
Glutaric Aciduria Type I Missed by Newborn Screening: Report of Four Cases from Three Families

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Case Reports
2.1. Case 1
2.2. Case 2
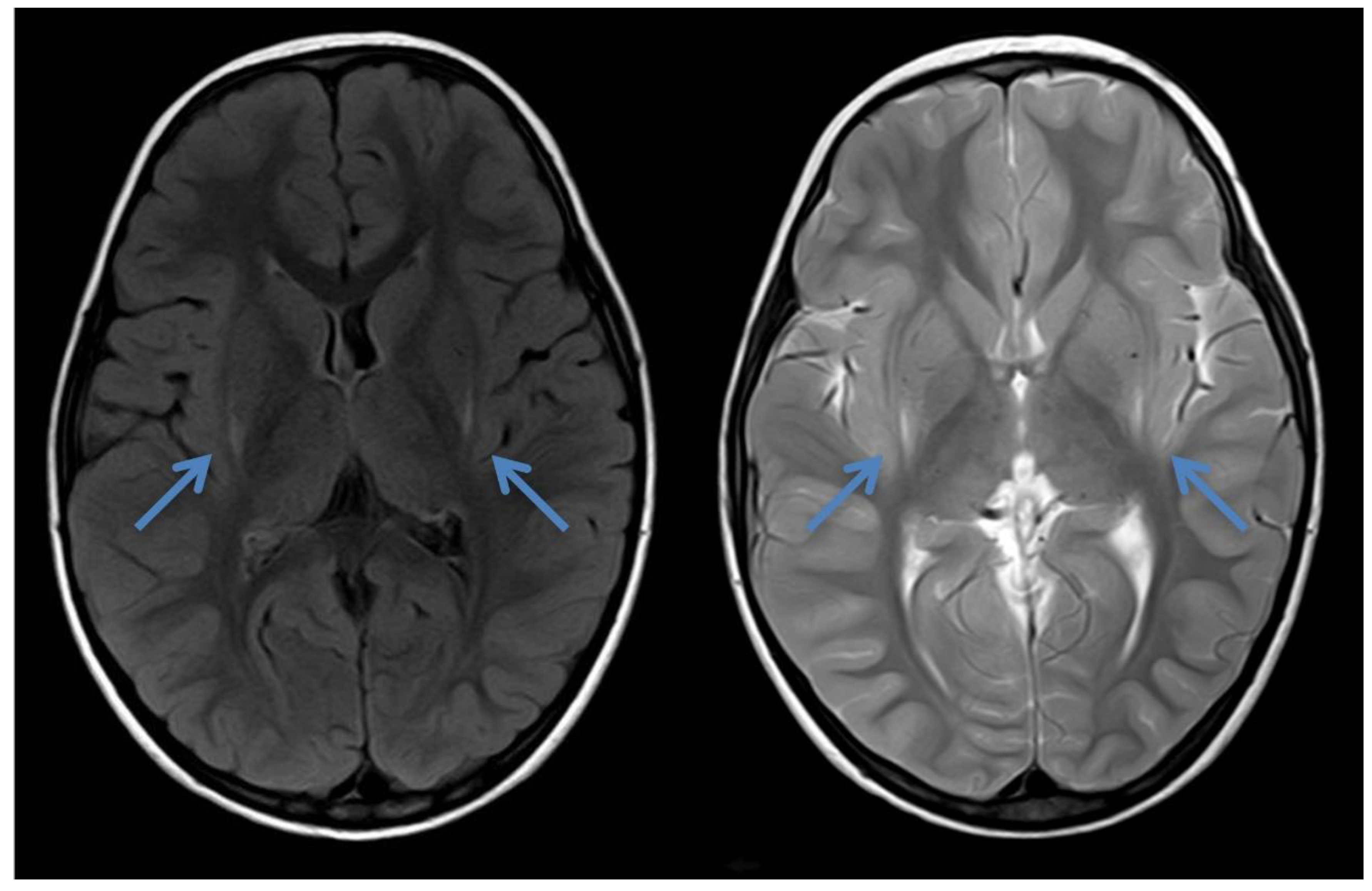
2.3. Case 3
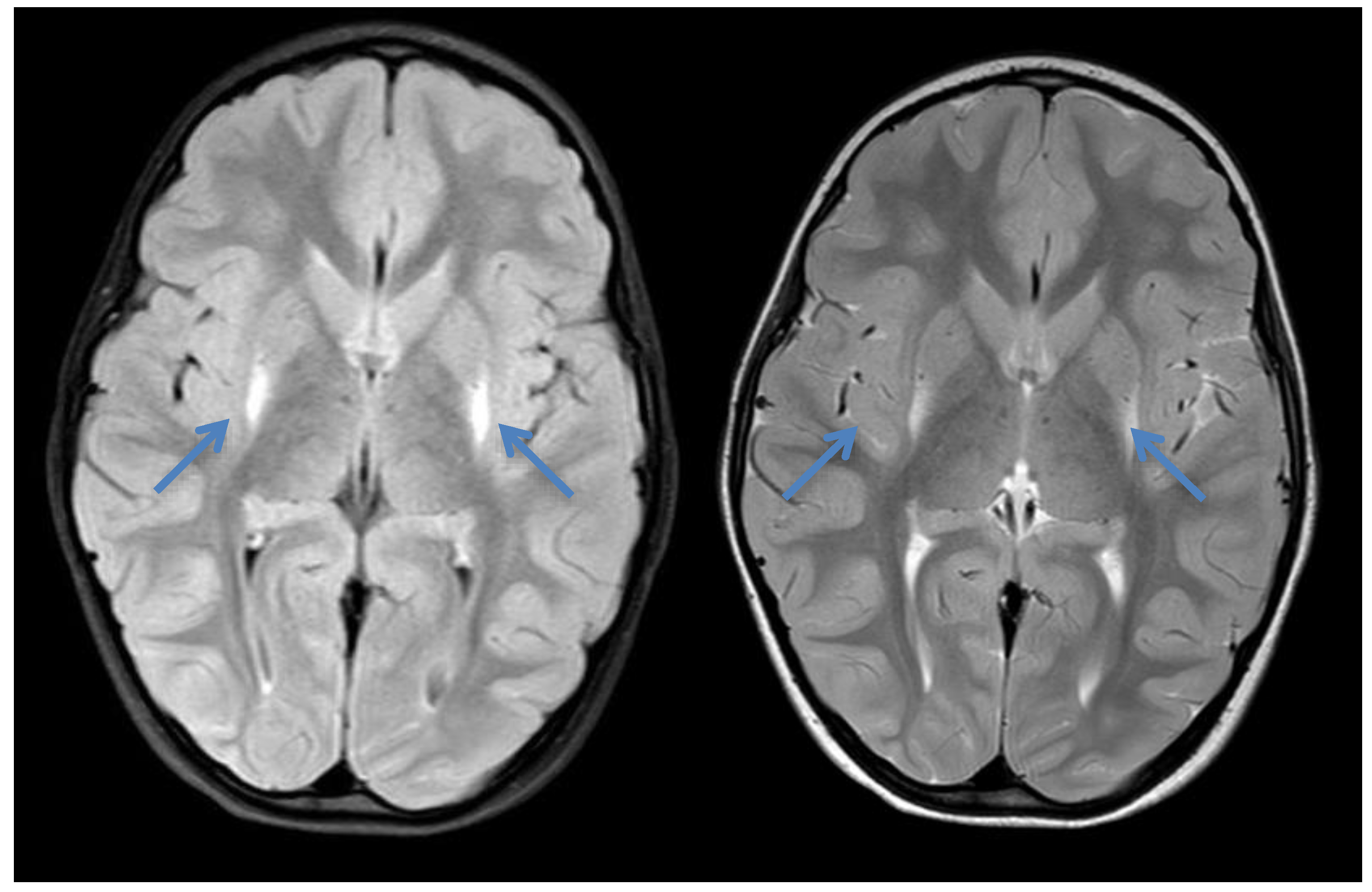
2.4. Case 4
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Heringer, J.; Boy, N.; Burgard, P.; Okun, J.G.; Kölker, S. Newborn Screening for Glutaric Aciduria Type I: Benefits and limitations. Int. J. Neonatal Screen. 2015, 1, 57–68. [Google Scholar] [CrossRef]
- Flott-Rahmel, B.; Falter, C.; Schluff, P.; Fingerhut, R.; Christensen, E.; Jakobs, C.; Musshoff, U.; Fautek, J.D.; Deufel, T.; Ludolph, A.; et al. Nerve cell lesions caused by 3-hydroxyglutaric acid: A possible mechanism for neurodegeneration in glutaric acidaemia I. Clin. Chim. Acta 1999, 283, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Baric, I.; Wagner, L.; Feyh, P.; Liesert, M.; Buckel, W.; Hoffmann, G.F. Sensitivity and specificity of free and total glutaric acid and 3-hydroxyglutaric acid measurements by stable-isotope dilution assays for the diagnosis of glutaric aciduria type I. J. Inher. Metab. Dis. 1999, 22, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Boy, N.; Mühlhausen, C.; Maier, E.M.; Heringer, J.; Assmann, B.; Burgard, P.; Dixon, M.; Fleissner, S.; Greenberg, C.R.; Harting, I.; et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: Second revision. J. Inherit. Metab. Dis. 2017, 40, 75–101. [Google Scholar] [CrossRef]
- Gallagher, R.C.; Cowan, T.M.; Goodman, S.I.; Enns, G.M. Glutaryl-CoA dehydrogenase deficiency and newborn screening: Retrospective analysis of a low excretor provides further evidence that some cases may be missed. Mol. Genet. Metab. 2005, 86, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Boy, N.; Mengler, K.; Heringer-Seifert, J.; Hoffmann, G.F.; Garbade, S.F.; Kölker, S. Impact of newborn screening and quality of therapy on the neurological outcome in glutaric aciduria type 1: A meta-analysis. Genet. Med. 2021, 23, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Heringer, J.; Boy, S.P.N.; Ensenauer, R.; Assmann, B.; Zschocke, J.; Harting, I.; Lücke, T.; Maier, E.M.; Mühlhausen, C.; Haege, G.; et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann. Neurol. 2010, 68, 743–752. [Google Scholar] [CrossRef]
- Smith, W.E.; Millington, D.S.; Koeberl, D.D.; Lesser, P.S. Glutaric acidemia, type I, missed by newborn screening in an infant with dystonia following promethazine administration. Pediatrics 2001, 107, 1184–1187. [Google Scholar] [CrossRef]
- Treacy, E.P.; Lee-Chong, A.; Roche, G.; Lynch, B.; Ryan, S.; Goodman, S.I. Profound neurological presentation resulting from homozygosity for a mild glutaryl-CoA dehydrogenase mutation with a minimal biochemical phenotype. J. Inherit. Metab. Dis. 2003, 26, 72–74. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Wiley, V.; Hammond, J.; Carpenter, K. Screening newborns for inborn errors of metabolism by tandem mass spectrometry. N. Engl. J. Med. 2003, 348, 2304–2312. [Google Scholar] [CrossRef]
- Christensen, E.; Ribes, A.; Merinero, B.; Zschocke, J. Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, L.-A.; Greene, C.L.; Strovel, E.; Rispoli-Joines, J.; Spector, E.; Woontner, M.; Scharer, G.; Enns, G.M.; Gallagher, R.; Zinn, A.B.; et al. The M405V allele of the glutaryl-CoA dehydrogenase gene is an important marker for glutaric aciduria typi I(GA-I) low excretors. Mol. Genet. Metab. 2016, 119, 50–56. [Google Scholar] [CrossRef] [PubMed]
- NIH/ClinVar. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/clinvar/variation/188872/ (accessed on 16 March 2021).
- Diagnostik, Therapie und Management der Glutarazidurie Typ I. Available online: https://www.awmf.org/uploads/tx_szleitlinien/027-018l_S3_Glutarazidurie_Typ_I_2016-07.pdf (accessed on 18 February 2021).
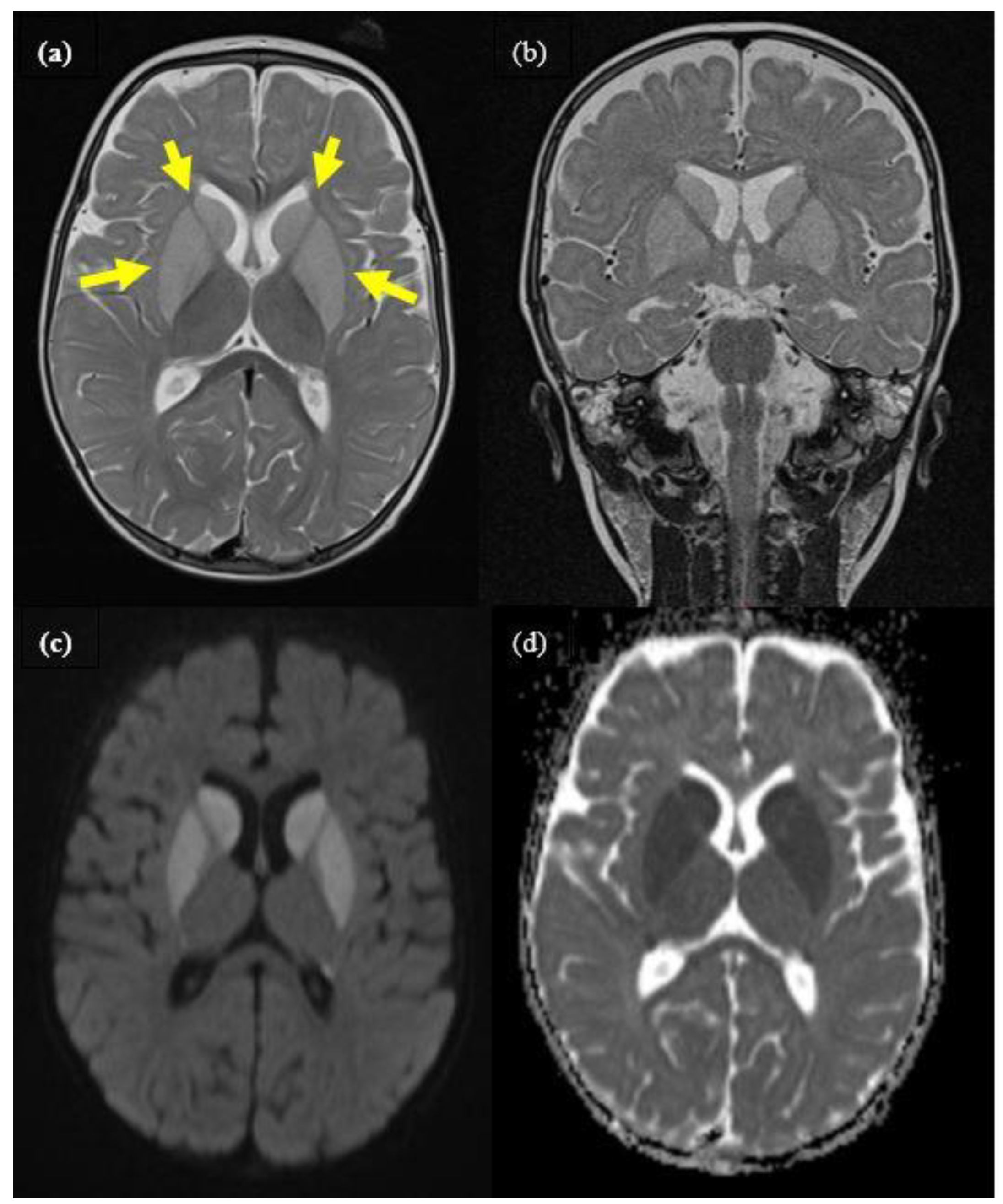
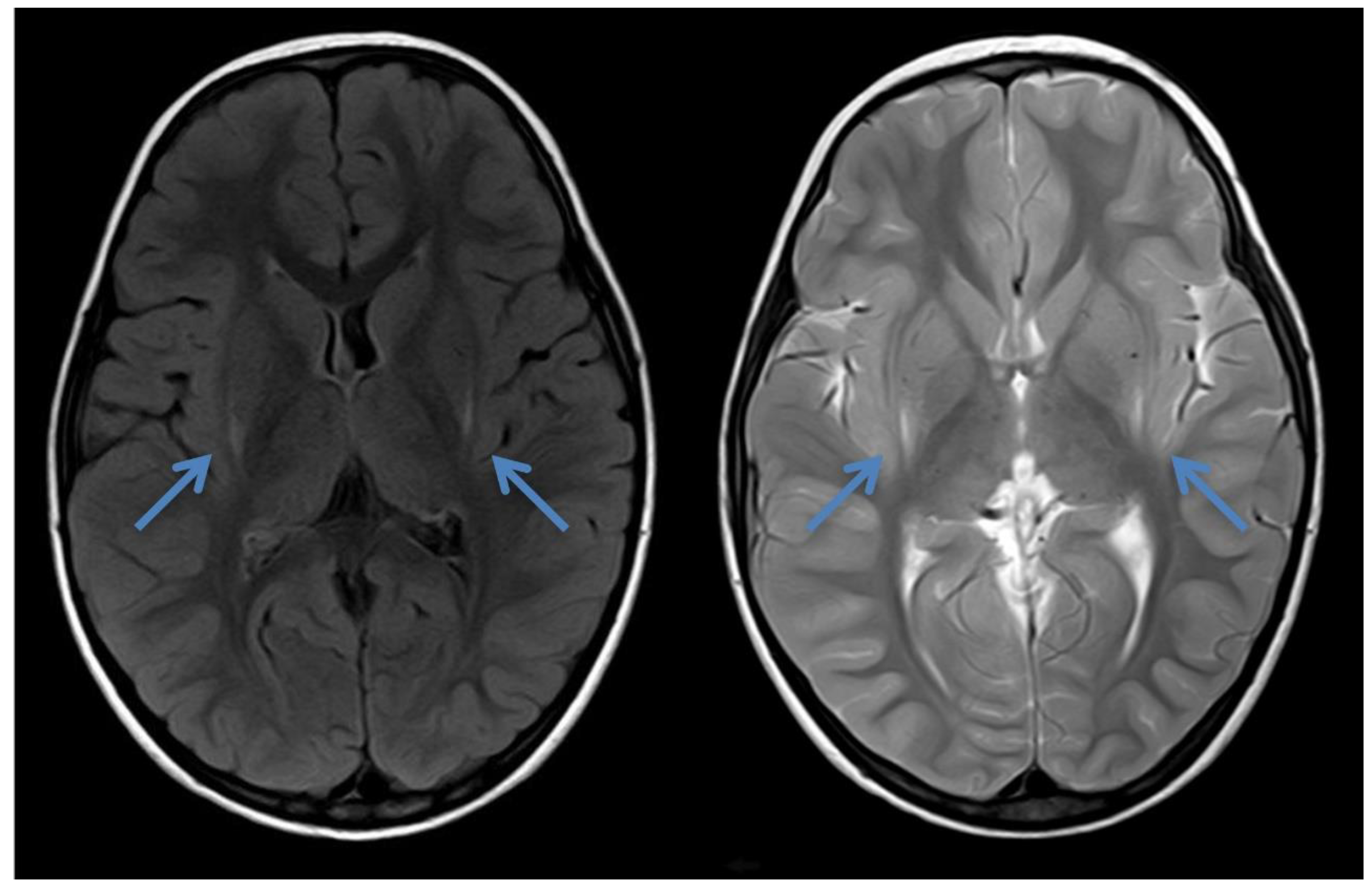
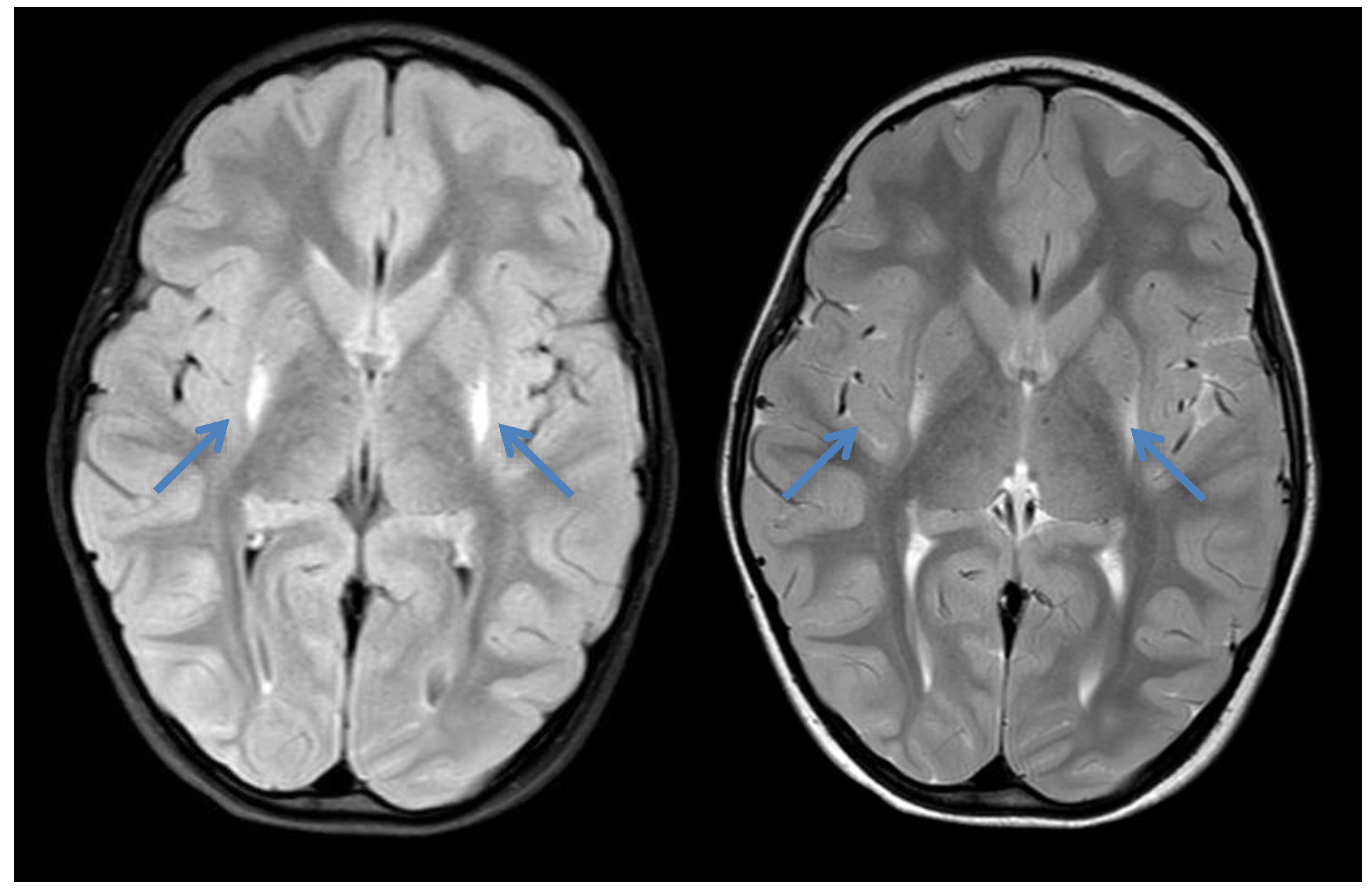

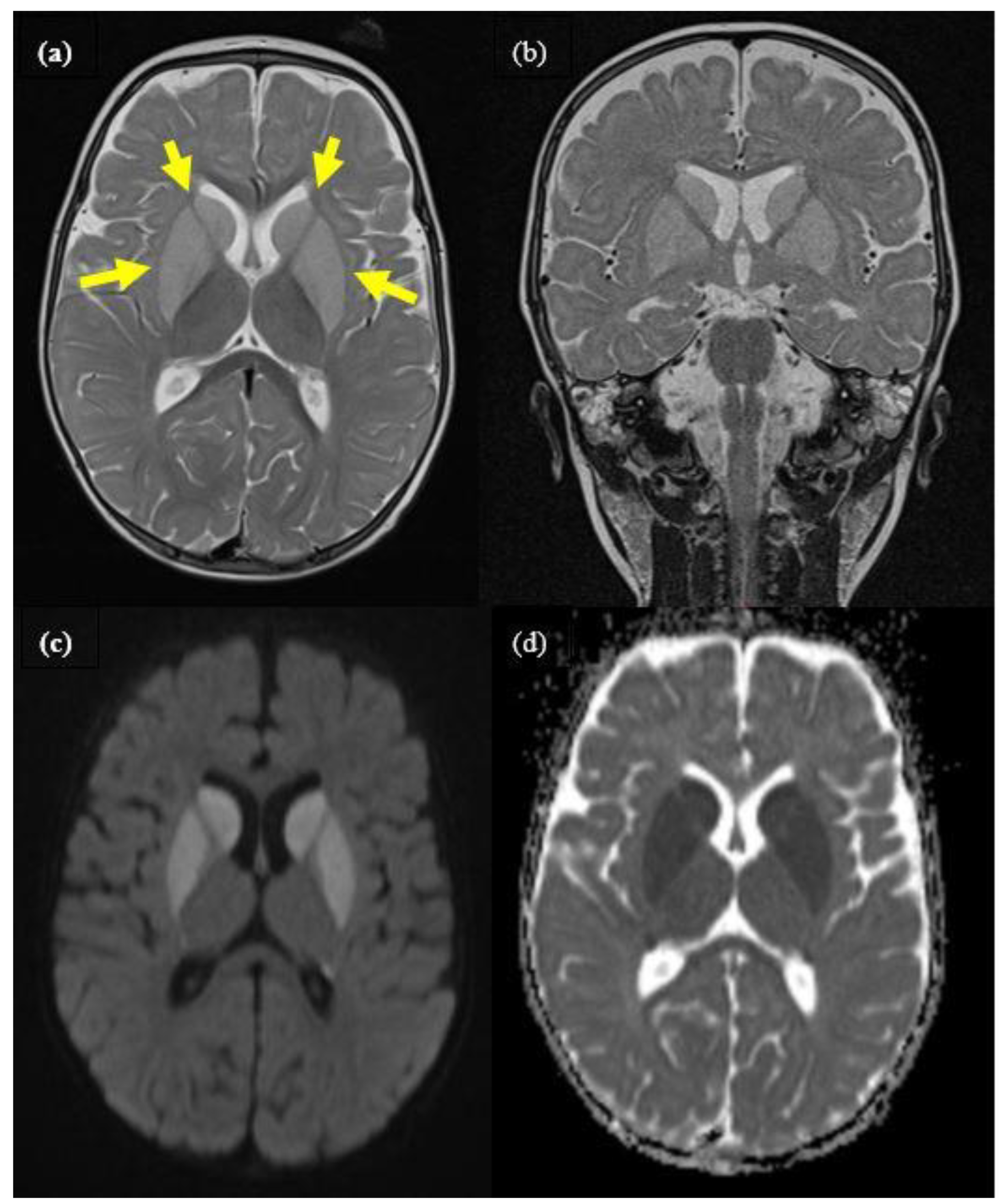{kind=link}
{kind=link}
{kind=link}
| Age | 4 d | Reference Range | 9 m | 10 m | 13 m | 15 m | 18 m | 24 m | Reference Range |
|---|---|---|---|---|---|---|---|---|---|
| DBS | |||||||||
| Free carnitine | 26 | 7–55 | 26 | 45 | 51 | 38 | 35 | 46 | 11–44 |
| Glutarylcarnitine | 0.5 | <0.53 | 0.03 | 0.04 | 0.04 | 0.06 | 0.01 | 0.04 | <0.06 |
| Urine | |||||||||
| Glutaric acid | n.d. | 26 | <10 | <10 | n.d. | n.d. | <10 | 0–20 | |
| 3-hydroxyglutaric acid | n.d. | 22 | 19 | 23 | n.d. | n.d. | 17 | 0–10 | |
| Age | 4 d | 2 y 8 m | 10 y | 11 y | Reference Range |
|---|---|---|---|---|---|
| DBS | |||||
| Free carnitine | n.r. | n.d | n.r. | 37.5 | 7.0–70.0 |
| Glutarylcarnitine | 0.34 | n.d. | 0.27 | 0.30 | <0.2 |
| C5DC/C2 | 0.01 | n.d. | n.r. | n.r. | <0.01 |
| C5DC/C4 | n.r. | n.d. | 1.29 | 5.09 | 0.02–0.32 |
| C5DC/C8 | 2.01 | n.d. | 9.25 | 6.73 | <8 |
| C5DC/C12 | n.r. | n.d. | 11.2 | 0.84 | 0.05–0.83 |
| C5DC/C16 | 0.13 | n.d. | n.r. | n.r. | <0.19 |
| Urine | |||||
| Glutaric acid | normal | normal | n.d. | 0–20 | |
| 3-hydroxyglutaric acid | normal | 13 | n.d. | 0–10 | |
| Age | 4 d | 4 y | 5 y | Reference Range |
|---|---|---|---|---|
| DBS | ||||
| Free carnitine | - | n.r. | 42.5 | 7.0–70.0 |
| Glutarylcarnitine | 0.19 | normal | 0.16 | <0.2 |
| C5DC/C2 | 0.01 | n.r. | n.r. | <0.01 |
| C5DC/C4 | 0.76 | n.r. | 0.54 | 0.02–0.32 |
| C5DC/C8 | 3.17 | 2.21 | 4.12 | <8 |
| C5DC/C12 | n.r | n.r. | 2.91 | 0.05–0.83 |
| C5DC/C16 | 0.05 | n.r. | n.r. | <0.19 |
| Urine | ||||
| Glutaric acid | n.d. | normal | n.d. | 0–20 |
| 3-hydroxyglutaric acid | n.d. | 14.2 | n.d. | 0–10 |
| Age | 47 h | 8 d | Reference Range | 21 m | Reference Range |
|---|---|---|---|---|---|
| DBS | |||||
| Free carnitine | 16.2 | 13.1 | 6–47 | n.r. | n.r |
| Glutarylcarnitine | 0.78 | 0.38 | <0.45 | 0.12 # | <0.09 |
| C5DC/C4 | 2.39 | 1.52 | <1.87 | n.r. | n.r. |
| C5DC/C8 | 5.97 | 7.5 | <5.60 | n.r. | n.r. |
| C5DC/C12 | 1.6 | 2.27 | <1.92 | n.r. | n.r. |
| Plasma | |||||
| Glutarylcarnitine | n.d. | n.d. | 0.35 | <0.23 | |
| Urine | |||||
| Glutaric acid | n.d. | n.d. | 0–20 | n.r. | 0–20 |
| 3-hydroxyglutaric acid | n.d. | n.d. | - | 8.66 | 0.17–1.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spenger, J.; Maier, E.M.; Wechselberger, K.; Bauder, F.; Kocher, M.; Sperl, W.; Preisel, M.; Schiergens, K.A.; Konstantopoulou, V.; Röschinger, W.; et al. Glutaric Aciduria Type I Missed by Newborn Screening: Report of Four Cases from Three Families. Int. J. Neonatal Screen. 2021, 7, 32. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7020032
Spenger J, Maier EM, Wechselberger K, Bauder F, Kocher M, Sperl W, Preisel M, Schiergens KA, Konstantopoulou V, Röschinger W, et al. Glutaric Aciduria Type I Missed by Newborn Screening: Report of Four Cases from Three Families. International Journal of Neonatal Screening. 2021; 7(2):32. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7020032
Chicago/Turabian StyleSpenger, Johannes, Esther M. Maier, Katharina Wechselberger, Florian Bauder, Melanie Kocher, Wolfgang Sperl, Martin Preisel, Katharina A. Schiergens, Vassiliki Konstantopoulou, Wulf Röschinger, and et al. 2021. "Glutaric Aciduria Type I Missed by Newborn Screening: Report of Four Cases from Three Families" International Journal of Neonatal Screening 7, no. 2: 32. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7020032