1. Introduction
Nanotechnology is one of the most promising strategies developed to improve drug delivery systems. Within this field, lipid-based formulations have attracted attention for the administration of poorly soluble bioactive substances, particularly in water and aqueous solutions. The numerous advantages of these nanosystems include biodegradability, biocompatibility, and high physicochemical stability. Their capacity for drug solubilization and cell adhesion are properties that enhance the bioavailability of drugs demonstrating irregular absorption [
1,
2]. An important parameter of drug delivery systems relates to no or low toxicity of the carrier itself either in vivo or in the environment as a by-product. Therefore, nanoparticles fabricated using an assembly of natural biomolecules, such as lipids, proteins, and carbohydrates are expected to be appropriate. Lipid-based systems are particularly useful for clinical applications. This is closely related to the ability of lipid particles to cross cell membranes directly due to the fact that lipids are one of the basic building blocks of cell membranes [
3,
4,
5,
6,
7,
8]. Drug delivery using lipid-based systems can mimic biological membranes because the translocation of natural cell membrane properties to synthetic systems combines the advantages of the membrane surface and flexibility of material chemistry. An additional benefit is that such a system can escape immune system elimination [
5]. Despite the membrane structure, the elasticity of nanoparticles also has an obvious impact on the ability to penetrate across the lipid bilayer as an effect of different morphological responses of hydrophilic and hydrophobic nanoparticles. Studies demonstrated that hydrophilic nanoparticles are flattened in the membrane plane, while hydrophobic nanoparticles are elongated during penetration. These results show the joint effects of elasticity and particle surface hydrophobicity on cellular uptake [
1,
2,
3,
4,
5,
6,
7,
8]. Effective delivery systems allow for the protecting of drugs from fast degradation, long-term in vivo retention, immune escape, targeted controlled drug release, and give the opportunity to easily cross specific barriers in vivo [
3,
4,
5,
6]. Other factors, such as size, surface charge, shape, lipid composition (headgroups, the length of the tail, and even the saturability of the lipid carriers), and elastic and curvature energies may also influence the barrier permeability and transport [
7,
8]. A lipid bilayer structure allows encapsulating both fat- and water-soluble molecules enabling the delivery of therapeutic agents with different physico-chemical properties. The unique properties of lipid-based systems, including their ability to facilitate extended circulation time, adhesion, and homologous targeting, have led to their various applications in the context of nanomedicine [
6,
7,
8,
9].
Currently, the knowledge of different routes of drug delivery is wide-spread, and the oral path is the most frequently chosen. That is why most of the pharmaceutical products are in the form of tablets or capsules. The major problem for this form is the poor solubility and low permeability of therapeutic substances. It is assumed that approximately 50% of medicinal substances are water-insoluble. This phenomenon contributes to the low bioavailability of drugs and reduces the likelihood of recovery. The huge list among literature positions contains a variety of methods that result in better absorption of the active substance. Bioavailability is also the first aspect of our interest. To increase bioavailability, scientists are currently focusing on the study of drugs based on emulsions, microemulsions, nanoparticles, and liposomes [
10,
11]. Nano- and microparticles, widely used for different biological purposes and cellular tracking, are intensively modified (by physico-chemical methods) to broaden their functionality and applicability in drug formulations.
The second aspect of our scientific interests is the widely understood biocompatibility. This term applies to many branches of medicine, pharmacy, and transplantation. In the case of the last one, the main problem is often not organ transplantation, but the lack of tissue compatibility between the donor and recipient. Transferring the organ causes the immune response of the recipient’s body as an effect of the action of its own unique tissue compatibility system. Such a response can contribute to the rejection of the transplant and can cause failure of this process. Hence, the innovation in transplantation was the use of immunosuppressive drugs reducing the immune response.
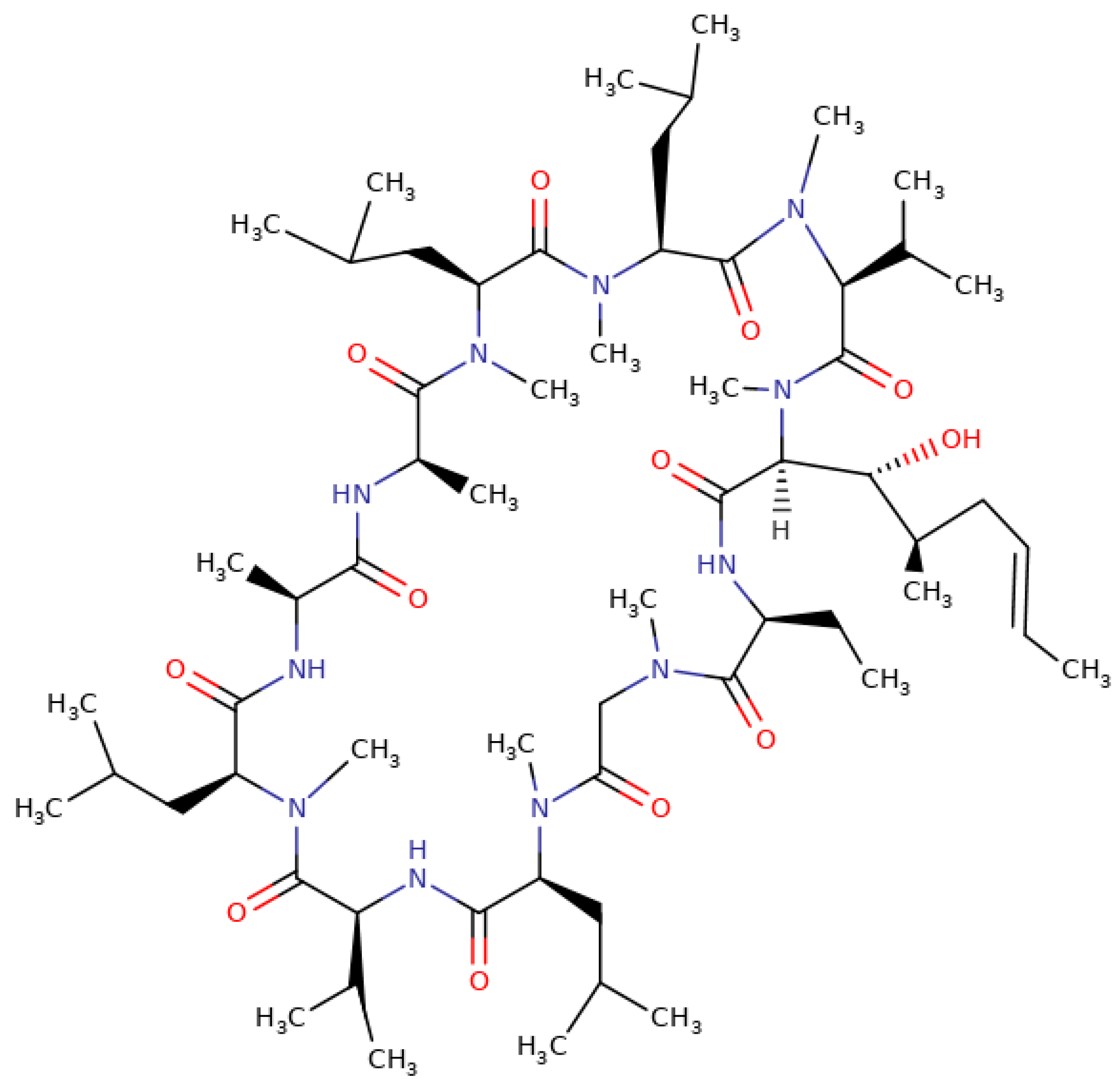
One such immunosuppressive drug is cyclosporine whose effectiveness was confirmed in 1978. Cyclosporine also exhibits a variety of biological activities, including anti-fungal, anti-inflammatory, and anti-parasitic properties. Therefore, this drug is very important not only in transplantation, but also in dermatology and ophthalmology. Additionally, the Food and Drug Administration (FDA) approved cyclosporine in 1997 as a means to treat psoriasis and rheumatoid arthritis. One of the varieties of cyclosporine is cyclosporine A (CsA). This is an organic chemical compound, a cyclic polypeptide consisting of 11 amino acids with the formula C
62H
111N
11O
12 (see
Figure 1). Both bioavailability and biocompatibility problems are associated with CsA. Principally from a medical point of view is CsA’s poor solubility in water, physiological fluids, and saturated hydrocarbons as an effect of its lipophilic structure. Due to the low solubility and variable bioavailability, the drug in its initial form is poorly absorbed orally. Over the last two decades, significant efforts have been made to design an ideal vehicle for CsA oral administration capable of providing improved absorption and reducing its adverse effects [
1,
2,
3,
4,
5,
6,
7,
8,
9]. CsA preparations must be used in different forms and delivered before a meal, as then they can reach a higher serum concentration. The amount of drug absorbed is also strongly influenced by changes in the method of formulation. An example of such a preparation is CsA microemulsions because these are characterized by better hydrophilicity, bioavailability, and faster action even after using a smaller dose of the drug compared to its original form. The maximum concentration of CsA occurs in the serum after only 1–2 h when taking the form of microemulsion compared to 1–6 h after capsule drug delivery. Different kinds of hydrophilic surfactants (hydrophile-lipophile balance (HLB) ≥ 12) and short-chain alcohols are used during microemulsion production for stabilization or to increase solubility [
11,
12]. The HLB scale, developed by Griffin in 1949 (currently, most frequently used in the range of 1–20), is essentially a ranking of how hydrophilic an amphiphilic molecule (e.g., a surfactant) is. The HLB value ranks the tendency of a substance to be hydrophilic or hydrophobic (lipophilic). Higher valued substances on the HLB scale are more hydrophilic and, thus, are more water soluble. Similarly, lower valued substances on the HLB scale are more lipophilic and, thus, more oil soluble. It is well-known that some components used in drug delivery systems are inactive but can increase drug durability, thus, improving the bioavailability [
11].
One method to increase drug absorption is through dissolving in a mixture of vegetable oils, fatty acid esters, or surfactants, e.g., transesterified polyethylene glycols and sorbitan fatty acid esters. Another efficacious methodology for improving the dissolution behavior of poorly-soluble drugs can be particle size reduction. This is because drug nanoparticles often show significant enhancements in dissolution as an effect of an increase in the available surface area and a decrease in the diffusion layer thickness. Suzuki and co-workers proved this on the basis of an accelerated dissolution rate of drugs using the Noyes-Whitney, the Prandtl, and the Ostwald-Freundlich equations [
13].
Chemicals with a solubility lower than 100 mg/mL (
Table 1) frequently show dissolution-limited absorption after their oral administration. CsA is classified by the Biopharmaceutics Classification System (BCS) as a BCS II drug due to its physicochemical properties (high lipophilicity, polar surface area, and molecular weight). The BCS defined, as BCS II, the drugs with low solubility and high membrane permeability, and with low bioavailability after oral administration, which may result in limited therapeutic potential. The limited absorption of such drugs can be notably increased even by slight improvements in the dissolution ratio. Thus, both liposomal and/or microemulsion CsA systems might enhance the oral absorption and administration, which might lead to better biopharmaceutical properties for CsA compared with the base CsA. This is important because long–term oral intake of base CsA does not guarantee complete absorption and causes nephro- and/or hepatotoxicity. The most promising are liposomal and/or o/w microemulsion medicinal preparations containing dissolved CsA prepared immediately before use. Soft capsules under the influence of decomposition with gastric juices also can form o/w emulsions, in which the oil phase with surfactants is responsible for the dissolution of the active substance and, as a result, the CsA complete absorption [
10,
11,
12].
From the literature, it is known that in addition to the dissolution rate, the form of the drug (crystalline or amorphous) is an additional parameter used for drug classification. Drugs in the amorphous state sometimes undergo crystallization during processing or storage, resulting in unstable formulations with low dissolution rates [
14]. S. Onoue and co-workers stated that crystalline CsA produced endothermal melting at 115 °C, but amorphous CsA exhibited thermal events at 128 °C. The transition during the endothermic events might be attributable to a solid-to-liquid transition at over 120 °C. On the other hand, in the case of CsA o/w emulsions, there was no thermal event in the DSC thermograms within the examined temperature range. This suggests that CsA in such a system might exist in a high-energy amorphous state with a homogeneous molecular interaction between CsA and the lipidic ingredient. A verification of appropriation to use the emulsion form is also that the amorphization of CsA might be effective for improvement of the wettability through a reduction in particle size and the absence of crystallinity [
14].
Based on the pharmacokinetic characteristic of CsA, it appears that its absorption rate is largely dependent on the drug dissolution rate. On the other hand, the difference in the colloidal stability might contribute to the variability in the oral absorption of CsA formulations. The large intra- and inter-individual differences in CsA absorption result in a reduction of the therapeutic effect and an increase in the risk of undesired side effects. The particle size and zeta potential analysis was mainly used as a simple approach allowing us to show the levels of modification and to check the colloidal stability. Many of the surface functionalities were applied, which allowed for the verification of a variety of approaches of the particle analysis. The particle size (by dynamic light scattering (DLS) and zeta potential (from electrophoretic mobility measurement) are convenient approaches to validate successful particle modification. The particle size reduction technique provides an increase in the surface area and a decrease in the diffusion layer of drug powders, leading to improvements in the dissolution rate. The high dispersibility of CsA formulations typically contributes to improvements in the wettability of CsA and in its bioavailability as a final effect.
The aim of this mini overview was the presentation the potential of analysis various cyclosporine (CsA) formulations with an emphasis on the physicochemical properties of cationic cyclosporine A colloidal formulations. Using an extensive literature review as a background in this study, emulsion or microemulsion systems that contained CsA with ethanol, oil, or
n-tetradecane and other biologically active substances to increase drug assimilation in the body were described. Cyclosporine A (CsA) liposomal or emulsion formulations were prepared using the o/w solvent method through a high-pressure homogenizer to reduce the droplet size and to obtain a narrow size distribution [
15,
16,
17]. As an antibacterial component, chitosan can be used [
16]. To ensure stable compositions, effective diameter and zeta potential measurements were used. Also, our work was focused on the development and characterization of different CsA formulations as suitable drug vehicles. These formulations can be available as dried powders that could be easily incorporated in conventional dosage forms to ensure long-term stability. On the other hand, these preparations should be artificial substance-free delivery systems; thus, their toxicity is expected to be diminished compared to the base CsA. An additional advantage for CsA microemulsion (or liposomes) use is that the pharmacokinetic behavior achieved can be easily projected, because the CsA absorption in such formulations is less bile-dependent. However, the low safety and efficacy correlation is still a problem as an effect of the narrow therapeutic CsA range. Not only drug particles but components of the formulations can be harmful for patient [
17]. Apart from these limitations, the different CsA forms are, to varying degrees, sensitive to storage conditions (temperature and humidity), and so instability should also be monitored. Therefore, in this paper, we aimed to describe the possibilities of full monitoring of CsA incorporated into different formulations developed to maximize the solubility, bioavailability, and stability.
We hope that this review will give a full description of the investigation options that may help avoid the problem of poor absorption of various drugs in the digestive system. The effectiveness of the modification depends on the component proportions, temperature, time, and strength of homogenization. It is important that the above modification does not lead to changes in the properties of the bulk phase of the material. Drug molecules have a tendency to recrystallize and the higher the degree of supersaturation, the lower the physical stability, leading to an increased tendency for drug precipitation. Therefore, the drug activity should be kinetically and/or thermodynamically controlled to avoid precipitation or sequestration within the micellar phase, as this could lead to incomplete drug absorption [
15].
2. Methodology
The following reagents are the most often used in the study of CsA-containing colloidal systems: cyclosporine A (CsA) 98.5% (HPLC), ethanol (Et) 96% p.a., cholesterol (Chol) 99%, dipalmitoylphosphocholine (DPPC) 99% (TLC vegetable oil (e.g., soybean oil (SO). Cyclosporine is one example of an active substance with low solubility in water [
1,
2,
3]. CsA comes in the form of a white powder or prismatic crystals with a molecular weight of 1202.6 g/mol. Deionized water is usually prepared using a Milli-Q purification system (Millipore, Burlington, MA, USA) (resistivity of 18.2 MΩcm).
2.1. CsA Solubility
The melting point of CsA compounds is 148–151 °C, while the solubility is relatively poor on the order of 27 µg/mL. CsA dissolves well in acetone, diethyl ether, and methanol, but is poorly soluble in water, physiological fluids, and saturated hydrocarbons. When heated, it decomposes, producing hazardous fumes of nitrogen oxides. The table below (
Table 1) contains detailed information on CsA solubility in several typical solvents [
10].
From the literature, it is well-known that the solubility parameter is temperature dependent but inversely proportional, for example about 100 μg/mL at 13 °C and only ~10 μg/mL at 38.5 °C [
18]. The solubility of CsA in pure water decreased with increased temperature as follows: about 46.1, 17.4, and 6.8 µg/mL at 15, 25, and 37 °C, respectively. This is most likely due to the intra-molecular H-bonds (produced by the NH groups), which adopt a more rigid configuration vs. the temperature increase. To explain this phenomenon, the conformational changes of the D-alanine (Dal) amino acid residue of drug should be taken into account. It loses hydration water with an increase in temperature, which affects the CsA conformation and, consequently, its solubility. CsA also lacks ionizable functional groups, so the manipulation of pH does not enhance its solubility. For the same reason, improving the solubility by salt formation is not feasible [
19].
2.2. CsA Bioavailability. Immunosuppression
In terms of pharmaceutical uses, an unfavorable feature of CsA associated with its low solubility is the low drug bioavailability, which can radically decrease the effectiveness of CsA administration. However, the development of drug delivery technology and deeper insight into the molecular nature of the drug have allowed researchers to obtain many promising CsA delivery systems. For instance, biodegradable polymeric nanoparticles appear to be hopeful candidates for oral administration [
20,
21,
22]. Researchers have not only improved the drug bioavailability, but also its overall therapeutic efficacy by targeting the lymphatic system and providing a persistent release of CsA. The results of our experiments dealing with liposomal CsA formulations are also promising, thus, opening up new possibilities of employing them as carriers for the oral delivery of drugs.
The main problem is that CsA is characterized by high instability of the pharmacokinetic parameters, and its bioavailability as an oral drug ranges from 10% to 89%. On the other hand, CsA demonstrates a narrow therapeutic index and a large number of side effects. The ratio of the oral dose taken to the blood content of the drug depends on many factors, and therefore it is difficult to determine the ideal mathematical equation for this drug’s effectiveness.
From the definition, the therapeutic range is the minimum concentration required to achieve maximal effectiveness to the concentration above which toxicity starts. If the blood concentration reaches a level below this range, the drug becomes ineffective; this can lead to rejection of the transplanted organ. On the other hand, the toxic concentration of the drug can cause nephro-, hepato-, or neurotoxicity. The optimal dose should offer the minimum blood concentration sufficient to achieve an immunosuppressive effect and should prevent potential toxic effects. From the literature, immunosuppression is achieved at 100 ng/mL in the whole blood, but the required concentration depends on the type of transplanted organ [
20,
21,
22]. Cyclosporine in the form of microemulsion is more bioavailable compared to an oil-based formulation. Based on the pharmacokinetic parameters, steps should be taken to stabilize the CsA dosage to minimize toxicity and improve the benefits [
10].
Additionally, micelles composed of polysaccharides (e.g., chitosan) can be a promising alternative to oral delivery vehicles as a result of their specific bioadhesive properties. Another system providing appropriate control of CsA release could be polymeric micro- and nanoparticles. The positively charged nano-formulations of CsA prepared using cationic polymers can improve its absorption rate and bioavailability as a result of the electrostatic interactions between the particles and negatively charged mucosal surfaces. A promising alternative to the commercial formulations might be solid lipid nanoparticles. As a drug carrier for the oral administration of CsA, they can exhibit a low variation in bioavailability.
2.3. Lipid-Based Liposomal Systems
Generally, lipid-based liposomes in water and in electrolyte solutions are prepared by injection of an alcohol solution [
23,
24,
25]. For example, by the slow injection of an ethanol solution of phospholipid (DPPC) into an aqueous solution and homogenization. Liposomes should be prepared at temperatures higher than the phospholipid phase transition temperature, but not exceeding about 50 °C. For DPPC, the phase transition temperature is equal to 41 °C, and so the temperature of preparation should be selected at about 45 °C. The dispersion in both water and electrolytes should be done in portions. Each portion should be homogenized while maintaining the homogenization parameters (e.g., 1 min at 10,000 rpm). The pH value of the systems should also be measured before and after homogenization. The same procedure is repeated for the system without CsA and CsA (1 mg). The incubation time required for high efficiency should be selected up to 15 min for cyclosporine liposomes, because further increases in the incubation time do not increase the inclusion efficiency considerably [
16].
2.4. Dynamic Light Scattering (DLS). Particle Size and Zeta Potential Determination
The most commonly used method to measure the particle size of various cyclosporine formulations is dynamic light scattering (DLS). In our research, the mean particle size of the different CsA formulations (liposomes, multilamellar vesicles, or o/w emulsions) were measured in five 1-min cycles by DLS using a Zeta Potential-Bi mass (Brookhaven, Preston, UK). For preparation, a typical oil/water emulsion was used with soybean oil as the oil phase. The amount of CsA was kept constant. The formulation was centrifuged at 10,000 rpm for 15 min. using a Heidolph Silent Crusher homogenizer. All measurements were performed at 20 °C or 37 °C (~physiological). The zeta potential was determined from the electrophoretic mobility by the same apparatus, but the samples were placed into cuvettes integrated with gold electrodes. Each sample was studied in five replicated measurements. The zeta potential was used to analyze different CsA modifications that can be difficult to study using conventional analysis techniques. The electrophoretic method is very useful with the ability to directly analyze nano- and microparticles. Chemically modified colloidal systems by the addition or attachment of different molecules can lead to a variety of functionalities.
2.5. Morphology. BAM Analysis
A very satisfactory method used to study biologically active substances is the monolayers testing methodology and, directly related with it is Brewster angle microscopy (BAM). In this field, our measurements, such methodologies are frequently used. To better characterize the DPPC-CsA and DPPC-Chol-CsA relationships in solution, the monolayer morphology can be investigated. Simultaneously, the Brewster angle microscopy (BAM) analysis of pure and mixed monolayers can be performed.
3. Physico-Chemical Characterization of Typical CsA Systems
3.1. Liposomal CsA Systems Stability
From the point of biopharmaceutics in transplantology, a more efficient and a safe delivery system with less toxicity is needed for immunosuppressive therapy. Cyclosporine is a neutral, high hydrophobic cyclic peptide, and the main component of liposomes are phospholipids. Each phospholipid has an intrinsic liquid crystalline phase. Liposome membranes are fluid above the characteristic individual temperature and are solid below it [
26]. Liposomes are widely used in clinical medicine for various purposes, e.g., selective drug delivery and diagnostic vehicles. Liposomes are highly specialized lipid spheres that can preferentially accumulate at sites of tumors, infection, and inflammation. CsA as a lipophilic drug appears to be a model candidate to include into liposomes. Intravenously supplied liposomes of CsA can lower drug side effects, as a result of its avoidance of the kidneys. CsA liposomes are favorably removed by the reticuloendothelial system and tend to accumulate in the spleen. A CsA liposomal carrier provides the possibility to attain the optimal drug level at sites of tissue rejection, as an effect of association of different parameters of the immune response and to decrease the drug exposure to sensitive organs. Studies of various drugs, not only CsA, showed that liposome-encapsulated drugs often have biodistribution and toxicity that are significantly different from the original drug [
27].
In some cases, when it is necessary to reduce the fluidity of the layer, cholesterol can be added to a certain extent. On the other hand, cholesterol, being a stabilizing agent of liposomes in the blood, limits the level of the drug incorporation. Therefore, a compromise between stability and the maximum drug uptake must be reached [
26]. Literature reports suggested that elevated levels of liposomes in the blood are more frequently seen for small liposomes (diameter ~200 nm). Thus, in order to increase blood circulation, the particle size must be kept narrow. The average particle size of the liposomal formulations prepared by Czogalla et al. was less than 200 nm, and the particles were found to be spherical in shape as confirmed by SEM. Research proved that all types of charged liposomes (positive, negative, and neutral) can be formulated as a stable lyophilized powder with a large entrapment effectiveness. However, due to a smaller size or due to interactions between the liposomal charge with plasma proteins, positive and neutral liposomes showed higher stability in the blood than negative liposomes. On the other hand, higher targeting was seen for the positive liposomes compared to the neutral and negative liposomes, giving the possibility of applying such formulations for liver and bone marrow transplantation [
27]. Positively charged liposomes could increase the penetration and drug availability; however, the potential of liposomes can be limited because of their short half-life on the surface (e.g., corneal). A technological challenge is to produce sterile liposomes on a large-scale. In pharmacokinetic studies of CsA liposomal systems, in addition to particle size, the absolute amount of lipids and the lipophilicity of the drug are critical factors. Based on the calorimetric ultrafiltration and titration method, the partition coefficient of CsA in the lipid bilayer/water system was evaluated to be 4000–4300 ± 600 [
27].
The particle size distribution of the liposomal formulations with or without CsA was estimated using a laser light scattering technique. The average particle size of the prepared liposome formulations (effective diameter, nm), turbidity (NTU), value of natural pH, polydispersion index (PDI), and zeta potential of exemplary systems are summarized in
Table 2. The order of particle size of the obtained CsA nanocarriers was CsA, CaCl
2 > CsA, H
2O > CsA, KCl, and it was difficult to decrease the particle size to <100 nm through high-pressure homogenization. On the other hand, the PDI of all carriers was near or much lower than 0.3, which can indicate relatively narrow particle-size distribution. CsA liposomes in electrolyte solution had a higher negative charge than CsA liposomes in water. A high zeta potential is necessary to maintain the in vitro stability of nanosuspensions. The liposomes with an absolute value of charge lower than 5 mV are not usually stable; however, the particle size and PDI of all vehicles had no significant changes within 24 h; thus, other mechanisms of stabilization should be considered [
2,
28].
In all cases, the mean particle diameter was below 600 nm, presenting a homogeneous size distribution and average values of surface charge. Regarding the particle size, turbidity, and zeta potential measurements of the CsA particles after their incubation in different mediums, the formulations in water and KCl solution presented a marked reduction in these three qualities. In contrast, in the CaCl2 solution, only the zeta potential decreased. The surface charge during 2 h measurements remained practically unchanged in all CsA formulations. The concentration of phospholipids in the CsA formulations was below the critical micelle concentration; therefore, the solubilizing effect of DPPC on CsA dissolution was likely limited. The particle size reduction approach provides an increase in the surface area and a decrease in the diffusion layer of drug powders, leading to an improvement in the dissolution rate. Based on the literature data and studies carried out by us, the following general conclusions can be drawn. Depending on the destination and application, if a small-sized system is needed, CsA in water or a KCl solution will be better; however, if liposome stability is the key decisive parameter, then the CsA in CaCl2 solution should be chosen.
3.2. Binding of CsA to Lipid Membranes (BAM Analysis)
The binding of CsA to lipid membranes follows classical hydrophobic effects and is accompanied by a positive enthalpy variation. This is rather uncommon because most peptide-lipid binding reactions require enthalpy-driven binding. Research often assumes that the CsA molecule occupies an area of 260 Å
2 in the lipid monolayer. During an increase in the CsA concentration, a model membrane perturbation is observable by changes in the lateral organization in the acyl chain region. As a result, the drug interacts mainly with the part of the fatty acyl chain proximal to the head group of the phospholipids. The characteristics of CsA interactions with biological membranes suggest a drug-dependent increase in the bilayer fluidity at temperatures below the main phase transition, but an increased spatial organization at temperatures above that point. On the other hand, the addition of cholesterol to the model membrane decreased the binding capacity of cyclosporine as a consequence of the acyl chain flexibility. The structure of a bilayer influences the nature and topology of CsA inclusions into monolayer. Different factors, e.g., the composition of the liposomal carriers of cyclosporine, the type of fatty acid chains, and, notably, the lipid head group, strictly affect the pharmacokinetic behavior. These conclusions have been proven on the basis of research described in detail in our earlier articles [
2,
29,
30,
31]. Therefore, to achieve the desired drug effect, the type and composition of the lipid formulation used as its carrier should be carefully considered.
The Brewster angle microscopy (BAM) analysis of a pure DPPC monolayer showed small characteristic and irregular domains, which appeared at surface pressure below 10 mN/m (
Figure 2). Observed structures are specific for the LE-LC phase transition, and they gradually change along with the progress of compression. After reaching a value of surface pressure of about 20 mN/m, the monolayer was homogenous. On the other hand, a pure CsA monolayer appeared to be homogenous and less condensed than a pure DPPC monolayer, and no domains were observed. The mixed monolayers of DPPC and CsA exhibited the formation of domains but smaller structures were observed if the amount of CsA in the monolayer was higher. Along with the increase in CsA content, the shape of the domains was more circular and they were arranged in points (
Figure 2). On the basis of our previous data, the π
coll for pure CsA was estimated at 25–26 mN/m and, at the higher surface pressure (about 30 mN/m), small domains appeared [
32]. In this study, the surface pressure-area isotherms and BAM images were used to investigate the effect of CsA on a model DPPC membrane for different molar ratios of ingredients (DPPC-CsA). Changes in isotherm shapes, their shifts, and also BAM images of domains appearing during the compression process were registered and analyzed. This type of research together with the thorough characterization of different CsA-based formulations may be helpful in better understanding CsA interactions with living cell membranes and should be planned each time for a given patient group because the exact explanation of the drug action requires not only knowing the physico-chemical characteristics but also an in-depth analysis at the molecular level.
In drug delivery systems, investigations regarding the ability to adhere to the cell wall should be conducted. In this type of research, Langmuir monolayer studies may be helpful as well as studies using the BAM technique. The negatively charged particulates or droplets should encounter electrostatic repulsion from the negatively charged layer of enterocytes. This is an unfavorable process for the absorption of CsA. However, CsA nanodispersions in lipid carriers may exhibit a large specific surface area and, thus, the possession of adhesion to the cell wall. These drugs are able to be solubilized in liposomes and microemulsions, which is very helpful for drug absorption. If the particle diameter is small (about 200 nm and less), this is helpful to increase the oral bioavailability. On the other hand, intestinal drug transport owing to lipid-based vehicles can be increased and can prolong the CsA absorption (approximately 6–8 h after administration), but lymphatic transport is slow and sporadic. The cyclosporine charged liposomal systems are useful for liver and bone marrow targeting and can, thereby, reduce the accumulation of the drug in the kidneys and the nephrotoxicity visible for base cyclosporine. In addition to CsA liposomes, properly prepared oil/water emulsions of CsA may also increase the bioavailability of this drug as will be discussed in more detail in the next chapter.
3.3. O/W Emulsions of CsA
The formulation of CsA can also be prepared as o/w emulsions. From the literature data, CsA powder can be predominantly amorphous in nature [
33]. In Wang et al.’s studies on the basis of SEM images, there were no clear crystalline particles visible. In another investigation, crystalline CsA powders indicated a tetragonal crystal form [
34]. As an effect, the kind of emulsion and its stability is strictly dependent on the CsA form. The stability of such systems can be defined on the basis of a regular shape and distributions of droplets or nanoparticles. Instead of micelles and liposomes, drug emulsion may be more convenient regarding the stability. On the other hand, with the increase in the drug/micelle (liposome) ratios, the micelles (liposomes) can become more unstable because the drug can be supersaturated in the core of the micelles. The high chemical potential of the drug in micelles (liposomes) or dissociation of the micelle particles could cause irreversible precipitation of CsA. Pharmacokinetically, the degree of supersaturation is a very important parameter and driving force for drug release because this also potentially affects its precipitation and absorption rates in the gastrointestinal tract.
The CsA suspended in soybean oil and cholesterol with or without the addition of ethanol formed an o/w emulsion where the average droplet sizes were in the range of 500–1500 nm according to the DLS analysis. The thermal behaviors of CsA formulations clarified using DSC showed that crystalline CsA produced a melting endothermic event at 115 °C, amorphous CsA exhibited thermal events at 128 °C, and the transition during the endothermic events might be attributable to a solid-to-liquid transition at over 120 °C [
35]. CsA in emulsion formulation might exist in a high-energy amorphous state with a homogeneous molecular interaction between CsA and the lipidic ingredient. In addition, the amorphization of CsA might be effective for improvement of the wettability through a reduction in particle size and the absence of crystallinity. Dollo and co-workers clarified the possible enhancement of dissolution behavior, the dissolution tests for CsA systems were conducted in distilled water [
36]. Poor dissolution behaviors were seen in both crystalline and amorphous CsA powders.
In the described studies, CsA showed fast dispersibility with the formation of emulsions as determined by dynamic light scattering. These observations show that formulation of emulsions for poorly soluble drugs, such as CsA, could lead to marked improvements in the dissolution properties. Emulsions and microemulsions as colloidal carriers can help to deliver drugs with poor aqueous solubility (e.g., CsA) and extensive first-pass metabolism, due to the special mechanisms. Particle size is a key parameter in defining the deposition pattern and bioavailability of the drug material delivered. In
Table 3, for the formulation characterization of CsA/soybean oil, cholesterol was presented (diameter size, zeta potential, pH, and polydispersity). According to the size-distribution diagram, there were mainly two peaks for the CsA particles ranging from 432 nm to 1580 nm. The micronized CsA with this size diameter was of a suitable size to avoid deposition possibly leading, for example, to effective delivery of the inhaled particles to the airway system organs [
37,
38]. The physicochemical properties of CsA were tested after introduction into o/w emulsions, as well as the emulsion stability at 20 and 37 °C using static (
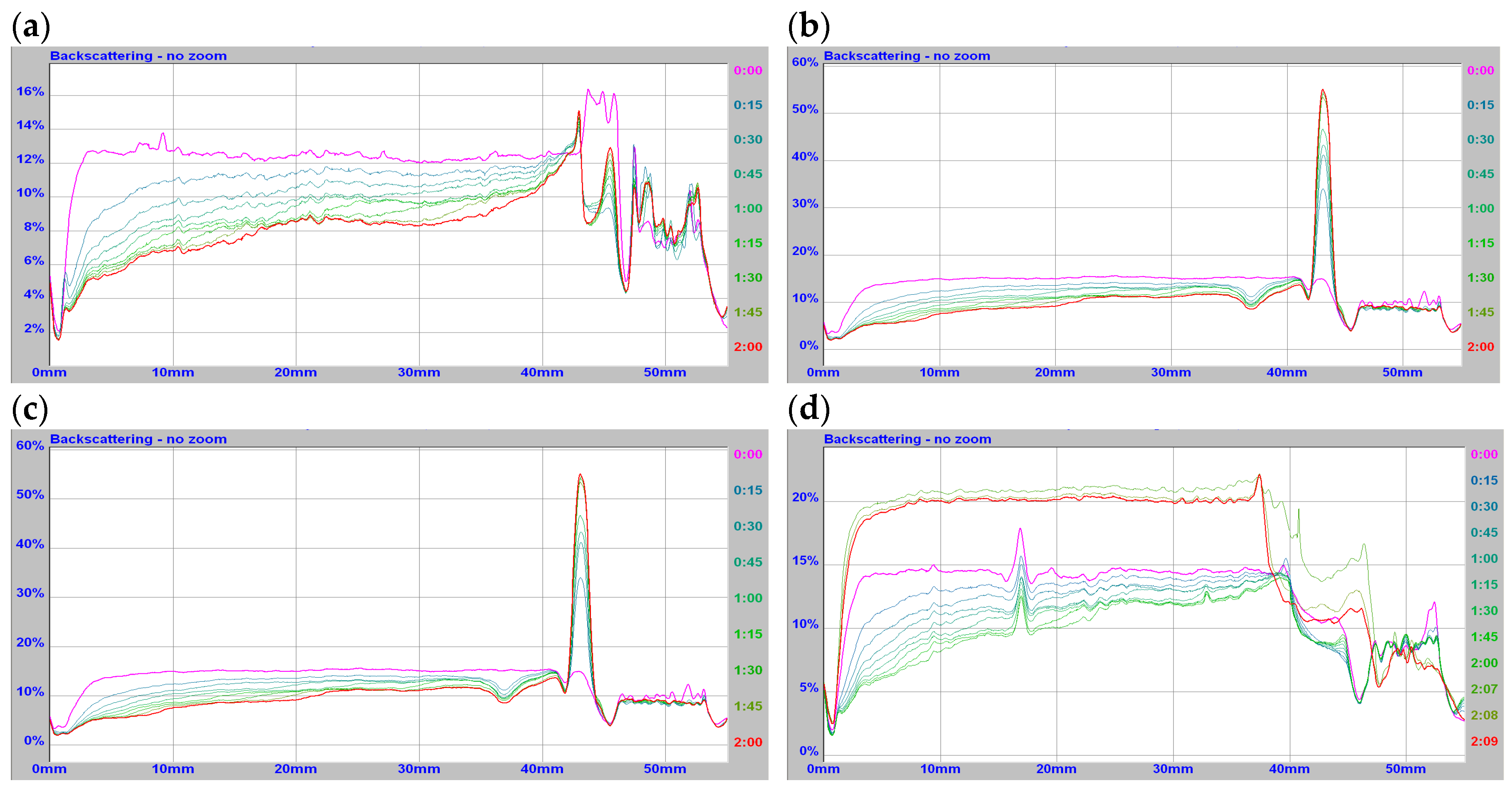
Figure 3a–d) and dynamic light scattering techniques. In
Figure 3, the representative relationships obtained for the simplest and the most complex CsA formulation are presented.
Drug carriers bind to the outer membrane of cells, followed by rapid internalization and shuttling to lymphocytes, where the size and surface charge are a key parameter. Thus, measurements of these two parameters (particle size and zeta potential) are very important for the drug pharmacokinetic characteristics. To increase the bioavailability of CsA, phospholipid DPPC and/or cholesterol (Ch) were used as components of the lipid phase, and ethanol (Et) was used to improve the solubility of the components. More stable systems were obtained at physiological temperatures with smaller diameter values and greater homogeneity confirmed by low polydispersity values and stable zeta potential. Among the obtained CsA systems, those with the highest stability can be subjected to pharmacokinetic tests to select the preparation with the highest drug effectiveness and the lowest toxicity. Static and dynamic light scattering analysis of CsA formulations suspended in soybean oil with other carriers demonstrated that the size distribution of CsA/oil was narrower than that of multicomponent CsA/oil, ethanol, or cholesterol. The average particle sizes of different multicomponent CsA formulations at 20 °C were not substantially different: minimum 1370 nm and maximum 1580 nm, respectively (
Figure 3). The high dispersibility of such CsA formulations can contribute to the improvement in the wettability of CsA. At physiological temperatures, the decrease of diameter was visible for all investigated formulations.
The size of the CsA emulsion droplets obtained was controlled by adjusting the composition, homogenization pressure, and temperature. CsA emulsions prepared with the addition of phospholipid in ethanol determined the improved bioavailability of the drug and can be used in poorly water-soluble CsA delivery systems. The increase in bioavailability can be due to the mechanism of increased solubilization. At physiological temperatures, more stable systems with smaller diameter values were obtained and greater homogeneity was confirmed by low polydispersity values and stable electrokinetic potential compared to these obtained at room temperature. The physicochemical properties of the obtained systems based on CsA proved the beneficial effects of the lipid phase on increasing the bioavailability of the drug. For example, CsA emulsions in the form of an aerosol of an appropriate diameter (less than 2 microns) would allow the distribution of the drug in the respiratory tract and achieve pulmonary concentration with sufficient immunosuppressive activity effective in the treatment of lung diseases.
3.4. CsA/Chitosan Systems. Transport across Membranes
As was mentioned earlier, studies using mono- or bilayer-imitating cell behavior provide a better prediction of the in vivo absorption of most drugs, including cyclosporine. Liposomes also play an important role in these processes. On the other hand, liposomes used as drug carriers have been shown to be stable when lyophilized in the presence of, for example, disaccharides [
26]. Considering this, some studies are carried out for systems with cholesterol, and the effect of antibacterial chitosan has also been studied. The binary or ternary monomolecular films of varying composition make it possible to produce coverage with pre-planned surface properties and check the transport across membranes. We recently described [
39] the possible behavior of CsA in a DPPC-Chol membrane to the full characteristics of such CsA formulations and their transport across membranes as illustrated previously (
Section 3.2). An additional component in these systems is the antibacterial chitosan. The prepared mixed layer and charge density dependent chitosan coating can possibly limit the excessive exposure of chitosan to the blood. As a result, a reduction in the effect of blood clot formation is possible. The monolayers morphology and behavior can provide detailed information on the packing, flexibility, and interactions for a better reference to the real conditions in vivo.
This type of hybrid coating allows the design of the structure of defined topography to play a major role in the interactions with living tissues. As a result of that, the problematic mechanisms of the possible immune response can include better recognition. The Langmuir technique permits us to determine the behavior of polypeptide CsA in contact with a model biological membrane built of its two main ingredients, phospholipid DPPC and cholesterol, especially at the pressure of the natural biological membrane (in the range of 25–30 mN/m). Due to the nature of drug, it is not possible to obtain a stable CsA monolayer at such pressure because it collapses near 25 mN/m. Therefore, the behavior of ternary DPPC-Chol-CsA films was examined depending on the drug amount of CsA at the surface pressure of 15 mN/m, regarded as the most suitable for such CsA/lipid-based systems. We found that cyclosporine significantly reduced the fluidity and packing of the tested biological membranes. At low CsA contents, domain formation was observed, and the highest values of excess Gibbs energy of mixing, which suggests repulsion of the film components and their partial miscibility. For higher CsA content, a homogeneous film structure was observed, proving an increase in the intermolecular attraction of the components in the monolayer. Langmuir–Blodgett films (LB) were formed on solid supports with adsorbed chitosan. These investigations are very important in terms of inhibiting platelet aggregation on the surface of, for example, implants. The multilayer hybrid systems technique is useful in the design and preparation of modern materials because it can combine widespread and multi-task utility properties.
From the literature, a moderate-supersaturation formulation resulted in optimal in vivo performance, maintaining a moderate degree of supersaturation over a longer period of time in the gastrointestinal tract, reducing the drug precipitation, and enhancing absorption. As was previously mentioned, the particle size is an important parameter, because it can influence the biopharmaceutical properties of nanoparticle formulation in the release and affects the possibility of a prolonged residence through particle mucoadhesion. The authors presented the effects of the different factors on the CsA/chitosan system properties. In such studies, chitosan molecules appear to be active at the interface in a specific way, resulting in a lower mean particle size [
16]. The polydispersity index is a measure of the particle size distribution within a sample. Mainly, the concentration of chitosan in the coating solution has a significant influence on the polydispersity index (
p < 0.05). CsA nanoparticles prepared with chitosan solution demonstrated a polydispersity value variability depending on the chitosan concentration. The assumption was the formation of a variable and solid chitosan coating layer or the formation of agglomerates during the preparation and/or freeze-drying of the nanoparticles at different chitosan concentrations [
16]. Many alternatives of in vitro models have attempted to mimic the physiological situation of the intestinal epithelium with its mucus layer; however, there is still a gap in simulating the gastric environment. For instance, strongly acidic pH or enzymes could modify the system properties before it reaches the intestinal barrier, thus, changing the rate of the drug permeability [
40].
3.5. Cyclosporine/Oil Phase/EtOH Formulations
Hermans et al. investigated cyclosporine A-chitosan systems obtained by the o/w emulsification method [
16]. To determine the influence of the formulation parameters on the particle size, polydispersity index, and zeta potential, a full factorial design was applied. The particle sizes ranged from 156 nm to 314 nm, and the polydispersity index values ranged from 0.07 to 0.56. All nanoparticles characterized positive zeta potential values. During in vivo experiments in rabbits, the therapeutic concentrations of CsA were revealed in the cornea and conjunctiva for at least 48 h after the local administration of chitosan-drug nanoparticles [
16]. Nanoparticles mostly loaded with chitosan preserved their positive zeta potential when dispersed in simulated tear fluid, confirming the possible electrostatic interactions between these particles and the negatively charged mucus layer at the eye. The in vitro release profile of cyclosporine A from chitosan-coated nanoparticles was found to be highly conditioned by the release medium used. In addition, cyclosporine A, as included in various chitosan preparations, also remained active against inflammation [
16].
In our research, synthetic n-tetradecane or natural soybean oil were used as the oil phase. Due to the hydrophobicity and extremely low solubility of CsA in water, a low concentration in the tear film, as well as a low corneal penetration were observed. A number of formulations designed to overcome this problem have been described in the literature. To solubilize CsA, various vegetable oils have been used, but the slow transfer of CsA to the corneal epithelium and side effects, such as blurred vision, itching, irritation, epithelial keratitis, and corneal toxicity, have limited their use. Liposomes and CsA o/w emulsions are simple and safe colloidal drug delivery systems to achieve optimal local pharmacological effects of CsA in specific organs with minimal side effects. An excess of CsA can cause irreversible morphological changes that result in progressive organ failure. While advances in the topical administration of CsA have been made, the issue of improvement in formulations in terms of the therapeutic concentrations at the corneal surface over a prolonged period of time is still open. Chitosan-coated CsA particles are preferred for muco/bioadhesive purposes as they show positive zeta potential values in simulated tear fluid. The positive charge of chitosan-CsA formulations can indicate the electrostatic interactions between these particles and the negatively charged mucus layer at the eye. In vitro tests showed little or no cytotoxic effect in spite of their cationic nature. Therefore, chitosan-coated CsA particles can be useful to achieve a longer residence time after instillations into the eye.
In the present study, novel formulations of CsA employing liposomes or o/w emulsion systems were designed for pharmacologic treatment. There was a marked improvement in the dissolution behavior of a nanoemulsified dispersion CsA compared with that of CsA powder. The obtained systems exhibited high dispersibility and particle distribution, and thus can be suitable not only in transplantation but also for oral absorption or inhalation therapy. The full physicochemical characteristics (
Figure 4) can help to obtain the CsA formulation, which might be an efficacious delivery option for immunosuppressive drugs, giving the possibility to provide an optimal local pharmacological action of CsA.
The studies demonstrated that the combined technology of liposomal and emulsion (microemulsion) systems could enhance the absorption and affectivity of CsA. From these findings, the nano-matrix formulation approach might be efficacious for developing different forms of CsA and other similar drugs with improved dissolution and absorption. The absolute zeta potential for all CsA preparations at both temperatures was below 12 mV, with a clear discrepancy between the particles prepared with the addition of n-tetradecane compared to soybean oil as the oil phase. Measurements performed in Td showed lower zeta potentials typically in the range of 1–5 mV. Only particles prepared with higher concentrated ethanol solution had zeta potential values near 10 mV. After increasing the physiological temperature, the negative zeta potential turned to positive, whereas the CsA/soybean oil system prepared with the higher ethanol concentration showed a positive zeta potential. The use of a higher ethanol concentration during nanoparticle preparation seemed to result in the particles being able to maintain their positive charge for a longer period of time. The capacity of ethanol dipoles for electrostatic interaction remains high. On the other hand, drug release is usually limited to the solubility of CsA in the respective media. This suggests that lipid-based drug-delivery systems may be more advantageous than others in improving the absorption of low solubility drugs, due to the multiple enhancement mechanisms of this process. Moderately supersaturated formulations provide a valuable alternative to other systems, resulting in optimal in vivo performance. The degree of supersaturation should be carefully designed during formulation to optimize the drug absorption. In the present studies, CsA was successfully incorporated into different emulsifying systems. The CsA colloidal formulations developed offer some advantages that might enhance the long-term stability of the final dosage form and reduce toxicity issues.
3.6. Process of CsA Absorption and Drug Release
The improved bioavailability is primarily due to two processes: effective absorption and targeted distribution of the drug. In the case of cyclosporine, the process of its absorption takes place in the liver and, to a lesser extent, in the gastrointestinal tract and the kidneys. However, the drug release process is mainly destination dependent because cyclosporine can be used in ophthalmic medications, orally administered, or applied externally on the skin. D’Souza’s review summarized the methods used to study drug release from nano-sized drug delivery systems. Additionally, the advantages and disadvantages of the methods are described, including the challenges with set-up and sampling. As no congruent standards exist, the drug release is currently assessed using a variety of methods. The most frequently used are: sample and separate method, continuous flow method, and the dialysis membrane method, as well as their combination with novel techniques, e.g., voltammetry and turbidimetry. The first of this method allows the direct measurement of drug release with simple set-up requirements. Continuous flow sampling is straightforward but time consuming. The dialysis membrane method appears to be methodologically easier, but it may not be suitable for drugs binding to the membrane. On the other hand, voltammetry and turbidimetry offer the possibility of real-time drug release measurement but may be restricted to certain types of drugs. The greatest efficacy was found for dialysis or its combination with the sample and separate method. More specialized techniques focus on connecting these methods with mathematical models that describe drug release mechanisms as well as facilitate the formulation development of nanoparticulate dosage forms [
41].
3.6.1. Oral CsA Delivery System
One method of CsA use is oral distribution. Beckerman et al. described studies of CsA for oral administration. The drug bioavailability was determined in vitro on the basis of T-cell proliferation. The important parameter was the CsA blood levels after oral intake. The authors concluded that the composition of formulated cyclosporine dispersion (e.g., lipid core component, used surfactants, and/or emulsifiers) had a major effect on the particle size of systems. This parameter, in turn, had an important influence on the human oral bioavailability. In other words, they found a strict correlation between the area under the curve (AUC) and C
max (indicators responsible for bioavailability and drug release) and the particle size as a function of time after the oral intake of different CsA capsules. AUC in pharmacokinetics means the area under the curve and is a measure of how much drug reaches a person’s bloodstream in a given period of time after a dose is given. Alternatively, C
max is the maximal concentration of drug [
42]. The in vitro release process of different CsA nanoparticles systems was described by Gupta et al. [
43]. They showed an interesting bi-phasic release with an initial burst effect. This effect was attributed to the CsA drug loaded on the particles surface. In the first hour, the drug release was about 21% and more. After, the drug release followed a steady profile similar to zero order release. Gupta’s study results indicate that zero order plots were not linear for all formulations, but rather, the first order was linear during the burst effect. Such linearity suggests that CsA release and circulate is diffusion controlled and also that these processes are limited by the drug particle dissolution and erosion of the polymer matrix [
43].
The intestinal metabolism may account for up to 50% of the delivery rate when CsA is orally administered; therefore, in some cases, nanoparticle system microemulsions of CsA will be optimal or liposomes may be found to improve the systemic absorption of CsA after oral administration. A liposomal system can also modify their tissue distribution helping to reduce the nephrotoxicity of the drug. The majority of emulsified formulations increase the surface area of the drugs to improve the solubilization behavior and membrane permeation, which might also result in bioavailability enhancement. The mechanism is complicated because digestive enzymes and bile salts are normally encountered in the gastrointestinal tract. Frequently, even the detailed in vitro drug release kinetics do not accurately reflect the in vivo kinetics. However, research with the use of a monolayer model imitating cell behavior comes to our aid and provides a better prediction of the in vivo absorption of the drug. This characteristic applies to most drugs used, as well as CsA.
Liposomal CsA may exhibit both bioequivalence with microemulsions and diminished inter-individual variations in bioavailability [
44]. The presence of a surface charge induces definite changes in the cyclosporine liposomes distribution in the different organs comparing to the free drug. There is a visible increase of accumulation in the liver and spleen and no substantial activity in the kidney, thyroid, and stomach. On the other hand, the type of liposome charge is very important in the pharmacokinetics of CsA because the residence time of positive liposomes in the blood at 24 h is two-fold greater than negative and neutral liposomes, and the incorporation of a positive charge in the lipid bilayer provides a very significant increase of the residence time of CsA liposomes in the blood compared to the free drug [
16,
45]. Another interesting observation is that the liposomes accumulated in the bones, albeit to a lesser extent (3–6%) in comparison to other body tissues, which indicates that the cyclosporine liposomes may be interacting with a blood component with a high affinity for bone marrow endothelium. In Keenan and co-worker’s research, nebulized CsA at a dose of up to about 4.0 mg/kg was administered to lung transplant patients for 6 months with no reports of nephro- or hepatotoxicity, while oral CsA at a similar dose for asthma treatment led to severe nephro- and hepatotoxicity [
46]. These clinical studies demonstrated that the lower systemic CsA concentration might produce a lower incidence of adverse events and that investigated CsA systems might provide an interesting therapy with a safety margin for treatment. The absorption rate of cyclosporine from the microemulsion is strictly connected with the drug solubility in the medium. CsA is usually solubilized in hydrophilic emulsifiers, but its tendency to diffuse from the solid lipid matrix is lower compared to the oily system (microemulsion and emulsion) [
17].
3.6.2. Ocular CsA Delivery System
In ophthalmology, CsA use was investigated as early as 1981, initially for administration after corneal graft transplantation [
45,
46,
47]. The eye possesses extremely efficient defense protective mechanisms (e.g., blinking, tearing, and tear film turnover) against external threats. However, such mechanisms allow the clearing of the ocular surface but also lead to poor drug penetration for topically applied drugs, typically below 5% [
47,
48]. From the time of CsA’s first use, several drug delivery strategies have been investigated to enhance its ocular bioavailability to achieve and/or improve disease management without the undesirable effects connected with oral administration. These include CsA nanoemulsion systems for the treatment of dry eye disease [
47,
48,
49].
The addition of sodium hyaluronate is an interesting solution for an ocular drug delivery system because it increases the viscosity of the emulsion, resulting in a longer residence time on the ocular surface than that of aqueous drops. For CsA ophthalmic applications, similarly as for oral drugs, the cationic o/w nanoemulsions act efficiently. Due to the net positive charge, the residence time and the ocular bioavailability of CsA are higher than with other formulations. For example, in a pharmacokinetic study on rabbit eyes, corneal exposure to CsA after a single dose was 1.84-times greater for CsA cationic emulsion than for anionic emulsion [
50]. Correspondingly, the corneal clearance of CsA was 57% less for the CsA cationic emulsion than for analogous anionic emulsion (0.8 g/h and 1.4 g/h, respectively). The residence time of the CsA cationic nanoemulsion was greater as an effect of electrostatic interactions between the positively charged droplets and the negatively charged mucus protein of the corneal epithelium [
49].
Kuwano et al. [
51] compared the pharmacokinetics of three CsA ocular formulations in rabbit eyes: a castor oil solution, a micellar solution, and an oil-in-water emulsion. This study reported that the AUC parameters describing the drug concentration in the period of 0–12 h of CsA emulsion and CsA micellar solution were, respectively, 9.2- and 28.5-fold higher than the same parameters of oil-CsA in the corneal stroma endothelium. The same trend was observed in the bulbar conjunctiva, wherein these parameters for analogous systems were 2.4-fold and 5.1-fold higher than the parameters of Oil-CsA, respectively [
51]. Chitosan use is also reasonable in ophthalmology because this polysaccharide and well-known cationic biopolymer possesses mucus-like bio adhesive properties and a favorable ocular tolerance [
52]. In colloidal system studies, Campos et al. described chitosan as a potential carrier for CsA [
53]. Basaran et al. [
54] made nanoparticles of chitosan by spray drying a hydro-ethanolic solution of CsA and chitosan; the nanoparticles were re-suspended in water and sterilized by autoclaving. A pharmacokinetic study after a single instillation of 500 μL in sheep eyes showed that the approximate CsA concentrations in the aqueous and vitreous humors were 30–40 ng/mL [
54]. In humans, the average drop size of typical commercial ophthalmic solutions is about 40 μL [
55]. So, although the concept of chitosan nanoparticles as CsA delivery vehicles appears promising, additional data and comparative studies demonstrating their efficacy and safety are required.
Shen et al. [
56] undertook an extensive pharmacokinetic study in rabbit ocular tissues. They demonstrated that these nanoparticles could deliver therapeutic tissue concentrations of CsA (cornea, conjunctiva, and iris-ciliary body) for up to 24 h post-installation. Li et al. [
57] investigated a CsA-loaded liposomal formulation coated with low-molecular-weight chitosan to confer bio-adhesiveness to liposomes. A pharmacokinetic evaluation in rabbits (single ocular administration of 100 μL) showed that the chitosan-coated particles resulted in a significantly greater CsA penetration, especially into the cornea, compared with non-coated liposomes. A safety study of liposomes suspended in a carbomer solution with a final CsA concentration of 0.2%, described by Mosallaei et al., showed no significant toxicity [
58].
New solutions include implant solid dosage forms that act as reservoirs for the sustained release of active ingredients. Presently, both biodegradable and non-biodegradable implants are available that can be implanted in several sites within the eye. Acton et al. [
59] tested a sub-conjunctival silicone implant of CsA in a red wolf. A novel approach was described by Eperon et al. [
60], wherein a PLGA CsA- and triamcinolone-loaded implant was attached to an intraocular lens. They concluded that the implants retained 60% of the CsA load, suggesting that these devices could support long-lasting CsA release for several additional months.
This approach is quite promising because it uses a safe polymer, the device can be sterilized by radiation, and it is implanted at the time of surgery. For two decades, more than 50 different approaches for the ocular delivery of CsA have been described. CsA remains a challenging compound to formulate in a suitable dosage form to devise an optimal ocular system [
61].
3.6.3. Skin CsA Delivery System
Essaghraoui et al. described interesting studies of skin CsA delivery systems. Two different nanocarriers were used and optimized: solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) where oleic acid was the liquid lipid. The developed nanoparticles showed mean sizes around 200 nm, a negative surface charge, as well as drug entrapment efficiencies around 85% and 70% for SLNs and NLCs, respectively [
62]. The amount of drug released from the lipid nanoparticles was evaluated using an in vitro dialysis bag technique to estimate the in vivo kinetics [
63] under different pH and temperature conditions to simulate physiological (pH 7.4, 37 °C) and skin (pH 5.5, 32 °C) environments over a period of 24 h. Under skin environmental conditions, less than 10% of the drug was released within the first 2 h, reaching about 25% and 35% after 6 h at 32 °C for SLNs and NLCs, respectively.
Evaluating the skin penetration and permeation kinetics are crucial steps in the characterization of formulations for skin administration [
64,
65,
66]. The assessment was performed using Franz diffusion cells, and pig ear skin was chosen as a model barrier due to its similarity, in morphology and function, to its human counterpart. CsA delivery from the control formulation (0.5 mg mL
−1 solution in olive oil) was 10- to 5-fold lower than that from the lipid nanoparticles (0.27 ± 0.03 µg/cm
2) at an equivalent CsA content. The CsA release kinetic profiles from both lipid nanoparticles in different pHs (7.4 and 5) were studied by analyzing the regression coefficients (r
2) and the adjusted correlation coefficient (r
2 and r
2adj) obtained after fitting into first order, Hixson–Crowell, Higuchi, and Korsmeyer–Peppas release kinetics models.
The last model was found to be the best. This mathematical model proposes a diffusion-controlled drug release profile for lipid nanoparticles [
65,
66] and elucidates the mechanism by interpretation of its release exponent (
n) [
65,
66,
67,
68]. In summing up, the developed SLN and NLC improved the drug’s aqueous solubility up to 95-fold. Skin permeation assays demonstrated that the lipid nanoparticles increased the topical bioavailability of CsA, and SLN evidenced lower cytotoxicity and transdermal permeation than NLC. Thus, the application of SLN was revealed to be the most promising targeted skin local delivery system for peptides [
63].








{kind=link}
{kind=link}
{kind=link}
{kind=link}