
Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant

 and
and
Abstract
:1. Introduction
2. Epidemiology in Japan
3. Neonatal Screening for PA in Japan: Issues to Be Addressed
3.1. Correlation between Genotypes and Clinical/Biochemical Phenotypes
3.2. Potential Risk of Cardiac Complications in Asymptomatic Patients Detected by Neonatal Screening
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Forny, P.; Höster, F.; Ballhausen, D.; Chakrapani, A.; Chapman, K.A.; Dionisi-Vici, C.; Dixon, M.; Grünert, S.C.; Grunewald, S.; Haliloglu, G.; et al. Guidelines for the diagnosis and management of methylmalonic acidaemia and propionic acidaemia: First revision. J. Inherit. Metab. Dis. 2021, 44, 566–592. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Jans, J.J.M.; Tas, S.Y.; Verhoeven-Duif, N.M.; van Hasselt, P.M. Pathophysiology of propionic and methylmalonic acidemias. Part 1: Complications. J. Inherit. Metab. Dis. 2019, 42, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; van Hasselt, P.M.; Jans, J.J.M.; Tas, S.Y.; Verhoeven-Duif, N.M. Pathophysiology of propionic and methylmalonic acidemias. Part 2: Treatment strategies. J. Inherit. Metab. Dis. 2019, 42, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sakamoto, O.; Matsubara, Y.; Kure, S.; Suzuki, Y.; Aoki, Y.; Yamaguchi, S.; Takahashi, Y.; Nishikubo, T.; Kawaguchi, C.; et al. Mutation spectrum of the PCCA and PCCB genes in Japanese patients with propionic acidemia. Mol. Genet. Metab. 2004, 81, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Yorifuji, T.; Kawai, M.; Muroi, J.; Mamada, M.; Kurokawa, K.; Shigematsu, Y.; Hirano, S.; Sakura, N.; Yoshida, I.; Kuhara, T.; et al. Unexpectedly high prevalence of the mild form of propionic acidemia in Japan: Presence of a common mutation and possible clinical implications. Hum. Genet. 2002, 111, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Heringer, J.; Valayannopoulos, V.; Lund, A.M.; Wijburg, F.A.; Freisinger, P.; Barić, I.; Baumgartner, M.R.; Burgard, P.; Burlina, A.B.; Chapman, K.A.; et al. Impact of age at onset and newborn screening on outcome in organic acidurias. J. Inherit. Metab. Dis. 2016, 39, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, K.; Nakajima, Y.; Tajima, G.; Watanabe, Y.; Hotta, Y.; Kataoka, T.; Kawade, Y.; Sugiyama, N.; Ito, T.; Kimura, K.; et al. Determination of methylmalonyl coenzyme A by ultra high-performance liquid chromatography tandem mass spectrometry for measuring propionyl coenzyme A carboxylase activity in patients with propionic acidemia. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1046, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Molema, F.; Langeveld, M.; Janssen, M.C.; Bosch, A.M.; van Spronsen, F.; Mulder, M.F.; Verhoeven-Duif, N.M.; Jans, J.J.M.; van der Ploeg, A.T.; et al. Retrospective evaluation of the Dutch pre-newborn screening cohort for propionic acidemia and isolated methylmalonic acidemia: What to aim, expect, and evaluate from newborn screening? J. Inherit. Metab. Dis. 2020, 43, 424–437. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.S.; Jans, J.J.M.; van der Ham, M.; van Hasselt, P.M.; Verhoeven-Duif, N.M. Understanding acute metabolic decompensateion in propionic and methylalonic acidemias: A deep metabolic phenotyping approach. Orphanet J. Rare Dis. 2020, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.; Haliloglu, G.; Karall, D.; Chapman, K.A.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 130–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pena, L.; Burton, B.K. Survey of health status and complications among propionic acidemia patients. Am. J. Med. Genet. A 2012, 158A, 1641–1646. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Valayannopoulos, V.; Touati, G.; Jais, J.P.; Rabier, D.; de Keyzer, Y.; Bonnet, D.; de Lonlay, P. Cardiomyopathies in propionic aciduria are reversible after liver transplantation. J. Pediatr. 2010, 156, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Valayannopoulos, V.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; Barić, I.; Karall, D.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2. The evolving clinical phenotype. J. Inherit. Metab. Dis. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, D.; Scholl-Bürgi, S.; Sass, J.O.; Sperl, W.; Schweigmann, U.; Stein, J.I.; Karall, D. Prolonged QTc intervals and decreased left ventricular contractility in patients with propionic acidemia. J. Pediatr. 2007, 150, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Hargreaves, I.; Krywawych, S.; Chalasani, A.; Land, J.M.; Davison, J.E.; Kwok, M.K.; Christov, G.; Karimova, A.; Ashworth, M.; et al. Successful reversal of propionic acidaemia associated cardiomyopathy: Evidence for low myocardial coenzyme Q10 status and secondary mitochondrial dysfunction as an underlying pathophysiological mechanism. Mitochondrion 2014, 17, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Bodi, I.; Grünert, S.C.; Becker, N.; Stoelzle-Feix, S.; Spiekerkoetter, U.; Zehender, M.; Bugger, H.; Bode, C.; Odening, K.E. Mechanisms of acquired long QT syndrome in patients with propionic academia. Heart Rhythm 2016, 13, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Riemersma, M.; Hazebroek, M.R.; Helderman-van den Enden, A.T.J.M.; Salomons, G.S.; Ferdinandusse, S.; Brouwers, M.C.G.; van der Ploeg, L.; Heymans, S.; Glatz, J.F.C.; van den Wijngaard, A.; et al. Propionic acidemia as a cause of adult-onset dilated cardiomyopathy. Eur. J. Hum. Genet. 2017, 25, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Cazorla, A.G.; Valayannopoulos, V.; Lund, A.M.; Burlina, A.B.; Sykut-Cegielska, J.; Wijburg, F.A.; Teles, E.L.; Zeman, J.; Dionisi-Vici, C.; et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: The initial presentation. J. Inherit. Metab. Dis. 2015, 38, 1041–1057. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Propionic Acidemia Detected by Neonatal Screening (n = 87) | Symptomatic Patients (n = 15) | |||
|---|---|---|---|---|
| Clinical phenotypes (n) | ||||
| None | 86 | Acute acidotic crisis, neonatal onset | 9 | |
| Acute acidotic crisis, infantile onset | 3 | |||
| Mental retardation | 1 | Mental retardation | 2 | |
| Syncope | 1 | |||
| Genetic variants (n) | ||||
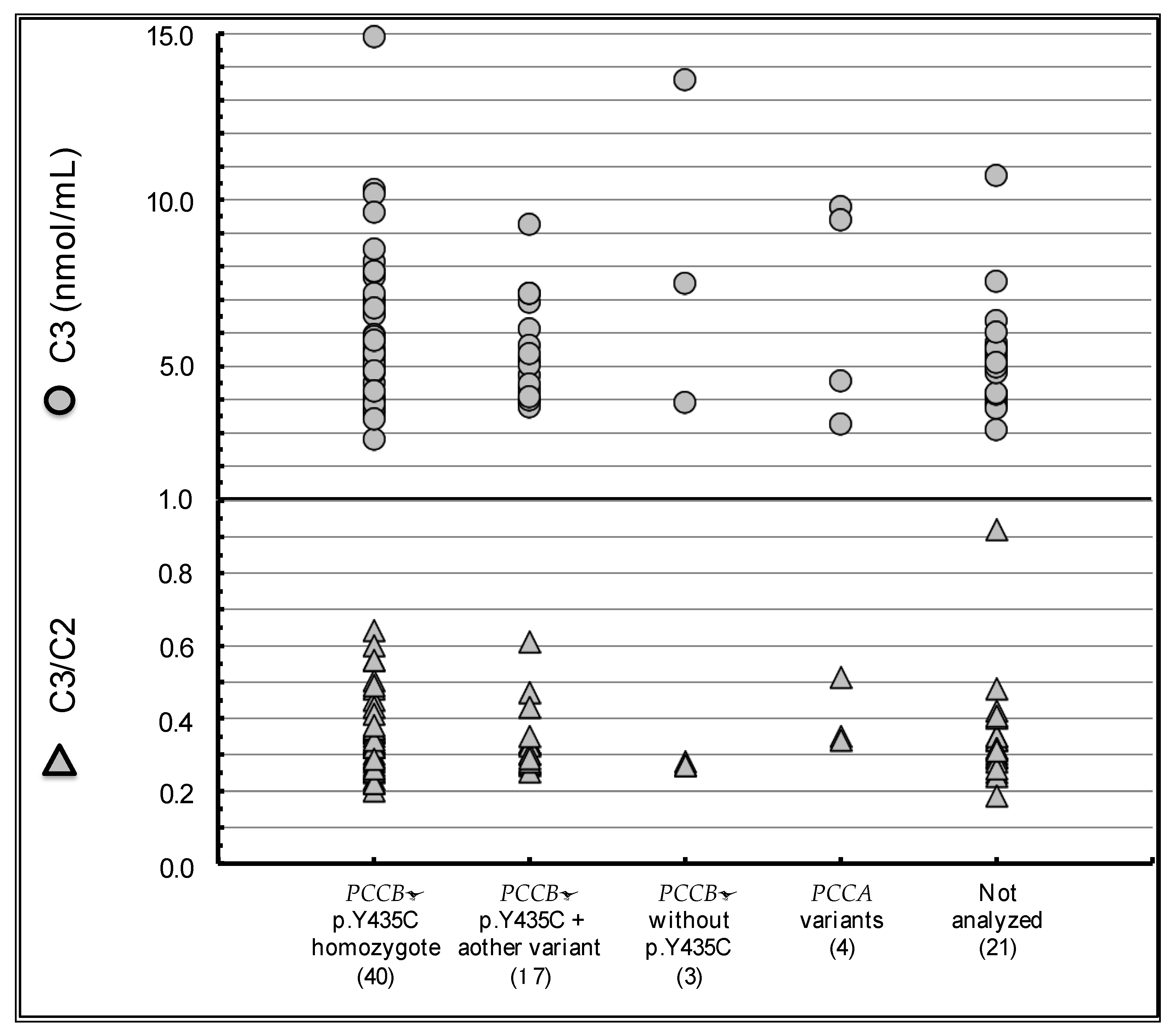
| PCCB | p.Y435C homozygote | 41 | p.T428I homozygote | 1 |
| p.Y435C + p.T428I | 5 | p.T428I + frameshift variant | 1 | |
| p.Y435C + another variant | 11 | Other biallelic variants | 2 | |
| p.Y435C heterozygote 1 | 4 | p.Y435C detected | 0 | |
| p.Y435C not detected 1 | 6 | |||
| PCCA | Biallelic variants detected | 5 | Biallelic variants detected | 5 |
| No information of genotypes | 15 | No information of genotypes | 6 | |
| Patients | Gene and Variant | Allele Frequency |
|---|---|---|
| Patients detected by neonatal screening (n = 58) | PCCB p.Y435C | 79/116 (68.1%) |
| PCCB p.T428I | 7/116 (6.0%) | |
| PCCB p.I430L | 3/116 (2.6%) | |
| PCCB p.S510del | 2/116 (1.7%) | |
| Symptomatic patients (n = 6) | PCCA p.L308fs | 2/11 (18.2%) |
| PCCA p.W559L | 2/11 (18.2%) |
| Clinical Phenotype | Number of Patients | Cardiac Complication | Cardiac Findings |
|---|---|---|---|
| Acute acidotic crisis, neonatal onset | 9 | 3 | Cardiomyopathy + ventricular tachycardia |
| Left ventricular dilatation + QT prolongation | |||
| Mild tricuspid regurgitation | |||
| Acute acidotic crisis, infantile onset | 3 | 2 | Left ventricular dilatation + mild mitral regurgitation |
| QT prolongation | |||
| Chronic symptoms only | 3 | 2 | QT prolongation |
| QT prolongation with syncope | |||
| Total | 15 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tajima, G.; Kagawa, R.; Sakura, F.; Nakamura-Utsunomiya, A.; Hara, K.; Yuasa, M.; Hasegawa, Y.; Sasai, H.; Okada, S. Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant. Int. J. Neonatal Screen. 2021, 7, 35. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7030035
Tajima G, Kagawa R, Sakura F, Nakamura-Utsunomiya A, Hara K, Yuasa M, Hasegawa Y, Sasai H, Okada S. Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant. International Journal of Neonatal Screening. 2021; 7(3):35. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7030035
Chicago/Turabian StyleTajima, Go, Reiko Kagawa, Fumiaki Sakura, Akari Nakamura-Utsunomiya, Keiichi Hara, Miori Yuasa, Yuki Hasegawa, Hideo Sasai, and Satoshi Okada. 2021. "Current Perspectives on Neonatal Screening for Propionic Acidemia in Japan: An Unexpectedly High Incidence of Patients with Mild Disease Caused by a Common PCCB Variant" International Journal of Neonatal Screening 7, no. 3: 35. https://0-doi-org.brum.beds.ac.uk/10.3390/ijns7030035