Isomerization and Properties of Isomers of Carbocyanine Dyes


N.M. Emanuel Institute of Biochemical Physics, Russian Academy of Sciences, Kosygin Str. 4, 119334 Moscow, Russia
*
Author to whom correspondence should be addressed.
Sci 2019, 1(1), 19; https://0-doi-org.brum.beds.ac.uk/10.3390/sci1010019
Submission received: 20 November 2018
/
Accepted: 26 November 2018
/
Published: 20 March 2019
Abstract
:One of the important features of polymethine (cyanine) dyes is isomerization about one of C–C bonds of the polymethine chain. In this review, spectral properties of the isomers, photoisomerization and thermal back isomerization of carbocyanine dyes, mostly meso-substituted carbocyanine dyes, are considered. meso-Alkyl-substituted thiacarbocyanine dyes are present in polar solvents mainly as cis isomers and, hence, exhibit no photoisomerization, whereas in nonpolar solvents, in which the dyes are in the trans form, photoisomerization takes place. In contrast, the meso-substituted dyes 3,3′-dimethyl-9-phenylthiacarbocyanine and 3,3′-diethyl-9-(2-hydroxy-4-methoxyphenyl)thiacarbocyanine occur as trans isomers and exhibit photoisomerization in both polar and nonpolar solvents. The behavior of these dyes may be explained by the fact that the phenyl ring of the substituent in their molecules can be twisted at some angle, removing the substituent from the plane of the molecule and reducing its steric effect on the conformation of the trans isomer. In some cases, photoisomerization of cis isomers of meso-substituted carbocyanine dyes is also observed (for some meso-alkyl-substituted dyes complexed with DNA and chondroitin-4-sulfate; for 3,3′-diethyl-9-methoxythiacarbocyanine in moderate polarity solvents). The cycle photoisomerization–thermal back isomerization of cyanine dyes can be used in various systems of information storage and deserves further investigation using modern research methods.
1. Introduction
Isomerization reactions, which involve rotation of a bulky molecular group around a body-fixed axis, often play a fundamental role in chemical and biological processes both in solution and in organized assemblies. In nature, the most important and known isomerization process is photoisomerization of 11-cis retinal, the chromophore of rhodopsin, which is a photoreceptive pigment for twilight vision [1,2]. Rotational isomerization is inherent in polyenic compounds, in particular, carotenoids and diphenylpolyenes [3]. Azo and azomethine dyes can also undergo cis-trans (or syn-anti) isomerization, while with more complex mechanisms [4,5]. The most abundant class of dyes that exhibit rotational isomerization is polymethine (cyanine) dyes.
At present, cyanine dyes attract great attention of researches in different fields of science, technology engineering, pharmacology and medicine [6]. In particular, these dyes are widely used in laser technology (for creating tunable laser media in the near IR spectral range, passive Q-switches of lasers and other devices of quantum electronics and optoelectronics) [7,8]. Cyanine dyes also attract interest due to the prospects of their use as spectral-fluorescent probes and labels in the study of various biological systems [9,10,11,12], and in biomedicine for the purposes of visualization of tissues and photodynamic therapy [6,13,14,15,16]. This is due to the unique properties of cyanine dyes, which are determined by the key features of their molecular structure. Various fields of applications of cyanine dyes determine the need for studying their photophysical and photochemical properties in solutions and molecularly organized media.
2. Structure and Spectral-Fluorescent Properties of Cyanine Dyes
Polymethine dyes contain conjugated polymethine chains (with an odd number of CH groups) with the general formula X(CH=CH)nCH=Y, where X and Y are terminal groups with heteroatoms (N, O, S, Se) or even C atoms (in case of carbocyclic azulene cyanines). The cationic type polymethine dyes are primarily cyanines, streptocyanines, rhodacyanines, hemicyanines, azacyanines, and styryl dyes, anionic polymethine dyes — oxonols. Neutral cyanines (merocyanines and ketocyanines) are subtypes of uncharged polymethine dyes.
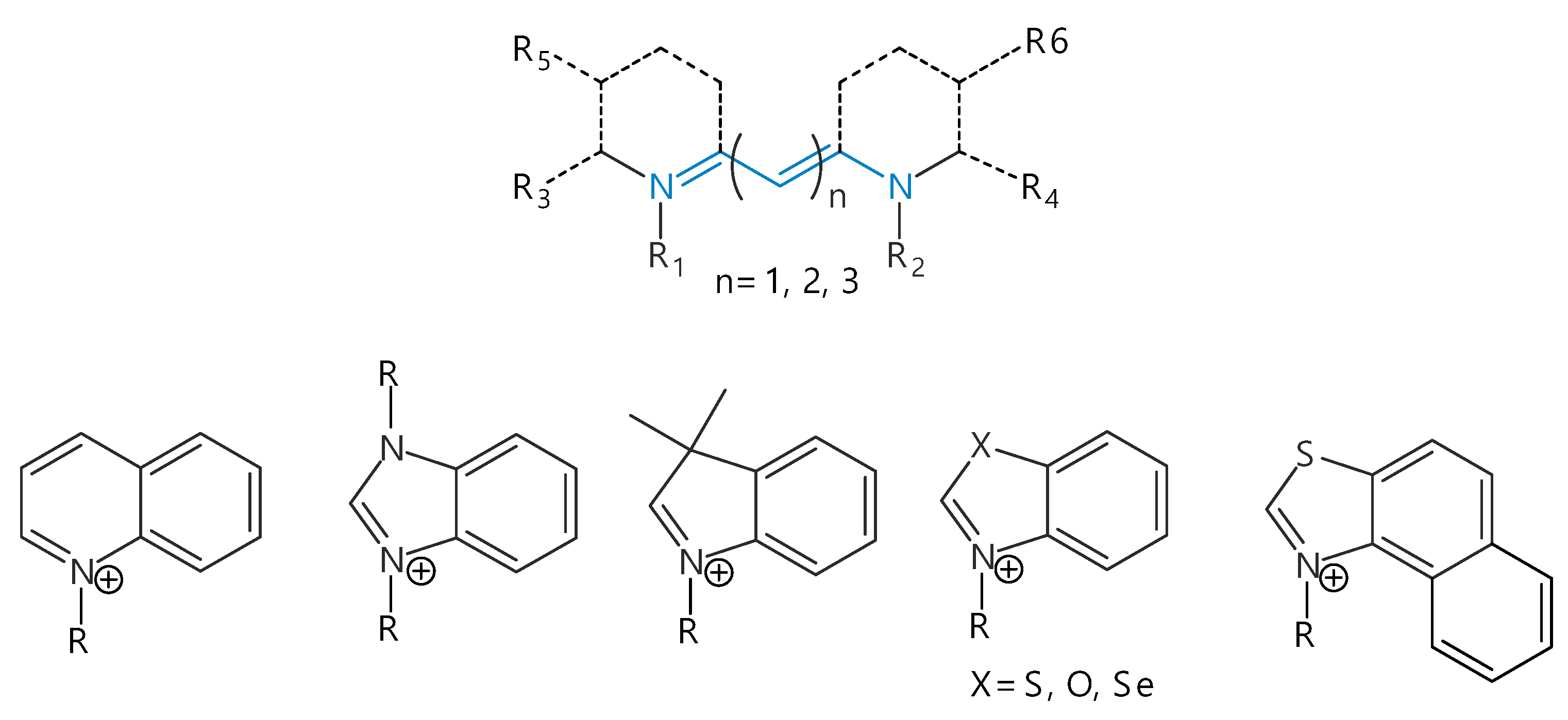
In this article, we confine ourselves to the most common subclass of polymethine dyes—carbocyanines with a positively charged chromophore with terminal nitrogen atoms. The nitrogen atoms can be a part of amino groups or terminal heterocycles B1 and B2 (quinolinyl, indolenyl, benzimidazolyl, benzothiazolyl, benzoxazolyl, and other heterocyclic residues, see Figure 1) [17,18]. Note that the total charge of such dyes can be not only positive, but also negative (or neutral) due to the presence of anionic substituents in heterocyclic residues.
The dye molecule usually carries a positive charge distributed over the polymethine chain. Due to complete π-electronic conjugation in the polymethine chain, these dyes have narrow absorption bands with high extinction coefficients (of the order of 105 L mol−1 cm−1) in the spectral range from 340 to 1400 nm [6,9,17,18]. Note that symmetric polymethine dyes are characterized by equalization of C–C bonds in the chromophore and approaching of their orders to one-and-a-half (polymethine state). From symmetry considerations, it follows that for symmetric cyanines (B1 = B2) this should lead to a uniform distribution of the electronic density along the polymethine chain and identical charges on the terminal groups (Figure 2). Hence, the electronic structure of symmetric cationic dyes can be represented by a superposition of two resonance structures [17].
Elongation of the polymethine chain in the symmetric dyes by one vinylene group results in a bathochromic shift of the absorption maximum by about 100–130 nm [17,18,19]. At present, there is a wide variety of cationic carbocyanines: monomethine cyanines, carbocyanines (or trimethine cyanines), dicarbocyanines (pentamethine cyanines), tricarbocyanines (heptamethine cyanines), etc.
As a rule, in their absorption spectra there is only one short-wavelength vibronic maximum as a characteristic shoulder at the edge of the main band. The presence of a system of alternating charges determines the high sensitivity of polymethine dyes to intermolecular interactions with the medium.
Due to the lack of rigidity of the molecule, which is characteristic of cyanine dyes with an open polymethine chain, various vibrations and rotations of individual fragments are possible in their molecules, first of all, torsions and rotations around various bonds of the polymethine chain of cyanines. This also determines the possibility of isomerization due to rotation around the C–C bonds of the polymethine chain. Photoisomerization (trans → cis or cis → trans) is a potent nonradiative channel for deactivation of the excited state of the molecules of polymethine dyes. This determines short lifetimes (from tens to hundreds of picoseconds) of their excited singlet state (S1), as well as low values of the quantum yields of fluorescence and intersystem crossing to the triplet state (S1 → T) [9,17,18]. Hence, the study of photoisomerization of polymethine dyes is a key to understanding the photophysical and photochemical processes occurring in their molecules after photoexcitation.
3. Isomerization of Carbocyanine Dyes
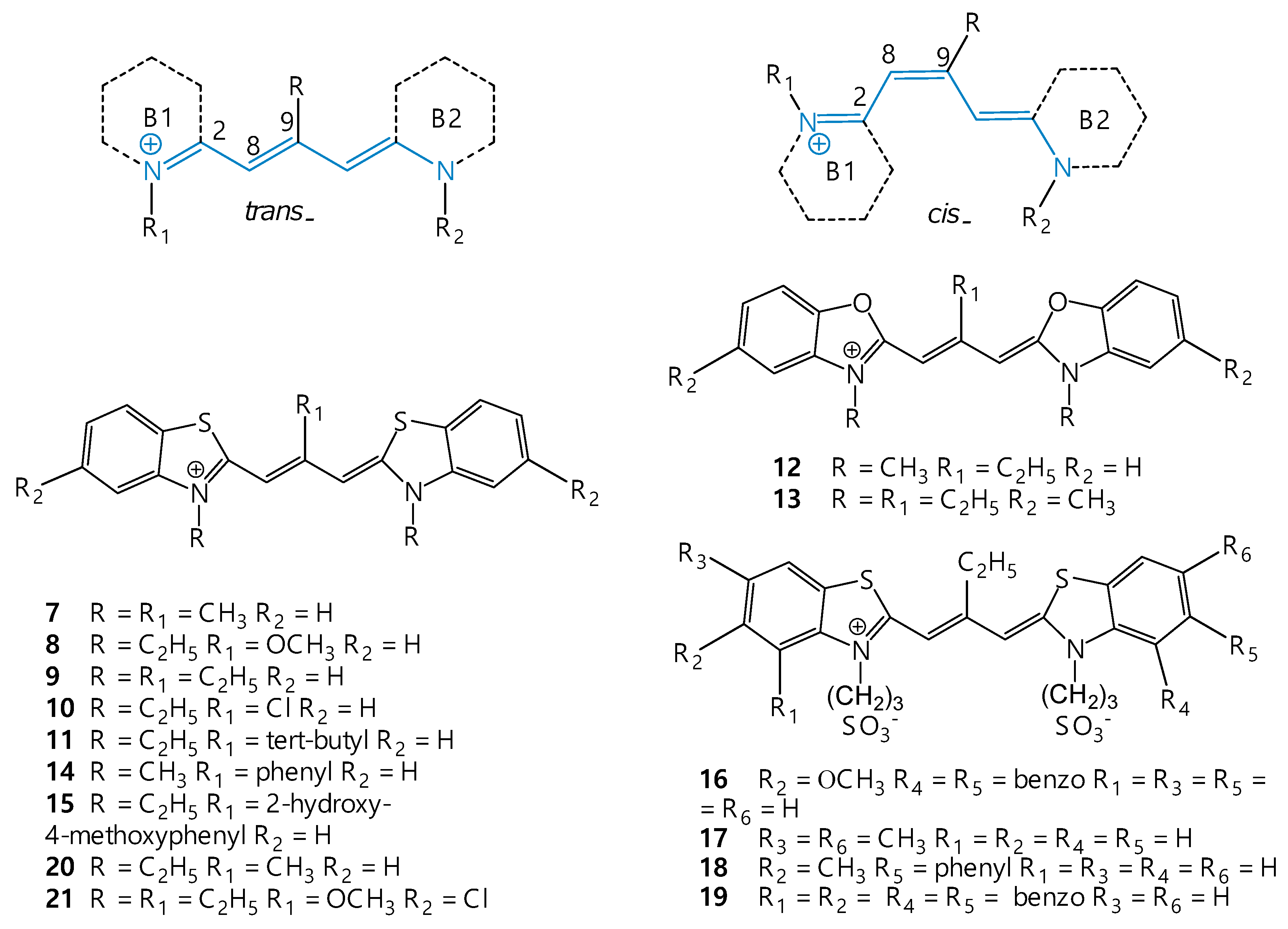
As noted above, one of the main paths of degradation of the energy of the S1 state of cyanines in low-viscosity media is usually trans → cis (or cis → trans) isomerization, performed by rotating a fragment of the dye molecule around one of the bonds of the polymethine chain by ~180° (also possible is internal conversion—the nonradiative transition S1 → S0 induced by vibrations and torsions). Figure 3 shows general molecular structures of the trans and cis isomers of the dyes.
The existence of photoisomers of polymethine dyes was first proven in 1965 by flash photolysis of dye solutions [20]. For example, for the unsubstituted cationic dye 3,3′-dimethylthiacarbocyanine perchlorate (1), the photoisomerization quantum yield (φi) is 0.24 in dichloromethane [21]. Upon photoexcitation of its analogue, 3,3′-diethylthiacarbocyanine iodide (2) in polar solvents (ethanol, dimethyl sulfoxide, acetonitrile), φi has a similar value (0.25 in methanol [22]); the contributions of fluorescence and intersystem crossing to the triplet state upon deactivation of excited singlet molecules are low.
Direct photoexcitation of carbocyanine dyes with no substituents in the polymethine chain (they are mainly present in solution in the form of trans isomers) leads to the formation of a short-lived cis photoisomer (see Equation (1)), which then undergoes back dark (thermal) isomerization to form the initial trans isomer (with a rate constant for 2 ki = 250 s−1 in ethanol [21]):
Upon flash photoexcitation of aerated solutions of the unsubstituted oxacarbocyanine dye 3,3′-diethyloxacarbocyanine iodide (3) in isopropanol and aqueous phosphate buffer solution (pH 7, 20 mmol L−1), the absorption and bleaching signals caused by photoisomerization and thermal back isomerization of the photoisomer formed was observed [22].
The criterion determining the assignment of the signals to the photoisomer was the absence of the effect of oxygen on the observed signals, since the photoisomers of cyanine dyes are formed, as a rule, from the S1 state of the dyes having a too short lifetime (hundreds of picoseconds) to be quenched with atmospheric oxygen [23]. The bleaching maximum (λmax = 480 nm) of the difference spectra of 3 approximately coincides with the absorption band of the dye (trans isomer), and the absorption maxima (λmax = 505 nm) are in the long-wavelength region. The assignment of the spectrum to the cis isomer is based on [24]. The value of ki for 3 does not depend on the excitation and registration wavelengths and is 9.0 s−1 (in aqueous phosphate buffer solution) [25].
4. Potential Surfaces of Isomerization of Cyanine Dyes
Historically, several models of potential surfaces of cyanine isomerization (surfaces corresponding to isomerization rotation around one of the C–C bonds of the polymethine chain) were considered in the literature. One of the first models was the Rulliere model [28], in which the potential surfaces calculated by Orlandi and Siebrand for isomerization of stilbene [29] were mechanically transferred to the case of isomerization of cyanines. However, it was later shown [30] that the potential wave functions for polyenes and cyanines are of different nature, despite some similarity of the shapes of the potential surfaces for the S1 state, so the results of Orlandi–Siebrand’s calculations are not applicable for cyanines.
On the basis of quantum-chemical calculations carried out for simple open-chain cyanine dyes (streptocyanines), two models of photoisomerization of cyanine dyes were proposed, taking into account the influence of solvent polarity in different ways [22]. According to these models, photoisomerization of cyanines occurs with activation energy barrier in the excited singlet state (S1) through the formation of an intermediate perpendicular (perp) form. The models also suggest an energy barrier for the reaction of thermal back isomerization of the dyes in the ground state (S0). According to model 1, photoisomers of thiacarbocyanine dyes are characterized by twisted, distorted configuration. This model assumes stabilization of intermediate perpendicular forms of dye photoisomers in polar solvents due to dipole-dipole interactions with the solvent [31]. Solvation of the perpendicular form should lead to a significant decrease in the corresponding energy barriers (minima on the S1 and S0 energy curves) [32]. In the absence of solvation of the perpendicular form, such effects are not expected [22,31]. In nonpolar solvents, a high energy barrier will prevent isomerization in the S1 state. In accordance with model 1, the rate of the back isomerization reaction of photoisomers of cyanine dyes in the ground state should strongly depend on the solvent polarity [31,32].
Model 2 proposed by Momicchioli et al. [30] assumes the formation of planar cis-isomers; dipole-dipole interactions with a solvent were not explicitly included in the calculations. The model assumes relatively small solvation effects on isomerization of cyanine dyes in polar media. In [22], the data on the photophysical properties and photoisomerization of dyes 2 and 3 in various solvents were analyzed.
The results obtained contradict the theoretical model of photoisomerization, in which the key role is played by the influence of solvent polarity (model 1) [22,31,32]. It was shown that, upon changing the dielectric constant of the solvent (ε = 2.2 to 32.7), no predicted effect of polarity on the S1 state potential barrier in the photoisomerization reaction was observed. The absorption spectra of the photoisomers of dyes 2 and 3 correspond to the planar structures of the molecules, which does not correspond to model 1 [22]. Model 2 [29] is in better agreement with experimental data and allows one to take into account the weak effects of solvent polarity on the Arrhenius parameters of dye 2 back isomerization. For the trans-to-perp transition, the activation energy was found to be ~17–19 kJ/mol, the transition of the cis isomer to the perpendicular state (cis-to-perp) is apparently barrier-free and depends on the energy of the viscous flow of the solvent. For dark processes of back isomerization of the photoisomers, an energy barrier of ~54–75 kJ/mol is characteristic, depending on cyanine and solvent [22].
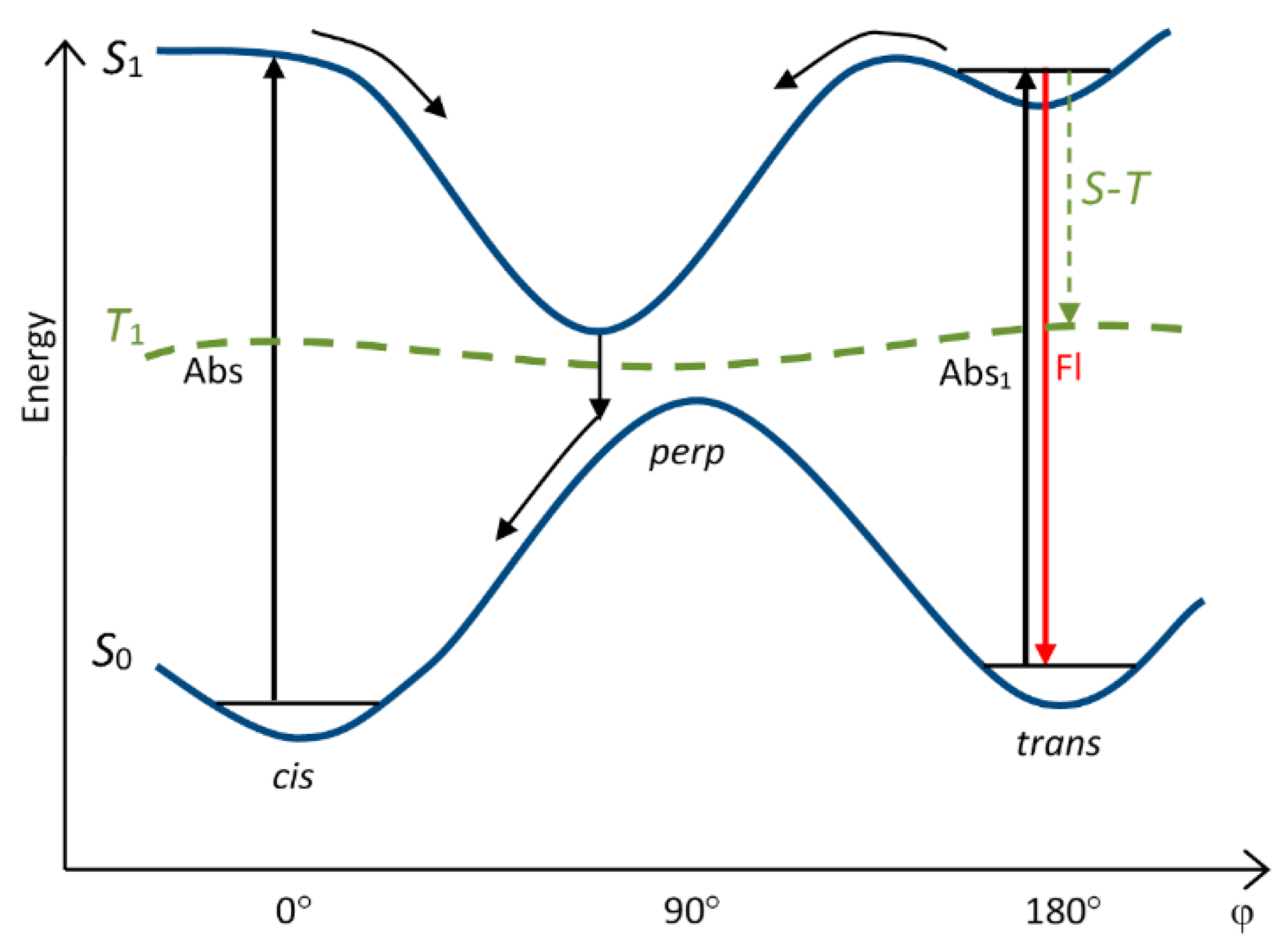
Now in the literature, a model of potential surfaces for cyanine dyes is used, starting from which, irrespective of the solvent, only the surface of S1 has a third minimum (turn by ~90°), which roughly corresponds to the top of the potential barrier of the surface of S0 having two minima (Figure 4) [30]. Upon photoexcitation, a molecule in the form of a trans isomer passes to the central minimum of S1, from which, when it reaches the surface of S0, it slips down to a minimum of cis-S0 (trans–cis photoisomerization) or trans-S0 (nonradiative degradation of the excitation energy). If the excited molecule is in the form of a cis isomer, then it also passes to the central minimum of S1, from which, when it reaches the surface of S0, it slips down to a minimum of cis-S0 (nonradiative degradation of the excitation energy) or trans-S0 (cis-trans photoisomerization). The quantum yields of photoisomerization and nonradiative degradation depend on the position of the minimum of S1 with respect to the maximum of S0. Theoretical calculations by the CS INDO method [30] and experimental verification of the consistency of this model with the isomerization process [22] showed its correspondence to cyanine isomerization.
For cyanines with no substituents in the polymethine chain, nonradiative relaxation of the excited singlet state (both trans- and cis-S1), occurring by movement along the potential surface of isomerization, leads to both cis and trans isomers of the dye. Thus, for these dyes, photoisomerization occurs upon photoexcitation of both trans and cis isomers.
In the case of most meso-substituted thiacarbocyanines (first of all, alkyl-substituted), the minimum potential energy of the dye in the S1 state corresponding to the perpendicular conformation is shifted with respect to the maximum of the energy barrier of the S0 state toward the cis isomer (Figure 5) [22,30].
For this reason, upon nonradiative relaxation of the S1 state of such dyes by moving along the S1 potential surface, the trans conformation of the dyes is not attained and photoexcitation into the absorption band of the cis isomer does not lead to the formation of the trans form, that is, the cis isomers of most meso-substituted thiacarbocyanines are not photoisomerized [21].
5. Effect of Solvent Viscosity on the Kinetics of Photoisomerization and Back (Thermal) Isomerization
The kinetics of isomerization of cyanines in solution is determined, first of all, by a potential barrier of rotation around the isomerizing bond. In solutions, the character of molecular environment is also an important factor, which can have an effect on isomerization of cyanine dyes. For isomerization processes with relatively low activation energies close to the activation energy of the viscous flow of the solvent (this is usually isomerization from the S1 state, that is, photoisomerization), it strongly depends on the viscosity of the medium (slows down sharply with increasing viscosity). Due to the very efficient photoisomerization process, the lifetime of the S1 state of cyanines in low-viscosity solvents is much shorter than the radiative lifetime and is usually hundreds of picoseconds or less. In high viscosity media, internal rotations in the cyanine molecule are hindered, which leads to a drop in the quantum yield of photoisomerization and an increase in the quantum yield of fluorescence, the fluorescence lifetime, and the quantum yield of the triplet state, since fluorescence and intersystem crossing to the triplet state compete with photoisomerization [30].
The influence of viscosity on ultrafast processes of cyanine dye photoisomerization was studied in a number of works. In particular, in [33] the formation of short-lived photoisomers of 1,1′-diethyl-4,4′-cyanine (4), 2,2′-trimethinequinocyanine (5, Pinacyanol) was studied after excitation with picosecond laser pulses. It was shown that the yield of photoisomers of the dyes strongly depends on viscosity: the relative concentration of the photoisomer decreases with an increase in the viscosity of the solvent (a mixture of glycerol and methanol). Studies using ultrafast femtosecond spectroscopy of the process of photoisomerization of the cationic monometinecyanine dye 1,1’-diethyl-2,2’-cyanine (6) in alcohols are reported in [34]. A lower quantum yield for the photoisomer was observed in more viscous solvents. The increase in viscosity results in slowing down intramolecular vibrations and torsions in the photoexcited cyanine molecule, including rotations of fragments of the dye molecule around C–C bonds of the polymethine chain leading to photoisomerization.
In [35], the kinetics of isomerization of molecules of indocarbocyanine dyes with substituents in positions 3 and 3’ of various length in solutions of alcohols (methanol–decanol series) was studied. The results indicate, in particular, that the nature of the reaction coordinate plays an important role in determining the dependence of photoisomerization on viscosity.
To take into account the effect of viscosity on the rate of the photoisomerization reaction, a number of theoretical approaches have been proposed. First of all, this is the approach based on the Kramers theory [36]:
where k is the rate constant, ωr and ωb are the frequency factors of the barrier on the potential energy surface in the vicinity of the reactants (ωr) and the top of the reaction barrier (ωb), respectively, γ—friction coefficient, m—effective mass, Ea is the activation energy of the reaction, and R is the universal gas constant.
It is common to assume that γ ∝ η (η is viscosity), and in the hydrodynamic framework at high viscosity, Equation (2) turns into:
Under these conditions, the reaction rate constant should be inversely proportional to the viscosity (η) of the solvent. However, for the isomerization reactions, this proportionality is usually not observed; it has been shown that the reaction rate constant with increasing solvent viscosity decreases much slower than Equation (3) predicts [37,38].
An explanation of the dependence of the isomerization rate constant on viscosity was also proposed in the theory of Grote and Hynes [39], which introduces the concept of frequency-dependent friction affecting the rate of chemical processes in the solvent phase. Due to the inclusion of frequency-dependent friction instead of constant friction, the theory of Grote and Hynes successfully predicts the rate constant of chemical reactions in viscous liquids.
A more detailed consideration of theoretical approaches describing the dependence of the isomerization kinetics on viscosity is beyond the scope of this review.
For isomerization processes, the following semi-empirical equation is also used [40,41,42]:
where Ea is the apparent activation energy of isomerization, E0 is the internal energy barrier of isomerization, Eη is the activation energy of a viscous flow, and α is a coefficient depending on the slope of the potential barrier in the transient state of the process (0 ≤ α ≤ 1). For photoisomerization, the coefficient α is larger than for the thermal back isomerization (in the latter case, a weak dependence of Ea on the viscosity is observed, which is manifested only in highly viscous solvents). In [42], for photoisomerization of dye 3 in different alcohols the values of α were found to be in the range of 0.54 to 0.76, and for thermal back isomerization α ~ 0.27–0.41.
Ea = E0 + αEη,
6. Effects of Ion Pair Formation on the Isomerization Processes of Cyanine Dyes
In polar solvents, cyanine dyes are present in the form of free ions (cations); in this case, the influence of polarity of the medium on the isomerization of polymethine dyes is small (see above) [22,42]. However, in weakly polar and nonpolar solvents, the dyes form ion pairs with a counterion, which can affect their photochemistry, in particular, the processes of photoisomerization and back isomerization of the resulting photoisomer.
In [43], photophysical and photochemical processes occurring upon flash photoexcitation in molecules of dyes 1 (iodide) and 2 (chloride) in a mixture of dimethyl sulfoxide (DMSO)–toluene of various compositions were studied. With a decrease in the DMSO content in the solution down to 3 vol %, the relative quantum yield of intersystem crossing of dye 1 to the triplet state increases by two orders of magnitude, which is accompanied by a decrease in the quantum yield of trans–cis photoisomerization. This effect is not observed for dye 2. The authors explain these results by the heavy atom effect upon the formation of ion pairs of the dye cation 1 with the iodide counterion under nonpolar conditions.
In [44], the effect of counterions (ClO4−, Cl−, I−) on photochemical processes (including isomerization) in molecules of cationic benzimidacyanine dyes was studied in acetonitrile–toluene mixtures. In low polarity media, in which the dyes formed ion pairs with counterions, an increase in the rate constant and a decrease in the activation energy of back isomerization of benzimidacyanines were observed. The effect was explained by the electrostatic influence of the counterion in ion pairs on the electronic density distribution in the dye cation, leading to a decrease in the activation barrier of the process [44]. The influence of the counterion on the transient (“twisted”) state of isomerization, which lowers its energy, is also possible.
7. Isomerization of Meso-Substituted Carbocyanine Dyes
While carbocyanine dyes, which do not have meso substituents in the polymethine chain, are present in solutions as trans isomers, meso-substituted thiacarbocyanine dyes in the crystalline state are usually present as cis isomers [45].
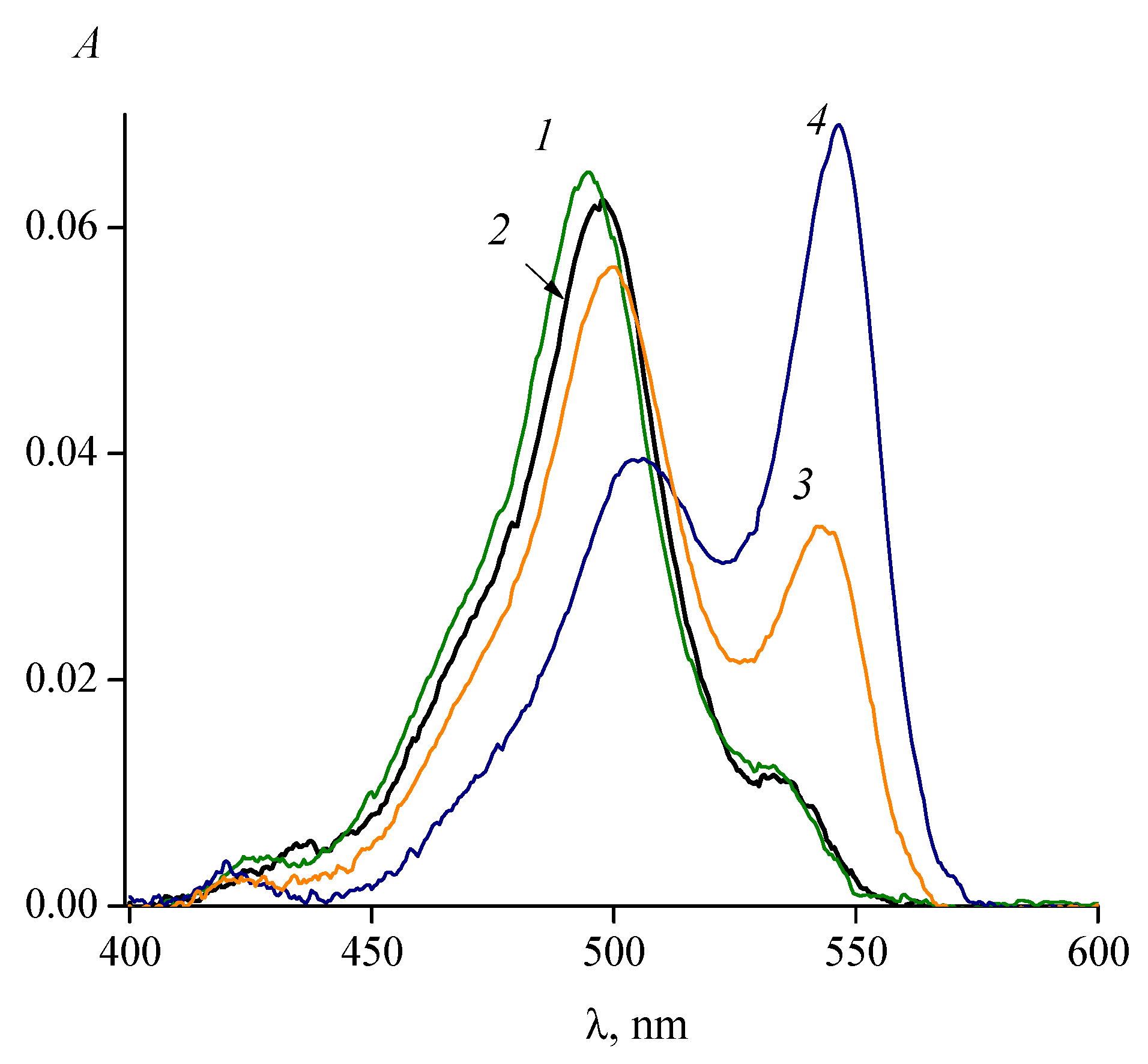
Bulky substituents in the meso (9) position of the polymethine chain create significant steric hindrances to the formation of the trans isomer (Figure 6). At low temperatures in solutions, the absorption bands of meso-substituted thiacarbocyanines are split into two closely spaced bands [46]. Simultaneous presence (in comparable concentrations) of all-trans and cis isomers can be observed [45,46]. In particular, it has been shown that 3,3′,9-trimethylthiacarbocyanine iodide (7; Cyan 2), having a meso-methyl substituent in the polymethine chain, is mainly in the cis form in polar solvents [47]. The equilibrium position depends strongly on the solvent polarity: the trans isomer predominates in low polarity media, while in highly polar solvents the trans isomer presents only as a small impurity to the cis isomer [26,45,46,47,48]. As an example, it was shown that 3,3′-diethyl-9-methoxythiacarbocyanine 8 in solvents of moderate polarity (ethyl acetate, isopropanol) is in the form of an equilibrium mixture of cis and trans isomers (Figure 7). The meso-substituted carbocyanine dye 3,3′,9-triethylthiacarbocyanine iodide (9) in isopropanol is in the form of a cis isomer, and in dioxane in the form of a trans isomer [49]. It was shown that 3,3′-diethyl-9-chlorothiacarbocyanine perchlorate (dye 10) is in the form of a cis isomer in water, and in the form of a trans isomer in a less polar solvent, isopropanol [50].
An increase in the effective volume of the substituent in the meso position of the polymethine chain of the molecule promotes an increase in the cis isomer content. It was established that 9-tert-butyl-3,3′-diethylthiacarbocyanine cation (11) exists both in solution and in the solid phase exclusively as a nonplanar di-cis form [26,47].
The simultaneous presence of trans and cis isomers in dye solutions is explained by distortion of the planar structure of the dye made by substituents in the meso position of the polymethine chain of the dyes. This leads to a significant increase in the energy of the trans isomer, which becomes close to that of the cis isomer [45,46,51]. The observed dynamic equilibrium depending on the solvent polarity is due to the electrostatic interaction of the dye cation with a counterion in ion pairs formed in low polarity media. The formation of ion pairs in low polarity media leads to a decrease in the energy of the trans isomer and its preferential formation, whereas in polar media the cis isomer is formed, which has a lower energy.
The different isomers present in the solution cause a difference in the photochemical properties of meso-substituted carbocyanine dyes in solvents of different polarity. In polar solvents, where the dyes are predominantly in the form of cis isomers, photoisomerization is generally not observed, while in low polarity solvents, in which the dyes are present as trans isomers, photoisomerization to form cis isomers occurs upon photoexcitation. This empirical rule is fulfilled by a series of thia- and oxacarbocyanines with bulky meso-substituents.
Upon flash photoexcitation of the carbocyanine dye 9, the signals corresponding to photoisomerization processes were not detected in the polar media (isopropanol), i.e., the cis isomer of 9 was not photoisomerized. In nonpolar dioxane solution, the signals corresponding to trans–cis photoisomerization and subsequent decay of the cis photoisomer of dye 9 were detected [50]. The thiacarbocyanine dye 10, which has Cl as a substituent in the meso-position of the polymethine chain, forms cis isomer by trans–cis photosomerization upon flash photolysis in isopropanol solution [50].
For meso-substituted oxacarbocyanine dyes 3,3′-dimethyl-9-ethyloxacarbocyanine (12) and 3,3′,9-triethyl-5,5′-dimethyloxacarbocyanine (13), the formation of cis photoisomers from their trans forms in aqueous solutions is also detected upon flash photoexcitation. The intensity of the signals of photoisomers of 12 and 13 is much lower than that for their unsubstituted analogue 3, being in the trans form, which can be explained by the presence of a significant admixture of cis isomers of the dyes (in equilibrium with trans isomers), whose photoisomerization does not occur [45,46].
It should be noted that the empirical rule under consideration also found exceptions. First of all, these include the case of two meso-substituted thiacarbocyanine dyes, 3,3′-dimethyl-9-phenylthiacarbocyanine iodide (14) and its analogue, 3,3′-diethyl-9-(2-hydroxy-4-methoxyphenyl)thiacarbocyanine iodide (15). For these dyes, the simple empirical rule that we are considering here is not satisfied. The analysis of the absorption, fluorescence, and fluorescence excitation spectra has shown that dyes 14 and 15 in solutions, regardless of the polarity of the solvent, are in the form of trans isomers [52]. Photoisomerization is observed for these dyes even in polar solvents. In particular, upon flash photoexcitation of 14 and 15 in ethanol, the difference spectra of cis photoisomers are observed, the bleaching maxima of which (λ = 560 and 568 nm, respectively) approximately coincide with the positions of the trans isomers in the stationary absorption spectra of the dyes. As in the case of meso-unsubstituted dye 2, which forms cis photoisomer upon photoexcitation [21,24], the stationary absorption spectra of the photoisomers of dyes 14 and 15 are shifted to the short-wavelength side (λmax ~ 550 and 558 nm, respectively).
Apparently, for dyes 14 and 15, the substituents in the meso position of the polymethine chain introduce less steric distortion into the structures of the trans isomers than alkyl substituents in the meso-alkyl-substituted dyes [52]. This is probably explained by twisting of the phenyl meso substituents in 14 and 15 relative to the dye chromophore plane, which sharply reduces their distorting effect on the planar structure of the dye chromophore and, hence, on the relative energy of the isomers.
Furthermore, for some meso-substituted thiacarbocyanines, photoisomerization of both trans and cis isomers is observed. As an example, dye 8 may be considered, for which both trans → cis and cis → trans photoisomerization processes were detected. In solvents of moderate polarity (ethyl acetate, isopropanol), depending on the photoexcitation wavelength, photoisomerization of either cis or trans isomer of 8 was observed, leading to the formation of trans or cis photoisomer, respectively [50]. The rate constants of thermal cis–trans transitions for ethyl acetate and isopropanol solution of 8 are given in Table 1. Since the cis and trans isomers in the solution of dye 8 are in equilibrium, the observed rate constant of the signal decay corresponds to the sum of the rate constants of cis → trans and trans → cis isomerization.
The rate constants of thermal cis–trans transitions for most meso-substituted thiacarbocyanine dyes were found to be much higher than for their unsubstituted analogs (see Table 1) [21,24]. Such an increase in the rate constants of the thermal back isomerization is due to steric hindrances, which create bulky groups of substituents in the meso position of the polymethine chain. The steric influence of the meso substituents leads to removing terminal heterocyclic groups of dye molecules from the plane, which results in distortion of the valence angles, an increase in the energy of the cis and trans isomers, and a corresponding decrease in the potential barrier of thermal isomerization in the ground state [21].
The observed rate constants for the decay of cis photoisomers of 14 and 15 were rather close to ki for the unsubstituted dye 2 (see Table 1), indicating a weak steric effect of meso substituents in 14 and 15 on the configuration of the cis photoisomer [52]. This also can be explained by the possibility of turning the phenyl ring of the substituent, which removes the substituent from the chromophore’s plane of the molecule and reduces its steric effect on the configuration of the isomers.
8. Isomerization of Cyanine Dyes in Structurally Organized Media
It is known that polymethine dyes can form noncovalent complexes with surfactants, polyelectrolytes, biomacromolecules [6,9,10,14,15,18,53,54,55,56,57]. The formation of such complexes, as a rule, hinders the intramolecular degrees of freedom of dye molecules leading to nonradiative dissipation of excitation energy, which results in a drop in the quantum yield of photoisomerization and an increase in the quantum yield of fluorescence [6,9,10,54]. In the case of meso-substituted polymethine dyes, which are characterized by dynamic cis–trans equilibrium depending on the medium, the formation of such complexes can significantly affect the cis–trans equilibrium.
In [55], photophysical and photochemical processes occurring in molecules of meso-substituted thiacarbocyanine dyes 3,3′-bis(γ-sulfopropyl)-5-methoxy-4′,5′-benzo-9-ethylthiacarbocyanine (16), 3,3′-bis(γ-sulfopropyl)-6,6′-dimethyl-9-ethylthiacarbocyanine (17), and 3,3′-bis(γ-sulfopropyl)-5-methyl-5′-phenyl-9-ethylthiacarbocyanine (18) in aqueous solutions in the presence of cationic (cetyltrimethylammonium bromide), anionic (sodium dodecyl sulfate, SDS) and neutral (Triton X-100) surfactants were studied.
In an aqueous solution in the absence of surfactants, dyes 16–18 are in the form of dimers in equilibrium with cis monomers and are not capable of photoisomerization. At surfactant concentrations above critical micelle concentration (CMC), the dimers decompose into monomers, and cis monomers are converted into the trans form upon solubilization of the dye molecules in the surfactant micelles. The resulting trans isomers of the dyes are capable of photoisomerization to form cis photoisomers.
The behavior of meso-substituted polymethine dyes 3,3′-di-(γ-sulfopropyl)-4,5,4′,5′-dibenzo-9-ethylthiacarbocyanine betaine (19, DEC) and 7 was studied in micellar systems of biological surfactants—bile salts sodium cholate, sodium deoxycholate, and sodium taurocholate [56]. Upon binding to micelles of bile salts, as in the case of synthetic surfactants, these dyes are converted into a trans form. Upon flash photolysis of aerated aqueous solutions of the dyes in the presence of micelles of bile salts and SDS, signals caused by photoisomerization of the trans isomers of the dyes and thermal back isomerization of the cis photoisomers formed were observed. The lifetimes of photoisomers of 7 and 19 in the presence of bile salt micelles were in the range of 60–190 μs. In deoxygenated solutions, the appearance of the triplet state of the dyes was also observed.
In [57], photophysical and photochemical properties of thiacarbocyanine dyes 2, 3,3′-diethyl-9-methylthiacarbocyanine iodide (20) and 3,3′,9-triethyl-5,5′-dichlorothiacarbocyanine iodide (21) were studied in the presence of water-soluble polyelectrolytes: polystyrene sulfonate (PSS), polyacrylic acid (PAA), and polymethacrylic acid (PMA). For 2 in the presence of PSS, a significant (tenfold) decrease in the quantum yield of trans–cis photoisomerization (accompanied by an increase in fluorescence and a moderate increase in the intersystem crossing to the triplet state) was observed.
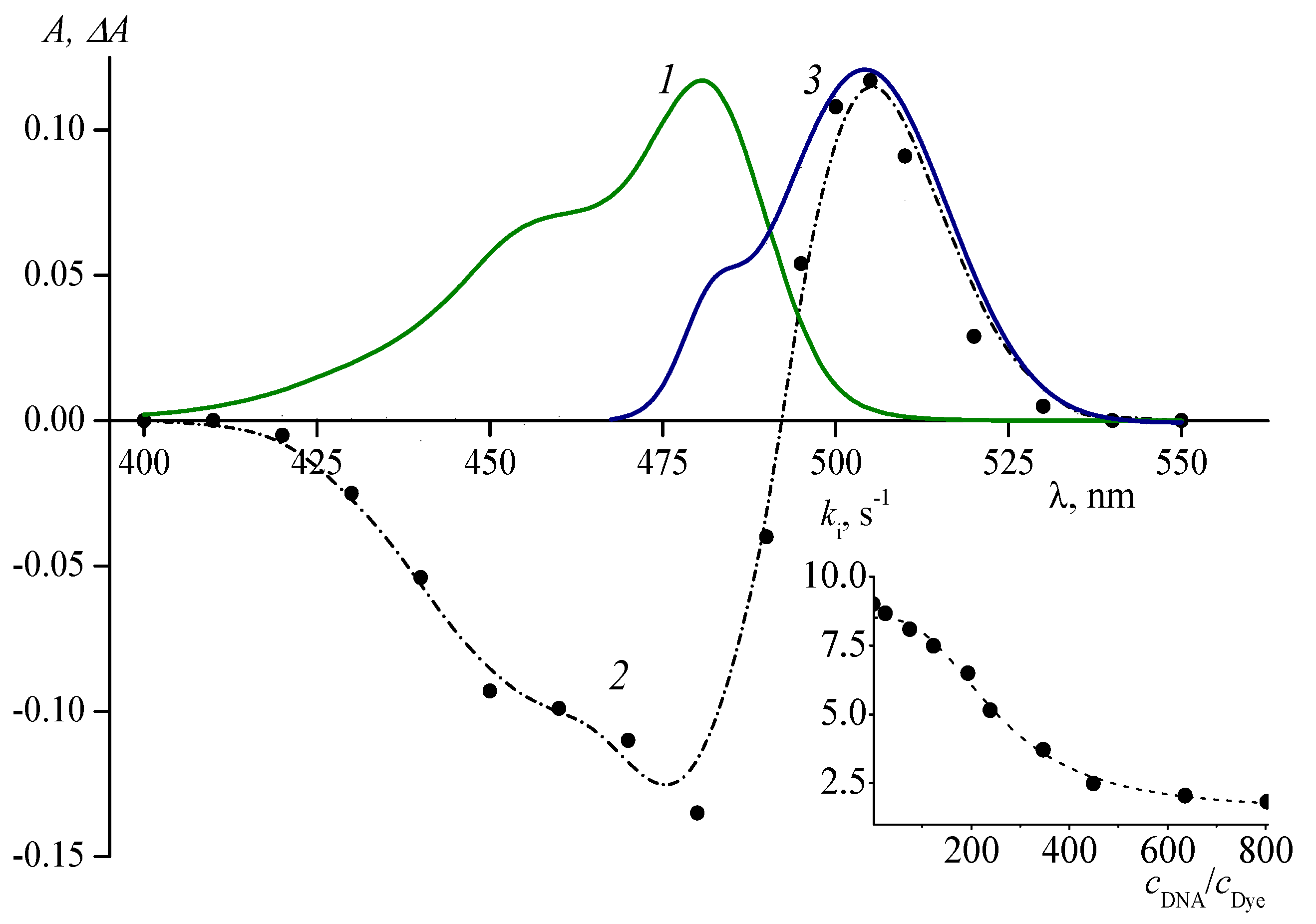
Besides surfactants and synthetic polyanions, cationic cyanine dyes are capable of forming noncovalent complexes with DNA. The interaction with DNA hampers the formation of photoisomers of meso-unsubstituted polymethine dyes. In [58], a drop in the yield of the photoisomer of dye 2 was observed upon the formation of its noncovalent complex with ds-DNA in a buffer solution. Upon flash photolysis of aerated solutions of meso-substituted thiacarbocyanine dyes 4, 9, 10, 14, 15 in the presence of DNA (with DNA concentration in the range of (2.3–5.0) × 10−4 mol L−1), signals of photoisomers were not observed [50,52,53]. Photoisomerization was also hampered by noncovalent interaction of oxacarbocyanine dyes 3, 12, 13 with DNA [25]. Upon flash photolysis of aerated solutions of 12 and 13 with DNA at cDNA = 4.4 × 10−5 mol L−1 and higher, the signals of photoisomers were not observed. For dye 3 with cDNA = 2.39 × 10−4 mol L−1, the absorption intensity of the photoisomer decreases 2.5 times, and its lifetime increases 5 times (Figure 8).
Thus, the interaction with DNA and synthetic polyanions hampers the formation of photoisomers in the case of a large number of cationic polymethine dyes, which is explained by the steric factor of complexation, which slows down the nonradiative processes of dissipation of excitation energy. Nevertheless, for some meso-substituted thiacarbocyanine dyes, the formation of photoisomers was enhanced upon the complexation. In particular, in [59], the photophysical and photochemical properties of the meso-methyl-substituted dye 20 were studied in aqueous solutions in the presence of PSS, PAA and PMA. Flash photolysis of 20 in the presence of PAA and PMA revealed a weak signal due to cis → trans photoisomerization of the dye bound to the polyelectrolyte in the cis form and subsequent dark decay of the resulting trans photoisomer (τ = 700 μs at 560 nm). This contradicts the rule that cis isomers of meso-alkyl-substituted carbocyanines are not photoisomerized (dye 20 is not photoisomerized in an aqueous solution without polyelectrolytes). Similarly, in [47], an enhancement of cis → trans photoisomerization of meso-methyl-substituted dyes 7 and 20 by noncovalent binding to DNA and chondroitin-4-sulfate in a buffer solution was shown. Upon flash photolysis of 7 and 20 in the presence of DNA and chondroitin-4-sulfate, signals due to the formation of trans photoisomers and their subsequent dark decay (τ = 3.9 and 2.2 ms for 7 in the presence of DNA and chondroitin-4-sulfate, respectively; τ = 0.7 ms for 20 in the presence of biopolymers, λreg = 560 nm) were observed. To explain the effect of stimulation of photoisomerization of meso-substituted cis isomers by complexation with polyelectrolytes, an assumption was made that the biopolymer matrix of polyelectrolyte affects the potential surfaces of photoisomerization of the dyes: deformation of the potential surface of S1 occurs with a shift of its minimum toward the trans isomer [47,59].
9. Conclusions
The use of polymethine dyes in various fields of science and technology requires a detailed study of their photophysical and photochemical properties. Recently, a lot of works have been published on photophysics and photochemistry of polymethine dyes (see, for example, [11,12,14,15,16,60,61,62]). Photoisomerization (followed by back isomerization of a photoisomer) is one of the main properties of polymethine dyes; at present, it is being studied in detail with the use of modern micro-, nano-, pico- and femtosecond techniques. While trans–cis (photo)isomerization of various compounds is widely used in molecular photoswitches, over the past decades, most works have been focused mainly on azobenzene derivatives [63], spiropyrans [64], diarylethenes [65], and stilbenes [66], and, until recently, this property of cyanine dyes has not been applied in practice. The use of cyanine photoisomerization in research is mainly for probing microviscosity of microheterogeneous media [67,68]. On the contrary, rigidization of the polymethine chain of dyes is often carried out by introducing “bridges” that suppress internal conversion and photoisomerization processes to increase the stability of the dyes and the fluorescence quantum yield [17,18,69,70]. It should be noted that the cycle photoisomerization–reverse dark isomerization of the photoisomer is completely reversible and, ideally, does not lead to decomposition of the dye molecule. Since the lifetime of the photoisomer, depending on the structure of the dye and the medium, can vary in very wide range (from seconds to picoseconds), this cycle can be used for transient recording and storage of information within different time ranges in modern information recording systems and devices. So we may expect in future widespread use of polymethine dyes as information storage media in various fields of science and technology.
Of great importance is also the dynamic cis–trans isomeric equilibrium, which strongly depends on the molecular microenvironment, observed for meso-substituted polymethine dyes. Since the photophysical and photochemical properties of the cis and trans isomers of such dyes are very different, these dyes can be widely used as probes for studying various molecularly organized and nanostructured systems. Indeed, a number of meso-substituted cyanine dyes have already been used as spectral-fluorescent probes for biomacromolecules (DNA, albumins, etc.) [71,72,73,74,75,76,77]. Next in turn is the use of polymethine dyes as kinetic probes, as it has been found recently that the lifetime of the photoisomers of meso-substituted oxonols (anionic polymethine dyes) [78] and squarylium cyanines [79] depend strongly on the polarity of the molecular microenvironment.
In summary, it can be noted that the study of (photo)isomerization and the properties of isomers of cyanine dyes using modern research methods is still relevant both for theory and for the practical application of cyanine dyes.
Author Contributions
P.P. and A.T. collected the data from the literature, analyzed the data, and wrote the manuscript. Both P.P. and A.T. contributed equally to the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Foundation for Basic Research (RFBR), grant number 16-03-00735. This work was performed under the Russian Federation State Assignment no. 001201253314.
Acknowledgments
The authors are grateful to B.I. Shapiro (Research Center Niikhimfotoproekt) for the supply of the polymethine dyes.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Kim, J.E.; Tauber, M.J.; Mathies, R.A. Wavelength dependent cis-trans isomerization in vision. Biochemistry 2001, 40, 13774–13778. [Google Scholar] [CrossRef]
- Kandori, H.; Shichida, Y.; Yoshizawa, T. Photoisomerization in rhodopsin. Biochemistry (Moscow) 2001, 66, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Zechmeister, L. Cis-trans isomerization and stereochemistry of carotenoids and diphenylpolyenes. Chem. Rev. 1944, 34, 267–344. [Google Scholar] [CrossRef]
- Herkstroeter, W.G. The Mechanism of Syn-Anti Isomerization of Azomethine Dyes. J. Am. Chem. Soc. 1973, 95, 8686–8691. [Google Scholar] [CrossRef]
- Liu, X.-M.; Jin, X.-Y.; Zhang, Z.-X.; Wang, J.; Bai, F.-Q. Theoretical study on the reaction mechanism of the thermal cis–trans isomerization of fluorine-substituted azobenzene derivatives. RSC Adv. 2018, 8, 11580–11588. [Google Scholar] [CrossRef]
- El-Shishtawy, R.M. Functional dyes, and some hi-tech applications. Int. J. Photoenergy 2009, 2009, 434897. [Google Scholar] [CrossRef]
- Pierce, B.M. Theoretical analysis of the third-order nonlinear optical properties of linear cyanines and polyenes. Proc. SPIE 1991, 1560, 148–161. [Google Scholar] [CrossRef]
- Pittman, M.; Plaza, P.; Martin, M.M.; Meyer, Y.H. Subpicosecond reverse saturable absorption in organic and organometallic solutions. Opt. Commun. 1998, 158, 201–212. [Google Scholar] [CrossRef]
- Levitus, M.; Ranjit, S. Cyanine dyes in biophysical research: The photophysics of polymethine fluorescent dyes in biomolecular environments. Q. Rev. Biophys. 2011, 44, 123–151. [Google Scholar] [CrossRef]
- Tatikolov, A.S. Polymethine dyes as spectral-fluorescent probes for biomacromolecules. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 55–90. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, J.; Xia, S.; Mazi, W.; Wan, S.; Mikesell, L.; Luck, R.L.; Liu, H. A near-infrared fluorescent probe based on a FRET rhodamine donor linked to a cyanine acceptor for sensitive detection of intracellular pH alternations. Molecules 2018, 23, 2679. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Massie, T.L.; Maeda, T.; Nakazumi, H.; Colyer, C.L. A long-wavelength fluorescent squarylium cyanine dye possessing boronic acid for sensing monosaccharides and glycoproteins with high enhancement in aqueous solution. Sensors 2012, 12, 5420–5431. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; James, N.S.; Chen, Y.; Dobhal, M.P. Cyanine dye-based compounds for tumor imaging with and without photodynamic therapy. Top. Heterocycl. Chem. 2008, 14, 41–74. [Google Scholar] [CrossRef]
- James, N.S.; Chen, Y.; Joshi, P.; Ohulchanskyy, T.Y.; Ethirajan, M.; Henary, M.; Strekowski, L.; Pandey, R.K. Evaluation of polymethine dyes as potential probes for near infrared fluorescence imaging of tumors: Part 1. Theranostics 2013, 3, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, Y.; Zhang, H.; Chen, X.; Zhao, N.; Tan, D.; Zhang, H.; Shi, C. The application of heptamethine cyanine dye DZ-1 and indocyanine green for imaging and targeting in xenograft models of hepatocellular carcinoma. Int. J. Mol. Sci. 2017, 18, 1332. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.-L.; Chang, C.-C.; Chung, C.-L.; Ko, W.-C.; Yang, C.-H.; Hsieh, C.-Y. Neuroprotective effects of platonin, a therapeutic immunomodulating medicine, on traumatic brain injury in mice after controlled cortical impact. Int. J. Mol. Sci. 2018, 19, 1100. [Google Scholar] [CrossRef]
- Ishchenko, A.A. Structure and spectral-luminescent properties of polymethine dyes. Russ. Chem. Rev. 1991, 60, 865–884. [Google Scholar] [CrossRef]
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2011. [Google Scholar] [CrossRef]
- Hamer, F.M.; Heilbron, I.M.; Reade, J.H.; Walls, H.N. Cyanine dyes and related compounds. J. Chem. Soc. 1932, 251–260. [Google Scholar] [CrossRef]
- McCartin, P.J. Observation of metastable geometrical isomers of cyanines by flash photolysis. J. Chem. Phys. 1965, 42, 2980–2981. [Google Scholar] [CrossRef]
- Khimenko, V.; Chibisov, A.K.; Gorner, H. Effects of alkyl substituents in the polymethine chain on the photoprocesses in thiacarbocyanine dyes. J. Phys. Chem. A. 1997, 101, 7304–7310. [Google Scholar] [CrossRef]
- Ponterini, G.; Momicchioli, F. Trans-cis photoisomerization mechanism of carbocyanines: Experimental check of theoretical models. Chem. Phys. 1991, 151, 111–126. [Google Scholar] [CrossRef]
- Krieg, M.; Redmond, R.W. Photophysical properties of 3,3′-dialkylthiacarbocyanine dyes in homogeneous solution. Photochem. Photobiol. 1993, 57, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Caselli, M.; Latterini, L.; Ponterini, G. Consequences of H-dimerization on the photophysics and photochemistry of oxacarbocyanines. Phys. Chem. Chem. Phys. 2004, 6, 3857–3863. [Google Scholar] [CrossRef]
- Pronkin, P.G.; Tatikolov, A.S. Photochemical properties of oxacarbocyanine dyes in solutions and in complexes with DNA. High Energy Chem. 2015, 49, 368–371. [Google Scholar] [CrossRef]
- Henrichs, P.M.; Gross, S. Conformational analysis of carbocyanine dyes with variable-temperature proton Fourier transform nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1976, 98, 7169–7175. [Google Scholar] [CrossRef]
- Aramendia, P.F.; Negri, R.M.; Roman, E.S. Temperature dependence of fluorescence and photoisomerization in symmetric carbocyanines. Influence of medium viscosity and molecular structure. J. Phys. Chem. 1994, 98, 3165–3173. [Google Scholar] [CrossRef]
- Rulliere, C. Laser action and photoisomerisation of 3,3′-diethyl oxadicarbocyanine iodide (DODCI): Influence of temperature and concentration. Chem. Phys. Lett. 1976, 43, 303–308. [Google Scholar] [CrossRef]
- Orlandi, G.; Siebrand, W. Model for the direct photo-isomerization of stilbene. Chem. Phys. Lett. 1975, 30, 352–354. [Google Scholar] [CrossRef]
- Momicchioli, F.; Baraldi, I.; Berthier, G. Theoretical study of trans-cis photoisomerism in polymethine cyanines. Chem. Phys. 1988, 123, 103–112. [Google Scholar] [CrossRef]
- Dietz, F.; Rentsch, S.K. On the mechanism of photoisomerization and the structure of the photoisomers of cyanine dyes. Chem. Phys. 1985, 96, 145–151. [Google Scholar] [CrossRef]
- Smedartchina, Z. The influence of a polar medium on the rate of fast photoprocesses. J. Photochem. 1985, 30, 13–23. [Google Scholar] [CrossRef]
- Sundstrh, V.; Gillbro, T. Viscosity-dependent isomerization yields of some cyanine dyes. A picosecond laser spectroscopy study. J. Phys. Chem. 1982, 86, 1788–1794. [Google Scholar] [CrossRef]
- Dietzek, B.; Tarnovsky, A.N.; Yartsev, A. Visualizing overdamped wavepacket motion: Excited-state isomerization of pseudocyanine in viscous solvents. Chem. Phys. 2009, 357, 54–62. [Google Scholar] [CrossRef]
- Åkesson, E.; Hakkarainen, A.; Laitinen, E.; Helenius, V.; Gillbro, T.; Korppi-Tommola, J.; Sundström, V. Analysis of microviscosity and reaction coordinate concepts in isomerization dynamics described by Kramers’ theory. J. Chem. Phys. 1991, 95, 6508–6523. [Google Scholar] [CrossRef]
- Kramers, H.A. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 1940, 7, 284–304. [Google Scholar] [CrossRef]
- Sumi, H.; Asano, T. Slow thermal isomerization in viscous solvents. J. Chem. Phys. 1995, 102, 9565–9573. [Google Scholar] [CrossRef]
- Murarka, R.K.; Bhattacharyya, S.; Biswas, R.; Bagchi, B. Isomerization dynamics in viscous liquids: Microscopic investigation of the coupling and decoupling of the rate to and from solvent viscosity and dependence on the intermolecular potential. J. Chem. Phys. 1999, 110, 7365–7375. [Google Scholar] [CrossRef]
- Grote, R.F.; Hynes, J.T. The stable states picture of chemical reactions. II. Rate constants for condensed and gas phase reaction models. J. Chem. Phys. 1980, 73, 2715–2732. [Google Scholar] [CrossRef]
- Sundstrom, V.; Gtllbro, T. Dynamics of the isomerization of trans-stilbene in n-alcohols studied by ultraviolet picosecond absorption recovery. Chem. Phys. Lett. 1984, 109, 538–543. [Google Scholar] [CrossRef]
- Åkesson, E.; Sundstrom, V.; Gillbro, T. Solvent-dependent barrier heights of excited-state photoisomerization reactions. Chem. Phys. Lett. 1985, 121, 513–522. [Google Scholar] [CrossRef]
- Ponterini, G.; Casselli, M. Photoisomerization dynamics of 3,3′-diethyloxacarbocyanine. Intramolecular and solvent viscosity effects. Ber. Bunsenges. Phys. Chem. 1992, 96, 564–573. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Voznyak, D.A.; Petrov, N.K.; Alfimov, M.V. The specific feature of photochemical processes in molecules of 3,3′-dialkylthiacarbocyanines in binary solvent mixtures. High Energy Chem. 2009, 43, 38–43. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Dzhulibekov, K.S.; Shvedova, L.A.; Kuzmin, V.A. Influence of “inert” counterions on the photochemistry of some cationic polymethine dyes. J. Phys. Chem. 1995, 99, 6525–6529. [Google Scholar] [CrossRef]
- Kolesnikov, A.M.; Mikhailenko, F.A. The conformations of polymethine dyes. Russ. Chem. Rev. 1987, 56, 275–287. [Google Scholar] [CrossRef]
- West, W.; Pearce, S.; Grum, F. Stereoisomerism in cyanine dyes-meso-substituted thiacarbocyanines. J. Phys. Chem. 1967, 71, 1316–1326. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Akimkin, T.M.; Pronkin, P.G.; Yarmoluk, S.M. Enhancement of photoisomerization of polymethine dyes in complexes with biomacromolecules. Chem. Phys. Lett. 2013, 556, 287–291. [Google Scholar] [CrossRef]
- Allmann, R.; Anis, H.-J.; Benn, R.; Grahn, W.; Olejnek, S.; Waskowska, A. Konformationsanalyse von polymethinen i. erstmaliger nachweis von di-, tri- und all-cis-konformationen bei sterisch gehinderten trimethincyaninen (carbocyaninen) der indolin- und benzothiazolreihe. Angew. Chem. Suppl. 1983, 22, 1147–1175. [Google Scholar] [CrossRef]
- Pronkin, P.G.; Tatikolov, A.S.; Anikovskii, M.Y.; Kuzmin, V.A. The study of cis-trans equilibrium and complexation with DNA of meso-substituted carbocyanine dyes. High Energy Chem. 2005, 39, 237–243. [Google Scholar] [CrossRef]
- Pronkin, P.G.; Tatikolov, A.S.; Sklyarenko, V.I.; Kuzmin, V.A. Photochemical properties of meso-substituted thiacarbocyanine dyes in solutions and in complexes with DNA. High Energy Chem. 2006, 40, 252–258. [Google Scholar] [CrossRef]
- Tocho, J.O.; Duchowicz, R.; Scaffardi, L.; Blimes, G.M.; Dipaolo, R.; Murphy, M. Spectroscopic properties of isomerizable cyanine dyes. Trends Phys. Chem. 1992, 3, 31–47. [Google Scholar]
- Pronkin, P.G.; Tatikolov, A.S. Influence of the interaction with DNA on the spectral-fluorescent and photochemical properties of some meso-substituted polymethine dyes. Spectrochim. Acta Part A 2018, 202, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Anikovsky, M.Y.; Tatikolov, A.S.; Pronkin, P.G.; Levin, P.P.; Sklyarenko, V.I.; Kuzmin, V.A. DNA effect on cis–trans equilibrium and fluorescent properties of 3,3′-diethyl-9-thiomethylthiacarbocyanine iodide in aqueous solution. High Energy Chem. 2003, 37, 398–404. [Google Scholar] [CrossRef]
- Armitage, B.A. Cyanine dye–DNA interactions: Intercalation, groove binding, and aggregation. Top. Curr. Chem. 2005, 253, 55–76. [Google Scholar] [CrossRef]
- Atabekyan, L.S.; Chibisov, A.K. Photoprocesses in aqueous solutions of 9-ethylthiacarbocyanine dyes in the presence of surfactants. High Energy Chem. 2007, 41, 91–96. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Pronkin, P.G. Photochemical processes in molecules of polymethine dye probes in the presence of bile salts. J. Appl. Spectrosc. 2018, in press. [Google Scholar]
- Slavnova, T.D.; Chibisov, A.K.; Gorner, H. Photoprocesses of thiacarbocyanine monomers, dimers, and aggregates bound to polyanions. J. Phys. Chem. A 2002, 106, 10985–10990. [Google Scholar] [CrossRef]
- Anikovsky, M.Y.; Tatikolov, A.S.; Kuzmin, V.A. Complex formation between 3,3′-diethylthiacarbocyanine iodide and DNA and its investigation in aqueous solution. Int. J. Photoenergy 1999, 1, 35–39. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Gorner, H. Photophysics of aggregated 9-methylthiacarbocyanine bound to polyanions. Chem. Phys. Lett. 2002, 357, 434–439. [Google Scholar] [CrossRef]
- Valandro, S.R.; Poli, A.L.; Correia, T.F.A.; Lombardo, P.C.; Schmitt, C.C. Photophysical behavior of isocyanine/clay hybrids in the solid state. Langmuir 2017, 33, 891–899. [Google Scholar] [CrossRef]
- Ghann, W.; Kang, H.; Emerson, E.; Oh, J.; Chavez-Gil, T.; Nesbitt, F.; Williams, R.; Uddin, J. Photophysical properties of near-IR cyanine dyes and their application as photosensitizers in dye sensitized solar cells. Inorg. Chim. Acta 2017, 467, 123–131. [Google Scholar] [CrossRef]
- Tahara, S.; Takeuchi, S.; Ohtani, H.; Tahara, T. Vibrational Wavepacket motion in ultrafast cyanine photoisomerization revealed by femtosecond stimulated Raman spectroscopy. In Proceedings of the International Conference on Ultrafast Phenomena, OSA, Santa Fe, NM, USA, 17–22 July 2016; Abstract Number UM2A.4. Optical Society of America: Washington, DC, USA, 2016. [Google Scholar] [CrossRef]
- Bleger, D.; Yu, Z.; Hecht, S. Toward optomechanics: Maximizing the photodeformation of individual molecules. Chem. Commun. 2011, 47, 12260–12266. [Google Scholar] [CrossRef]
- Kaiser, C.; Halbritter, T.; Heckel, A.; Wachtveitl, J. Thermal, photochromic and dynamic properties of water-soluble spiropyrans. ChemistrySelect 2017, 2, 4111–4123. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Bai, F.-Q.; Li, L.; Zhang, H.-X. Synthesis of novel sulfonamide azoles via C–N cleavage of sulfonamides by azole ring and relational antimicrobial study. New J. Chem. 2015, 39, 1634–1642. [Google Scholar] [CrossRef]
- Yao, H.-H.; Cheng, H.-H.; Cheng, C.-H.; Lin, C.-K.; Yang, J.-S.; Chen, I.C. Charge-transfer and isomerization reactions of trans-4-(N-arylamino)stilbenes. Phys. Chem. Chem. Phys. 2016, 18, 28164–28174. [Google Scholar] [CrossRef]
- Chmyrov, V.; Spielmann, T.; Hevekerl, H.; Widengren, J. Trans-cis isomerization of lipophilic dyes probes membrane microviscosity in biological membranes and in live cells. Anal. Chem. 2015, 87, 5690–5697. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.M.; Tatikolov, A.S.; Costa, S.M.B. The effect of anionic, cationic and neutral surfactants on the photophysics and isomerization of 3,3′-diethylthiacarbocyanine. Phys. Chem. Chem. Phys. 2001, 3, 4325–4332. [Google Scholar] [CrossRef]
- Henary, M.; Mojzych, M. Stability and reactivity of polymethine dyes in solution. In Heterocyclic Polymethine Dyes; Strekowski, L., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 221–238. [Google Scholar] [CrossRef]
- Bricks, J.L.; Kachkovskii, A.D.; Slominskii, Y.L.; Gerasov, A.O.; Popov, S.V. Molecular design of near infrared polymethine dyes: A review. Dyes Pigments 2015, 121, 238–255. [Google Scholar] [CrossRef]
- Yarmoluk, S.M.; Kovalska, V.B.; Lukashov, S.S.; Slominskii, Y.L. Interaction of cyanine dyes with nucleic acids. XII.β-substituted carbocyanines as possible fluorescent probes for nucleic acids detection. Bioorg. Med. Chem. Lett. 1999, 9, 1677–16789. [Google Scholar] [CrossRef]
- Armitage, B.A. Cyanine dye–nucleic acid interactions. In Heterocyclic Polymethine Dyes; Strekowski, L., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 11–29. [Google Scholar] [CrossRef]
- Panova, I.G.; Sharova, N.P.; Dmitrieva, S.B.; Poltavtseva, R.A.; Sukhikh, G.T.; Tatikolov, A.S. Use of a cyanine dye as a probe for albumin and collagen in the extracellular matrix. Anal. Biochem. 2007, 361, 183–189. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Akimkin, T.M.; Kashin, A.S.; Panova, I.G. Meso-substituted polymethine dyes as efficient spectral and fluorescent probes for biomacromolecules. High Energy Chem. 2010, 44, 224–227. [Google Scholar] [CrossRef]
- Akimkin, T.M.; Tatikolov, A.S.; Yarmoluk, S.M. Spectral and fluorescent study of the interaction of cyanine dyes Cyan 2 and Cyan 45 with DNA. High Energy Chem. 2011, 45, 222–228. [Google Scholar] [CrossRef]
- Bychkova, A.V.; Pronkin, P.G.; Sorokina, O.N.; Tatikolov, A.S.; Rosenfeld, M.A. Study of protein coatings cross-linked via the free-radical mechanism on magnetic nanoparticles by the method of spectral and fluorescent probes. Colloid J. 2014, 76, 387–394. [Google Scholar] [CrossRef]
- Gorobets, M.G.; Wasserman, L.A.; Vasilyeva, A.D.; Bychkova, A.V.; Pronkin, P.G.; Bugrova, A.E.; Indeykina, M.I.; Shilkina, N.G.; Konstantinova, M.L.; Kononikhin, A.S.; et al. Modification of human serum albumin under induced oxidation. Dokl. Biochem. Biophys. 2017, 474, 231–235. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Costa, S.M.B. Medium effects on the isomerization of an anionic polymethine dye. Chem. Phys. Lett. 2007, 440, 73–78. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Costa, S.M.B. Photophysical and aggregation properties of a long-chain squarylium indocyanine dye. J. Photochem. Photobiol. A Chem. 2001, 140, 147–156. [Google Scholar] [CrossRef]
Figure 1.
Generalized molecular structure of cyanine dyes and examples of terminal heterocycles B1 and B2.
Figure 1.
Generalized molecular structure of cyanine dyes and examples of terminal heterocycles B1 and B2.

Figure 2.
Resonance structures of polymethine (cyanine) dyes.

Figure 3.
Generalized molecular structures of trans and cis isomers of unsubstituted carbocyanine dyes and structural formulas of cyanines 1–6.
Figure 3.
Generalized molecular structures of trans and cis isomers of unsubstituted carbocyanine dyes and structural formulas of cyanines 1–6.

Figure 4.
General scheme of the potential surfaces of isomerization of cyanine dyes (only S0, S1, and T1 surfaces are shown).
Figure 4.
General scheme of the potential surfaces of isomerization of cyanine dyes (only S0, S1, and T1 surfaces are shown).

Figure 5.
Scheme of the potential surfaces of isomerization of meso-substituted cyanine dyes (only S0, S1, and T1 surfaces are shown).
Figure 5.
Scheme of the potential surfaces of isomerization of meso-substituted cyanine dyes (only S0, S1, and T1 surfaces are shown).

Figure 6.
Generalized molecular structures of trans and cis isomers of of meso-substituted carbocyanine dyes and structural formulas of cyanines 7–21.
Figure 6.
Generalized molecular structures of trans and cis isomers of of meso-substituted carbocyanine dyes and structural formulas of cyanines 7–21.

Figure 7.
Absorption spectra of dye 8 (3,3′-diethyl-9-methoxythiacarbocyanine iodide; cdye = 1.1 × 10−6 mol L−1) in (1) acetonitrile, (2) isopropanol, (3) mixture of isopropanol and dioxane (1:1), and (4) dioxane.
Figure 7.
Absorption spectra of dye 8 (3,3′-diethyl-9-methoxythiacarbocyanine iodide; cdye = 1.1 × 10−6 mol L−1) in (1) acetonitrile, (2) isopropanol, (3) mixture of isopropanol and dioxane (1:1), and (4) dioxane.

Figure 8.
(1) Absorption spectrum of oxacarbocyanine dye 3 (cdye = 1.17 × 10−6 mol L−1) in a phosphate buffer solution in the presence of DNA (cDNA = 7.5 × 10−5 mol L−1), (2) difference absorption spectrum of the dye photoisomer (cdye = 6.2 × 10−7 mol L−1) obtained by flash photolysis in the presence of DNA, and (3) reconstruction of the photoisomer absorption spectrum. Inset: dependence of the decay kinetics of the photoisomer of dye 3 corresponding to back dark isomerization on the relative concentration of DNA.
Figure 8.
(1) Absorption spectrum of oxacarbocyanine dye 3 (cdye = 1.17 × 10−6 mol L−1) in a phosphate buffer solution in the presence of DNA (cDNA = 7.5 × 10−5 mol L−1), (2) difference absorption spectrum of the dye photoisomer (cdye = 6.2 × 10−7 mol L−1) obtained by flash photolysis in the presence of DNA, and (3) reconstruction of the photoisomer absorption spectrum. Inset: dependence of the decay kinetics of the photoisomer of dye 3 corresponding to back dark isomerization on the relative concentration of DNA.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Thermal back isomerization rate constants (ki, s−1) for photoisomers of carbocyanine dyes.
| Dye | R | Solvent | ki, s−1 | Ref. |
|---|---|---|---|---|
| Thiacarbocyanine dyes | ||||
| 2 | – | ethanol | 250 | [21] |
| 8 | OCH3 | isopropanol | 1.7 × 106 | [50] |
| 9 | C2H5 | dioxane | ~5 × 103 | |
| 10 | Cl | isopropanol | 1.6 × 103 | |
| 14 | C6H5 | ethanol | 340 | [52] |
| 15 | C7H6OH | 865 | ||
| 20 | SCH3 | isopropanol | 5 × 105 | [53] |
| Oxacarbocyanine dyes | ||||
| 3 | – | aqueous solutions | 9.0 | [25] |
| 11 | C2H5 | 1.5 × 104 | ||
| 12 | C2H5 | 1 × 104 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pronkin, P.; Tatikolov, A. Isomerization and Properties of Isomers of Carbocyanine Dyes. Sci 2019, 1, 19. https://0-doi-org.brum.beds.ac.uk/10.3390/sci1010019
AMA Style
Pronkin P, Tatikolov A. Isomerization and Properties of Isomers of Carbocyanine Dyes. Sci. 2019; 1(1):19. https://0-doi-org.brum.beds.ac.uk/10.3390/sci1010019
Chicago/Turabian StylePronkin, Pavel, and Alexander Tatikolov. 2019. "Isomerization and Properties of Isomers of Carbocyanine Dyes" Sci 1, no. 1: 19. https://0-doi-org.brum.beds.ac.uk/10.3390/sci1010019