scholar] [CrossRef]
- Pierpont, E.I.; Eisengart, J.B.; Shanley, R.; Nascene, D.; Raymond, G.V.; Shapiro, E.G.; Ziegler, R.S.; Orchard, P.J.; Miller, W.P. Neurocognitive Trajectory of Boys Who Received a Hematopoietic Stem Cell Transplant at an Early Stage of Childhood Cerebral Adrenoleukodystrophy. JAMA Neurol. 2017, 74, 710–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallack, E.J.; Turk, B.; Yan, H.; Eichler, F.S. The Landscape of Hematopoietic Stem Cell Transplant and Gene Therapy for X-Linked Adrenoleukodystrophy. Curr. Treat. Options Neurol. 2019, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Turk, B.; Mallack, E.; Adang, L.; Becker, C.; Eichler, F.; Haren, K.V.; Hollandsworth, K.; Kurtzberg, J.; Kwon, J.; Lund, T.; et al. Consensus Guidelines: MRI Surveillance of Children with Presymptomatic Adrenoleukodystrophy. In Proceedings of the AAN 2019 Annual Meeting, Philadelphia, PA, USA, 8 May 2019. [Google Scholar]
- Regelmann, M.O.; Kamboj, M.K.; Miller, B.S.; Nakamoto, J.M.; Sarafoglou, K.; Shah, S.; Stanley, T.L.; Marino, R. Adrenoleukodystrophy: Guidance for Adrenal Surveillance in Males Identified by Newborn Screen. J. Clin. Endocrinol. Metab. 2018, 103, 4324–4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eng, L.; Regelmann, M.O. Adrenoleukodystrophy in the era of newborn screening. Curr. Opinion Endocrinol. Diabetes Obes. 2020, 27, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Huffnagel, I.C.; Laheji, F.K.; Aziz-Bose, R.; Tritos, N.A.; Marino, R.; Linthorst, G.E.; Kemp, S.; Engelen, M.; Eichler, F. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J. Clin. Endocrinol. Metab. 2019, 104, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, P.; Raymond, G.V.; Moser, A.B.; Kharkar, S.; Bezman, L.; Moser, H.W. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J. Pediatr. 2005, 146, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Mallack, E.J.; Turk, B.R.; Yan, H.; Price, C.; Mlis, M.D.; Moser, A.B.; Becker, C.; Hollandsworth, K.; Adang, L.; Vanderver, A.; et al. MRI Surveillance of Boys with X-linked Adrenoleukodystrophy Identified by Newborn Screening: Meta-analysis and Consensus Guidelines. J. Inherit. Metab. Dis. 2020. [Google Scholar] [CrossRef]
- Shulman, D.I.; Palmert, M.R.; Kemp, S.F. Adrenal insufficiency: Still a cause of morbidity and death in childhood. Pediatrics 2007, 119, e484–e494. [Google Scholar] [CrossRef] [Green Version]
- Recommended Uniform Screening Panel. Available online: https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html (accessed on 9 November 2020).
- Hubbard, W.C.; Moser, A.B.; Tortorelli, S.; Liu, A.; Jones, D.; Moser, H. Combined liquid chromatography-tandem mass spectrometry as an analytical method for high throughput screening for X-linked adrenoleukodystrophy and other peroxisomal disorders: Preliminary findings. Mol. Genet. Metab. 2006, 89, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, W.C.; Moser, A.B.; Liu, A.C.; Jones, R.O.; Steinberg, S.J.; Lorey, F.; Panny, S.R.; Vogt, R.F., Jr.; Macaya, D.; Turgeon, C.T.; et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): Validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol. Genet. Metab. 2009, 97, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Theda, C.; Gibbons, K.; Defor, T.E.; Donohue, P.K.; Golden, W.C.; Kline, A.D.; Gulamali-Majid, F.; Panny, S.R.; Hubbard, W.C.; Jones, R.O.; et al. Newborn screening for X-linked adrenoleukodystrophy: Further evidence high throughput screening is feasible. Mol. Genet. Metab 2014, 111, 55–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, B.H.; Bradley, S.E.; Adams, D.J.; D’Aco, K.; Erbe, R.W.; Fong, C.; Iglesias, A.; Kronn, D.; Levy, P.; Morrissey, M.; et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: Diagnostic protocol, surveillance protocol and treatment guidelines. Mol. Genet. Metab. 2015, 114, 599–603. [Google Scholar] [CrossRef]
- Kemper, A.R.; Brosco, J.; Comeau, A.M.; Green, N.S.; Grosse, S.D.; Jones, E.; Kwon, J.M.; Lam, W.K.; Ojodu, J.; Prosser, L.A.; et al. Newborn screening for X-linked adrenoleukodystrophy: Evidence summary and advisory committee recommendation. Genet. Med. 2017, 19, 121–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assembly Bill No. 1559. Available online: https://leginfo.legislature.ca.gov/faces/billNavClient.xhtml?bill_id=201320140AB1559 (accessed on 9 November 2020).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Matteson, J.; Rinaldo, P.; Tortorelli, S.; Currier, R.; Sciortino, S. The Clinical Impact of CLIR Tools toward Rapid Resolution of Post-Newborn Screening Confirmatory Testing for X-linked Adrenoleukodystrophy. Int. J. Neonatal Screen. 2020, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Laboratory Integrated Reports. Available online: https://clir.mayo.edu/ (accessed on 9 November 2020).
- Wiens, K.; Berry, S.A.; Choi, H.; Gaviglio, A.; Gupta, A.; Hietala, A.; Kenney-Jung, D.; Lund, T.; Miller, W.; Pierpont, E.I.; et al. A report on state-wide implementation of newborn screening for X-linked Adrenoleukodystrophy. Am. J. Med Genet. Part A 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Clinard, K.; Young, S.P.; Rehder, C.W.; Fan, Z.; Calikoglu, A.S.; Bali, D.S.; Bailey, D.B., Jr.; Gehtland, L.M.; Millington, D.S.; et al. Evaluation of X-Linked Adrenoleukodystrophy Newborn Screening in North Carolina. JAMA Netw. Open 2020, 3, e1920356. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Li, H.; Hagar, A.F.; Jerris, S.C.; Wittenauer, A.; Wilcox, W. Newborn Screening for X-Linked Adrenoleukodystrophy in Georgia: Experiences from a Pilot Study Screening of 51,081 Newborns. Int. J. Neonatal Screen. 2020, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Amorosi, C.A.; Myskova, H.; Monti, M.R.; Argarana, C.E.; Morita, M.; Kemp, S.; Dodelson de Kremer, R.; Dvorakova, L.; Oller de Ramirez, A.M. X-linked adrenoleukodystrophy: Molecular and functional analysis of the ABCD1 gene in Argentinean patients. PLoS ONE 2012, 7, e52635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ligtenberg, M.J.; Kemp, S.; Sarde, C.O.; van Geel, B.M.; Kleijer, W.J.; Barth, P.G.; Mandel, J.L.; van Oost, B.A.; Bolhuis, P.A. Spectrum of mutations in the gene encoding the adrenoleukodystrophy protein. Am. J. Hum. Genet. 1995, 56, 44–50. [Google Scholar] [PubMed]
- Klouwer, F.C.; Berendse, K.; Ferdinandusse, S.; Wanders, R.J.; Engelen, M.; Poll-The, B.T. Zellweger spectrum disorders: Clinical overview and management approach. Orphanet J. Rare Dis. 2015, 10, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmermans, S.; Buchbinder, M. Patients-in-waiting: Living between sickness and health in the genomics era. J. Health Soc. Behav. 2010, 51, 408–423. [Google Scholar] [CrossRef] [PubMed]
- Schwan, K.; Youngblom, J.; Weisiger, K.; Kianmahd, J.; Waggoner, R.; Fanos, J. Family Perspectives on Newborn Screening for X-Linked Adrenoleukodystrophy in California. Int. J. Neonatal Screen. 2019, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Taneja, K.K.; Kalra, V.; Behari, M.; Aneja, S.; Bansal, S.K. Genomic profiling identifies novel mutations and SNPs in ABCD1 gene: A molecular, biochemical and clinical analysis of X-ALD cases in India. PLoS ONE 2011, 6, e25094. [Google Scholar] [CrossRef] [PubMed]
- The ALD Mutation Database. Available online: https://adrenoleukodystrophy.info/mutations-and-variants-in-abcd1 (accessed on 17 February 2021).



{kind=link}
{kind=link}
| Males | Females | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Pathogenic | VUS | No Variant | All Males | Pathogenic | VUS | No Variant | All Females | All Cases | |
| Screen Positive | 32 | 70 | 52 | 154 | 52 | 77 | 72 | 201 | 355 |
| Cases with Plasma VLCFA a Performed | 27 | 59 | 44 | 130 | 27 | 53 | 54 | 134 | 264 |
| Resolved | 32 | 70 | 52 | 154 | 52 | 77 | 72 | 201 | 355 |
| ALD (Male) | 32 | 60 | 3 b | 95 | - | - | - | - | 95 |
| ALD heterozygous female | - | - | - | - | 52 | 54 | 4 c | 110 | 110 |
| No disorder | 0 | 7 | 33 | 40 | 0 | 12 | 41 | 53 | 92 |
| Zellweger Spectrum Disorder | 0 | 0 | 7 | 7 | 0 | 1 | 15 | 16 | 23 |
| Other disorder | 0 | 0 | 6 | 6 | 0 | 2 | 4 | 6 | 12 |
| Could not be determined | 0 | 3 | 3 | 6 | 0 | 8 | 8 | 16 | 22 |
| Before Cutoff Change | After Cutoff Change | |||||
|---|---|---|---|---|---|---|
| (n = 876,131) | (n = 978,500) | |||||
| Resolution | Count | Birth Prevalence | Count | Birth Prevalence | ||
| (1 in) | (per 1,000,000) | (1 in) | (per 1,000,000) | |||
| ALD (male) | 61 | 7181 | 139 | 34 | 14,390 | 69 |
| ALD heterozygous female | 59 | 7424 | 135 | 51 | 9593 | 104 |
| Zellweger Spectrum Disorder | 6 | 146,022 | 7 | 17 | 57,559 | 17 |
| Other disorder | 6 | 146,022 | 7 | 6 | 163,083 | 6 |
| All disorders | 132 | 6637 | 151 | 108 | 9060 | 110 |
| Year 1 | Year 2 | Year 3+ | Total Cases a | |
|---|---|---|---|---|
| Reports with Signs of Adrenal Involvement (% of total reports) | ||||
| Abnormal ACTH test | 8 (13%) | 7 (13%) | 1 (6%) | 14 (20%) |
| Poor weight gain, hyperpigmentation, or adrenal insufficiency | 2 (3%) | 1 (2%) | 1 (6%) | 4 (6%) |
| Treated with hydrocortisone (or other glucocorticoid) | 2 (3%) | 3 (6%) | 1 (6%) | 5 (7%) |
| Reports with Signs of Cerebral Involvement (% of total reports) | ||||
| Abnormal MRI | 0 (0%) | 3 (6%) | 2 (11%) | 5 (7%) |
| Behavior changes, seizures, or other neurologic problems | 0 (0%) | 1 (2%) | 1 (6%) | 1 (1%) |
| Treated with HSCT | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Total Reports Submitted | 63 | 52 | 18 | 71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matteson, J.; Sciortino, S.; Feuchtbaum, L.; Bishop, T.; Olney, R.S.; Tang, H. Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up. Int. J. Neonatal Screen. 2021, 7, 22. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7020022
Matteson J, Sciortino S, Feuchtbaum L, Bishop T, Olney RS, Tang H. Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up. International Journal of Neonatal Screening. 2021; 7(2):22. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7020022
Chicago/Turabian StyleMatteson, Jamie, Stanley Sciortino, Lisa Feuchtbaum, Tracey Bishop, Richard S. Olney, and Hao Tang. 2021. "Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up" International Journal of Neonatal Screening 7, no. 2: 22. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7020022