Mass Spectrometry-Based Methodologies for Targeted and Untargeted Identification of Protein Covalent Adducts (Adductomics): Current Status and Challenges

 and
and
Abstract
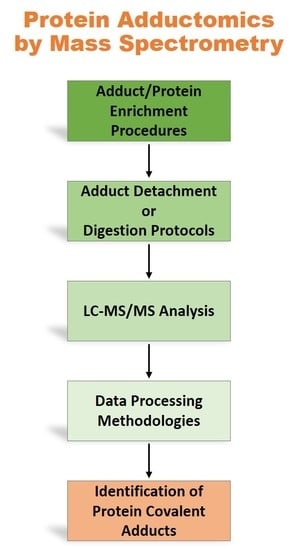
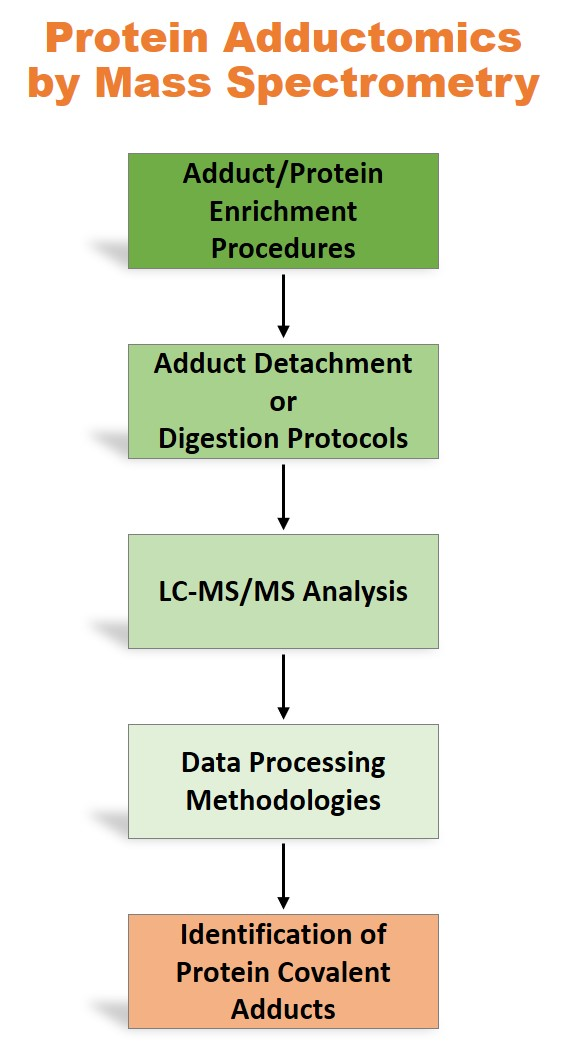
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
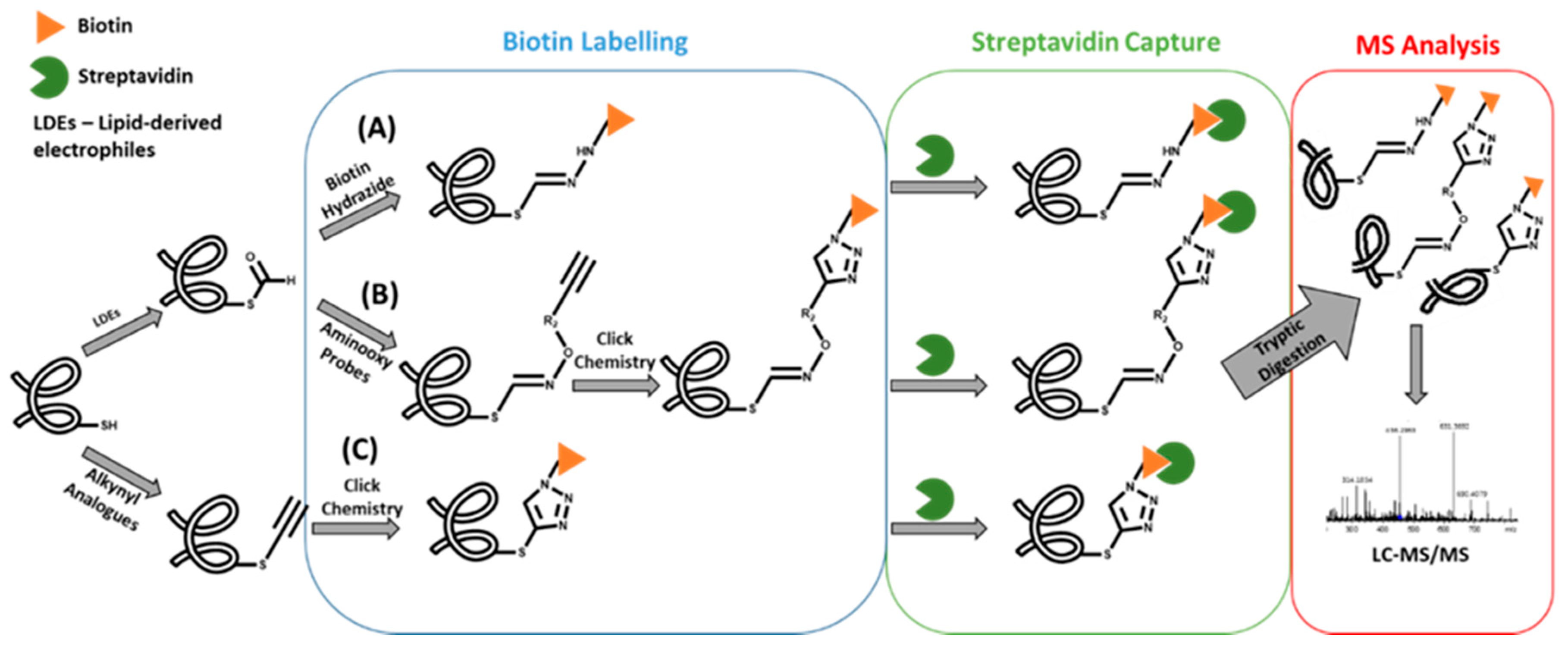
2. Sample pre-treatment and adducts enrichment
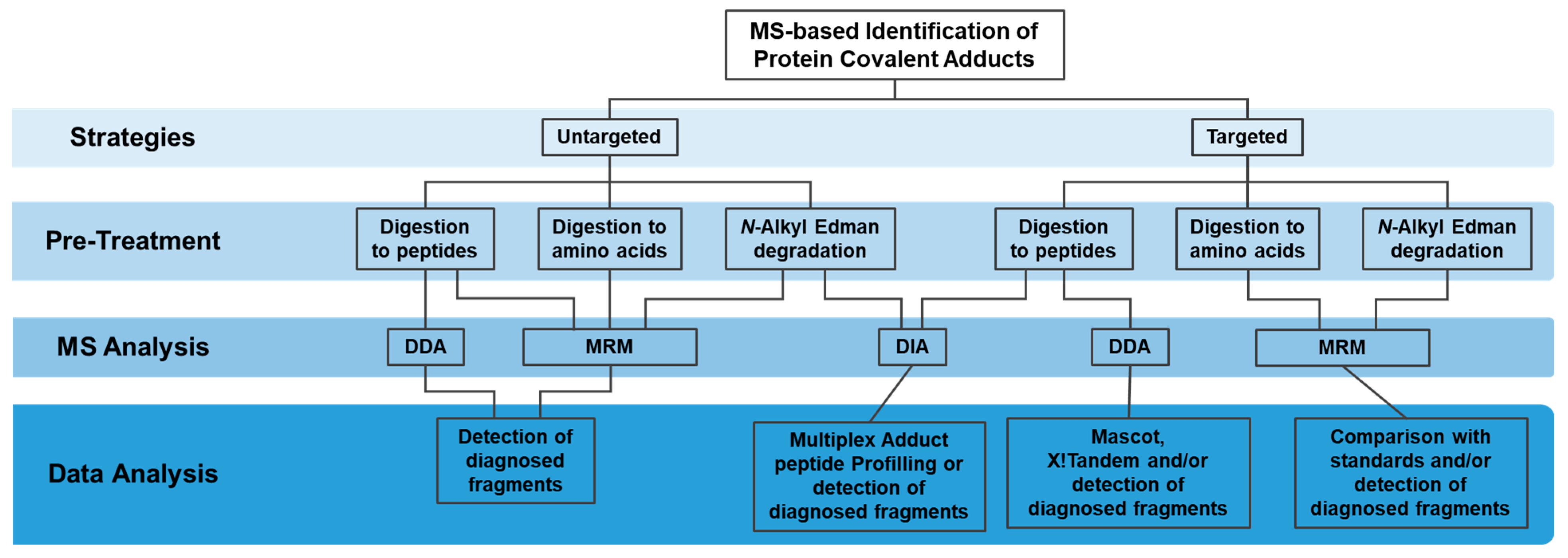
3. MS Data Acquisition
4. MS Data Processing and Protein Covalent Adducts Identification
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BChE | butyrylcholinesterase |
| DDA | data dependent analysis |
| DIA | data independent analysis |
| FITC | fluorescein isothiocyanate |
| Hb | hemoglobin |
| HRMS | high resolution mass spectrometry |
| HAS | Human serum albumin |
| LDEs | lipid-derived electrophiles |
| MDI | 4,4′-methylenediphenyl diisocyanate |
| MRM | multiple reaction monitoring |
| SRM | selected reaction monitoring |
References
- Barone, E.; Head, E.; Butterfield, D.A.; Perluigi, M. HNE-modified proteins in Down syndrome: Involvement in development of Alzheimer disease neuropathology. Free Radic. Biol. Med. 2017, 111, 262–269. [Google Scholar] [CrossRef]
- Xiao, L.; Patrick, D.M.; Aden, L.A.; Kirabo, A. Mechanisms of isolevuglandin-protein adduct formation in inflammation and hypertension. Prostaglandins Other Lipid Mediat. 2018, 139, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Antoniak, D.T.; Duryee, M.J.; Mikuls, T.R.; Thiele, G.M.; Anderson, D.R. Aldehyde-modified proteins as mediators of early inflammation in atherosclerotic disease. Free Radic. Biol. Med. 2015, 89, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Jaeschke, H. Mechanisms of Inflammatory Liver Injury and Drug-Induced Hepatotoxicity. Curr. Pharmacol. Rep. 2018, 4, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Hardt, U.; Larsson, A.; Gunnarsson, I.; Clancy, R.M.; Petri, M.; Buyon, J.P.; Silverman, G.J.; Svenungsson, E.; Grönwall, C. Autoimmune reactivity to malondialdehyde adducts in systemic lupus erythematosus is associated with disease activity and nephritis. Arthritis Res. Ther. 2018, 20, 36. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T. Reactions of electrophiles with nucleophilic thiolate sites: relevance to pathophysiological mechanisms and remediation. Free Radic. Res. 2016, 50, 195–205. [Google Scholar] [CrossRef]
- Escher, B.I.; Hackermüller, J.; Polte, T.; Scholz, S.; Aigner, A.; Altenburger, R.; Böhme, A.; Bopp, S.K.; Brack, W.; Busch, W.; et al. From the exposome to mechanistic understanding of chemical-induced adverse effects. Environ. Int. 2017, 99, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, S.M. Redefining environmental exposure for disease etiology. NPJ Syst. Biol. Appl. 2018, 4, 30. [Google Scholar] [CrossRef]
- Nunes, J.; Martins, I.L.; Charneira, C.; Pogribny, I.P.; de Conti, A.; Beland, F.A.; Marques, M.M.; Jacob, C.C.; Antunes, A.M.M. New insights into the molecular mechanisms of chemical carcinogenesis: in vivo adduction of histone H2B by a reactive metabolite of the chemical carcinogen furan. Toxicol. Lett. 2016, 264, 106–113. [Google Scholar] [CrossRef]
- Sabbioni, G.; Turesky, R.J. Biomonitoring Human Albumin Adducts: The Past, the Present, and the Future. Chem. Res. Toxicol. 2017, 30, 332–366. [Google Scholar] [CrossRef]
- Hammond, T.G.; Meng, X.; Jenkins, R.E.; Maggs, J.L.; Castelazo, A.S.; Regan, S.L.; Bennett, S.N.; Earnshaw, C.J.; Aithal, G.P.; Pande, I.; Kenna, J.G.; Stachulski, A.V.; Park, B.K.; Williams, D.P. Mass spectrometric characterization of circulating covalent protein adducts derived from a drug acyl glucuronide metabolite: multiple albumin adductions in diclofenac patients. J. Pharmacol. Exp. Ther. 2014, 350, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Lee, S.H.; Oe, T. An LC/ESI-SRM/MS method to screen chemically modified hemoglobin: simultaneous analysis for oxidized, nitrated, lipidated, and glycated sites. Anal. Bioanal. Chem. 2016, 408, 5379–5392. [Google Scholar] [CrossRef]
- Soreghan, B.A.; Yang, F.; Thomas, S.N.; Hsu, J.; Yang, A.J. High-throughput proteomic-based identification of oxidatively induced protein carbonylation in mouse brain. Pharm. Res. 2003, 20, 1713–1720. [Google Scholar] [CrossRef]
- Vila, A.; Tallman, K.A.; Jacobs, A.T.; Liebler, D.C.; Porter, N.A.; Marnett, L.J. Identification of protein targets of 4-hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 2008, 21, 432–444. [Google Scholar] [CrossRef]
- Kim, H.Y.; Tallman, K.A.; Liebler, D.C.; Porter, N.A. An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Mol. Cell. Proteomics 2009, 8, 2080–2089. [Google Scholar] [CrossRef]
- Spiess, P.C.; Deng, B.; Hondal, R.J.; Matthews, D.E.; van der Vliet, A. Proteomic profiling of acrolein adducts in human lung epithelial cells. J. Proteomics 2011, 74, 2380–2394. [Google Scholar] [CrossRef]
- Codreanu, S.G.; Kim, H.Y.; Porter, N.A.; Liebler, D.C. Biotinylated probes for the analysis of protein modification by electrophiles. Methods Mol. Biol. 2012, 803, 77–95. [Google Scholar]
- Chen, Y.; Cong, Y.; Quan, B.; Lan, T.; Chu, X.; Ye, Z.; Hou, X.; Wang, C. Chemoproteomic profiling of targets of lipid-derived electrophiles by bioorthogonal aminooxy probe. Redox Biol. 2017, 12, 712–718. [Google Scholar] [CrossRef]
- Windsor, K.; Genaro-Mattos, T.C.; Miyamoto, S.; Stec, D.F.; Kim, H.Y.; Tallman, K.A.; Porter, N.A. Assay of protein and peptide adducts of cholesterol ozonolysis products by hydrophobic and click enrichment methods. Chem. Res. Toxicol. 2014, 27, 1757–1768. [Google Scholar] [CrossRef]
- Huang, Z.; Ogasawara, D.; Seneviratne, U.I.; Cognetta, A.B., 3rd; Am Ende, C.W.; Nason, D.M.; Lapham, K.; Litchfield, J.; Johnson, D.S.; Cravatt, B.F. Global Portrait of Protein Targets of Metabolites of the Neurotoxic Compound BIA 10-2474. ACS Chem. Biol. 2019, 14, 192–197. [Google Scholar] [CrossRef]
- Lin, P.H.; Yang, H.J.; Hsieh, W.C.; Lin, C.; Chan, Y.C.; Wang, Y.F.; Yang, Y.T.; Lin, K.J.; Lin, L.S.; Chen, D.R. Albumin and hemoglobin adducts of estrogen quinone as biomarkers for early detection of breast cancer. PLoS ONE 2018, 13, e0201241. [Google Scholar] [CrossRef]
- Meier, B.W.; Gomez, J.D.; Zhou, A.; Thompson, J.A. Immunochemical and proteomic analysis of covalent adducts formed by quinone methide tumor promoters in mouse lung epithelial cell lines. Chem. Res. Toxicol. 2005, 18, 1575–1585. [Google Scholar] [CrossRef]
- Bellamri, M.; Wang, Y.; Yonemori, K.; White, K.K.; Wilkens, L.R.; Le Marchand, L.; Turesky, R. Biomonitoring an albumin adduct of the cooked meat carcinogen 2-amino-1-methyl-6-phenylimidazo [4,5-b]pyridine in humans. Carcinogenesis 2018, 39, 1455–1462. [Google Scholar] [CrossRef]
- Wang, Y.; Villalta, P.W.; Peng, L.; Dingley, K.; Malfatti, M.A.; Turteltaub, K.W.; Turesky, R.J. Mass Spectrometric Characterization of an Acid-Labile Adduct Formed with 2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and Albumin in Humans. Chem. Res. Toxicol. 2017, 30, 705–714. [Google Scholar] [CrossRef]
- Pastorelli, R.; Guanci, M.; Cerri, A.; Negri, E.; La Vecchia, C.; Fumagalli, F.; Mezzetti, M.; Cappelli, R.; Panigalli, T.; Fanelli, R.; Airoldi, L. Impact of inherited polymorphisms in glutathione S-transferase M1, microsomal epoxide hydrolase, cytochrome P450 enzymes on DNA, and blood protein adducts of benzo(a)pyrene-diolepoxide. Cancer Epidemiol. Biomarkers Prev. 1998, 7, 703–709. [Google Scholar]
- Pastorelli, R.; Catenacci, G.; Guanci, M.; Fanelli, R.; Valoti, E.; Minoia, C.; Airoldi, L. 3,4 Dichloroaniline haemoglobin adducts in humans: preliminary data on agricultural workers exposed to propanil. Biomarkers 1998, 3, 227–233. [Google Scholar] [CrossRef]
- Sabbioni, G.; Dongari, N.; Sepai, O.; Kumar, A. Determination of albumin adducts of 4,4’-methylenediphenyl diisocyanate in workers of a 4,4’-methylenedianiline factory. Biomarkers. 2016, 4, 1–8. [Google Scholar] [CrossRef]
- McCoy, L.F.; Scholl, P.F.; Schleicher, R.L.; Groopman, J.D.; Powers, C.D.; Pfeiffer, C.M. Analysis of aflatoxin B1-lysine adduct in serum using isotope-dilution liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2203–2210. [Google Scholar] [CrossRef]
- Carlsson, H.; Rappaport, S.M.; Törnqvist, M. Protein Adductomics: Methodologies for Untargeted Screening of Adducts to Serum Albumin and Hemoglobin in Human Blood Samples. High Throughput 2019, 8, E6. [Google Scholar] [CrossRef]
- Funk, W.E.; Li, H.; Iavarone, A.T.; Williams, E.R.; Riby, J.; Rappaport, S.M. Enrichment of cysteinyl adducts of human serum albumin. Anal. Biochem. 2010, 400, 61–68. [Google Scholar] [CrossRef]
- Chung, M.K.; Grigoryan, H.; Iavarone, A.T.; Rappaport, S.M. Antibody enrichment and mass spectrometry of albumin-Cys34 adducts. Chem. Res. Toxicol. 2014, 27, 400–407. [Google Scholar] [CrossRef]
- Grigoryan, H.; Edmands, W.; Lu, S.S.; Yano, Y.; Regazzoni, L.; Iavarone, A.T.; Williams, E.R.; Rappaport, S.M. Adductomics Pipeline for Untargeted Analysis of Modifications to Cys34 of Human Serum Albumin. Anal. Chem. 2016, 88, 10504–10512. [Google Scholar] [CrossRef]
- Yano, Y.; Grigoryan, H.; Schiffman, C.; Edmands, W.; Petrick, L.; Hall, K.; Whitehead, T.; Metayer, C.; Dudoit, S.; Rappaport, S.M. Untargeted adductomics of Cys34 modifications to human serum albumin in newborn dried blood spots. Anal. Bional. Chem. 2019. [Epub ahead of print]. [Google Scholar] [CrossRef]
- Shibata, T.; Shimizu, K.; Hirano, K.; Nakashima, F.; Kikuchi, R.; Matsushita, T.; Uchida, K. Adductome-based identification of biomarkers for lipid peroxidation. J. Biol. Chem. 2017, 292, 8223–8235. [Google Scholar] [CrossRef]
- Yoshitake, J.; Shibata, T.; Shimayama, C.; Uchida, K. 2-Alkenal modification of hemoglobin: Identification of a novel hemoglobin-specific alkanoic acid-histidine adduct. Redox Biol. 2019, 101115. [Google Scholar] [CrossRef]
- Grilo, N.M.; Antunes, A.M.M.; Caixas, U.; Marinho, A.T.; Charneira, C.; Oliveira, M.C.; Marques, M.M.; Pereira, S.A. Monitoring abacavir bioactivation in humans: screening for an aldehyde metabolite. Toxicol Lett. 2013, 219, 59–64. [Google Scholar] [CrossRef]
- Charneira, C.; Grilo, N.M.; Pereira, S.A.; Godinho, A.L.A.; Monteiro, E.C.; Marques, M.M.; Antunes, A.M.M. N-terminal valine adduct from the anti-HIV drug abacavir in rat hemoglobin as evidence for abacavir metabolism to a reactive aldehyde in vivo. Br. J. Pharmacol. 2012, 167, 1353–1361. [Google Scholar] [CrossRef]
- Caixas, U.; Antunes, A.M.M.; Marinho, A.T.; Godinho, A.L.; Grilo, N.M.; Marques, M.M.; Oliveira, M.C.; Branco, T.; Monteiro, E.C.; Pereira, S.A. Evidence for nevirapine bioactivation in man: searching for the first step in the mechanism of nevirapine toxicity. Toxicology. 2012, 301, 33–39. [Google Scholar] [CrossRef]
- Törnqvist, M.; Osterman-Golkar, S.; Kautiainen, A.; Jensen, S.; Farmer, P.B.; Ehrenberg, L. Tissue doses of ethylene oxide in cigarette smokers determined from adduct levels in hemoglobin. Carcinogenesis 1986, 7, 1519–1521. [Google Scholar] [CrossRef]
- Bergmark, E.; Calleman, C.J.; He, F.S.; Costa, L.G. Determination of Hemoglobin Adducts in Humans Occupationally Exposed to Acrylamide. Toxicol. Appl. Pharmacol. 1993, 120, 45–54. [Google Scholar] [CrossRef]
- Bergmark, E. Hemoglobin Adducts of Acrylamide and Acrylonitrile in LaboratoryWorkers, Smokers and Nonsmokers. Chem. Res. Toxicol. 1997, 10, 78–84. [Google Scholar] [CrossRef]
- Tareke, E.; Rydberg, P.; Karlsson, P.; Eriksson, S.; Törnqvist, M. Analysis of acrylamide, a carcinogen formed in heated foodstuffs. J. Agric. Food Chem. 2002, 50, 4998–5006. [Google Scholar] [CrossRef]
- Carlsson, H.; Von Stedingk, H.; Nilsson, U.; Törnqvist, M. LC-MS/MS screening strategy for unknown adducts to N-terminal valine in hemoglobin applied to smokers and nonsmokers. Chem. Res. Toxicol. 2014, 27, 2062–2070. [Google Scholar] [CrossRef]
- Gesslbauer, B.; Kuerzl, D.; Valpatic, N.; Bochkov, V.N. Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry. Antioxidants 2018, 7, E116. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, Y. Comprehensive analysis of protein modifications by top-down mass spectrometry. Circ. Cardiovasc. Genet. 2011, 4, 711. [Google Scholar] [CrossRef]
- Gan, J.; Zhang, H.; Humphreys, W.G. Drug-Protein Adducts: Chemistry, Mechanisms of Toxicity, and Methods of Characterization. Chem. Res. Toxicol. 2016, 29, 2040–2057. [Google Scholar] [CrossRef]
- Pathak, K.V.; Bellamri, M.; Wang, Y.; Langouët, S.; Turesky, R.-J. 2-Amino-9H-pyrido[2,3-b]indole (AαC) Adducts and Thiol Oxidation of Serum Albumin as Potential Biomarkers of Tobacco Smoke. J. Biol. Chem. 2015, 290, 16304–16318. [Google Scholar] [CrossRef]
- Peng, L.; Dasari, S.; Tabb, D.L.; Turesky, R.J. Mapping serum albumin adducts of the food-borne carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by data-dependent tandem mass spectrometry. Chem. Res. Toxicol. 2012, 25, 2179–2193. [Google Scholar] [CrossRef]
- Mathews, T.P.; Carter, M.D.; Johnson, D.; Isenberg, S.L.; Graham, L.A.; Thomas, J.D.; Johnson, R.C. High-Confidence Qualitative Identification of Organophosphorus Nerve Agent Adducts to Human Butyrylcholinesterase. Anal. Chem. 2017, 89, 1955–1964. [Google Scholar] [CrossRef]
- Michalski, A.; Cox, J.; Mann, M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 2011, 10, 1785–1793. [Google Scholar] [CrossRef]
- Meng, X.; Jenkins, R.E.; Berry, N.G.; Maggs, J.L.; Farrell, J.; Lane, C.S.; Stachulski, A.V.; French, N.S.; Naisbitt, D.J.; Pirmohamed, M.; Park, B.K. Direct evidence for the formation of diastereoisomeric benzylpenicilloyl haptens from benzylpenicillin and benzylpenicillenic acid in patients. J. Pharmacol. Exp. Ther. 2011, 338, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Howarth, A.; Earnshaw, C.J.; Jenkins, R.E.; French, N.S.; Back, D.J.; Naisbitt, D.J.; Park, B.K. Detection of drug bioactivation in vivo: mechanism of nevirapine-albumin conjugate formation in patients. Chem. Res. Toxicol. 2013, 26, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lawrenson, A.S.; Berry, N.G.; Maggs, J.L.; French, N.S.; Back, D.J.; Khoo, S.H.; Naisbitt, D.J.; and Park, B.K. Abacavir forms novel cross-linking abacavir protein adducts in patients. Chem. Res. Toxicol. 2014, 27, 524–535. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.; Shiao, T.C.; Roy, R.; Sleno, L. Absolute quantitation of NAPQI-modified rat serum albumin by LC-MS/MS: monitoring acetaminophen covalent binding in vivo. Chem. Res. Toxicol. 2014, 27, 1632–1639. [Google Scholar] [CrossRef] [PubMed]
- Sabbioni, G.; Vanimireddy, L.R.; Lummus, Z.L.; Bernstein, D.I. Comparison of biological effects with albumin adducts of 4,4’-methylenediphenyl diisocyanate in workers. Arch. Toxicol. 2017, 91, 1809–1814. [Google Scholar] [CrossRef]
- Aldini, G.; Regazzoni, L.; Orioli, M.; Rimoldi, I.; Facino, R.M.; Carini, M. A tandem MS precursor-ion scan approach to identify variable covalent modification of albumin Cys34: a new tool for studying vascular carbonylation. J. Mass Spectrom. 2008, 43, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Witort, E.; Capaccioli, S.; Becatti, M.; Fiorillo, C.; Batignani, G.; Pavoni, V.; Piccini, M.; Orioli, M.; Carini, M.; Aldini, G.; Lulli, M. Albumin Cys34 adducted by acrolein as a marker of oxidative stress in ischemia-reperfusion injury during hepatectomy. Free Radic. Res. 2016, 50, 831–839. [Google Scholar] [CrossRef]
- Carlsson, H.; Aasa, J.; Kotova, N.; Vare, D.; Sousa, P.F.M.; Rydberg, P.; Abramsson-Zetterberg, L.; Törnqvist, M. Adductomic Screening of Hemoglobin Adducts and Monitoring of Micronuclei in School-Age Children. Chem. Res. Toxicol. 2017, 30, 1157–1167. [Google Scholar] [CrossRef]
- Law, K.P.; Lim, Y.P. Recent advances in mass spectrometry: data independent analysis and hyper reaction monitoring. Expert Rev. Proteomics 2013, 10, 551–566. [Google Scholar] [CrossRef]
- Bruderer, R.; Bernhardt, O.M.; Gandhi, T.; Miladinović, S.M.; Cheng, L.Y.; Messner, S.; Ehrenberger, T.; Zanotelli, V.; Butscheid, Y.; Escher, C.; Vitek, O.; Rinner, O.; Reiter, L. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol. Cell. Proteomics 2015, 14, 1400–1410. [Google Scholar] [CrossRef]
- Porter, C.J.; Bereman, M.S. Data-independent-acquisition mass spectrometry for identification of targeted-peptide site-specific modifications. Anal. Bioanal. Chem. 2015, 407, 6627–6635. [Google Scholar] [CrossRef]
- Zhang, H.; Gan, J.; Shu, Y.Z.; Humphreys, W.G. High-resolution mass spectrometry-based background subtraction for identifying protein modifications in a complex biological system: detection of acetaminophen-bound microsomal proteins including argininosuccinate synthetase. Chem. Res. Toxicol. 2015, 28, 775–781. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Beavis, R.C. Using the global proteome machine for protein identification. Methods Mol. Biol. 2006, 328, 217–228. [Google Scholar]
- Craig, R.; Beavis, R.C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Han, Y.; Ma, B.; Zhang, K. SPIDER: software for protein identification from sequence tags with de novo sequencing error. Proc. IEEE Comput. Syst. Bioinform. Conf. 2004, 206–215. [Google Scholar] [CrossRef]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat. Methods. 2017, 14, 513–520. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, W.; Cobb, M.H.; Zhao, Y. PTMap--a sequence alignment software for unrestricted, accurate, and full-spectrum identification of post-translational modification sites. Proc. Natl. Acad. Sci. USA 2009, 106, 761–766. [Google Scholar] [CrossRef]
- Fu, Y.; Xiu, L.Y.; Jia, W.; Ye, D.; Sun, R.X.; Qian, X.H.; He, S.M. DeltAMT: a statistical algorithm for fast detection of protein modifications from LC-MS/MS data. Mol. Cell. Proteomics 2011, 10, M110.000455. [Google Scholar] [CrossRef]
- Savitski, M.M.; Nielsen, M.L.; Zubarev, R.A. ModifiComb, a new proteomic tool for mapping substoichiometric post-translational modifications, finding novel types of modifications, and fingerprinting complex protein mixtures. Mol. Cell. Proteomics 2006, 5, 935–948. [Google Scholar] [CrossRef]
- Horlacher, O.; Lisacek, F.; Müller, M. Mining Large Scale Tandem Mass Spectrometry Data for Protein Modifications Using Spectral Libraries. J. Proteome Res. 2016, 15, 721–731. [Google Scholar] [CrossRef]
- Egertson, J.D.; MacLean, B.; Johnson, R.; Xuan, Y.; MacCoss, M.J. Multiplexed peptide analysis using data-independent acquisition and Skyline. Nat. Protoc. 2015, 10, 887–903. [Google Scholar] [CrossRef]
- Tsou, C.C.; Avtonomov, D.; Larsen, B.; Tucholska, M.; Choi, H.; Gingras, A.C.; Nesvizhskii, A.I. DIA-Umpire: comprehensive computational framework for data independent acquisition proteomics. Nat. Methods 2015, 12, 258–264. [Google Scholar] [CrossRef]
- Ting, Y.S.; Egertson, J.D.; Bollinger, J.G.; Searle, B.C.; Payne, S.H.; Noble, W.S.; MacCoss, M.J. PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods 2017, 14, 903–908. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar] [CrossRef]
- Nunes, J.; Morello, J.; Nunes, C.; Gouveia-Fernandes, S.; Serpa, J.; Antunes, A.M.M. Biomarkers of chemically-induced cancer: the acrylamide case study. In Proceedings of the 12th National Organic Chemistry Meeting and 5th National Medicinal Chemistry Meeting, Coimbra, Portugal, January 2018. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, J.; Charneira, C.; Morello, J.; Rodrigues, J.; Pereira, S.A.; Antunes, A.M.M. Mass Spectrometry-Based Methodologies for Targeted and Untargeted Identification of Protein Covalent Adducts (Adductomics): Current Status and Challenges. High-Throughput 2019, 8, 9. https://0-doi-org.brum.beds.ac.uk/10.3390/ht8020009
Nunes J, Charneira C, Morello J, Rodrigues J, Pereira SA, Antunes AMM. Mass Spectrometry-Based Methodologies for Targeted and Untargeted Identification of Protein Covalent Adducts (Adductomics): Current Status and Challenges. High-Throughput. 2019; 8(2):9. https://0-doi-org.brum.beds.ac.uk/10.3390/ht8020009
Chicago/Turabian StyleNunes, João, Catarina Charneira, Judit Morello, João Rodrigues, Sofia A. Pereira, and Alexandra M. M. Antunes. 2019. "Mass Spectrometry-Based Methodologies for Targeted and Untargeted Identification of Protein Covalent Adducts (Adductomics): Current Status and Challenges" High-Throughput 8, no. 2: 9. https://0-doi-org.brum.beds.ac.uk/10.3390/ht8020009