Mediation of FoxO1 in Activated Neuroglia Deficient for Nucleoside Diphosphate Kinase B during Vascular Degeneration

 and
and 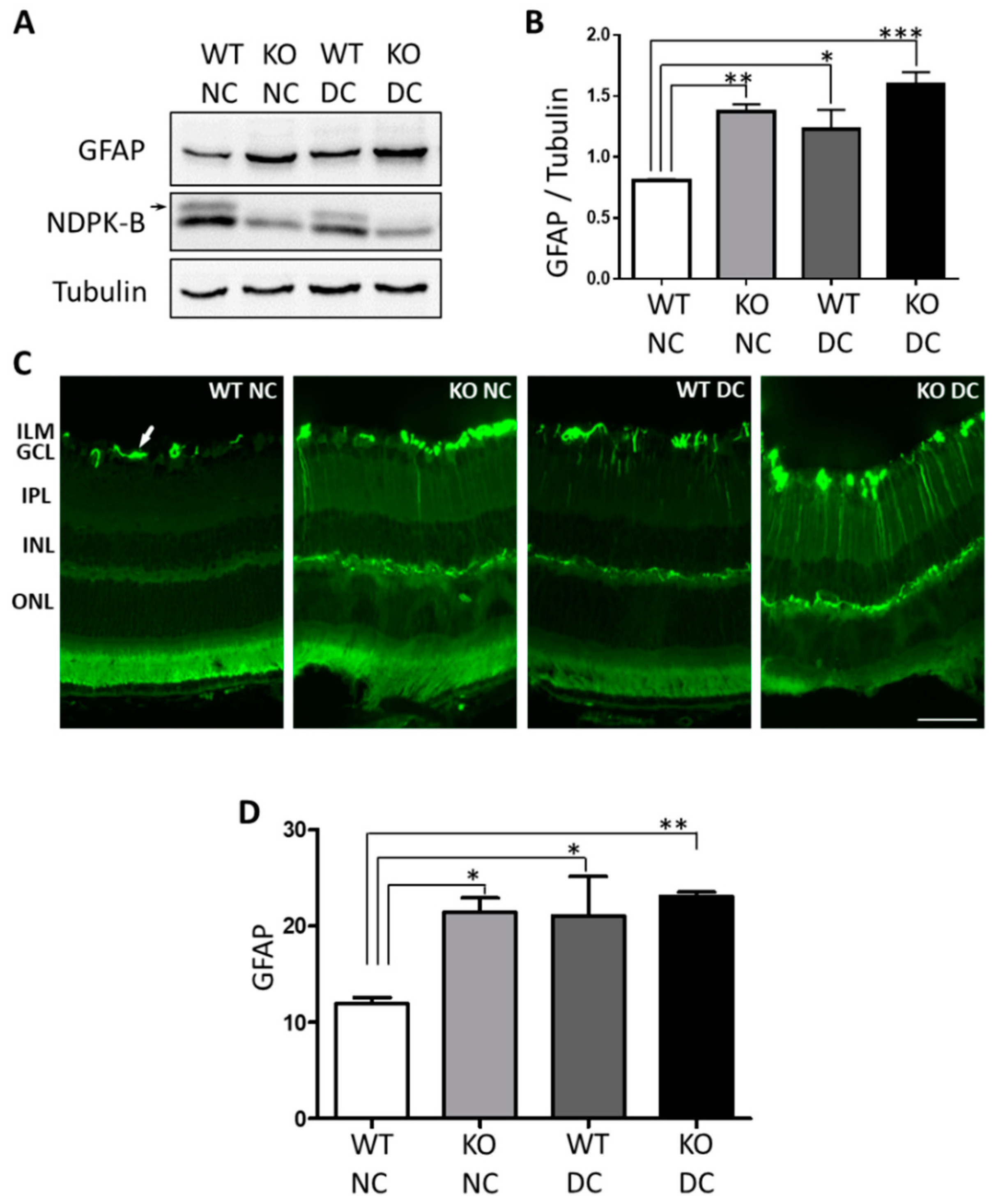{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Immunofluorescence and Quantification
2.3. Cell Culture and High Glucose Stimulation
2.4. siRNA Mediated FoxO1 Knockdown in Müller Cells
2.5. Western Blot
2.6. Quantitative Real Time PCR
2.7. Statistical Analysis
3. Results
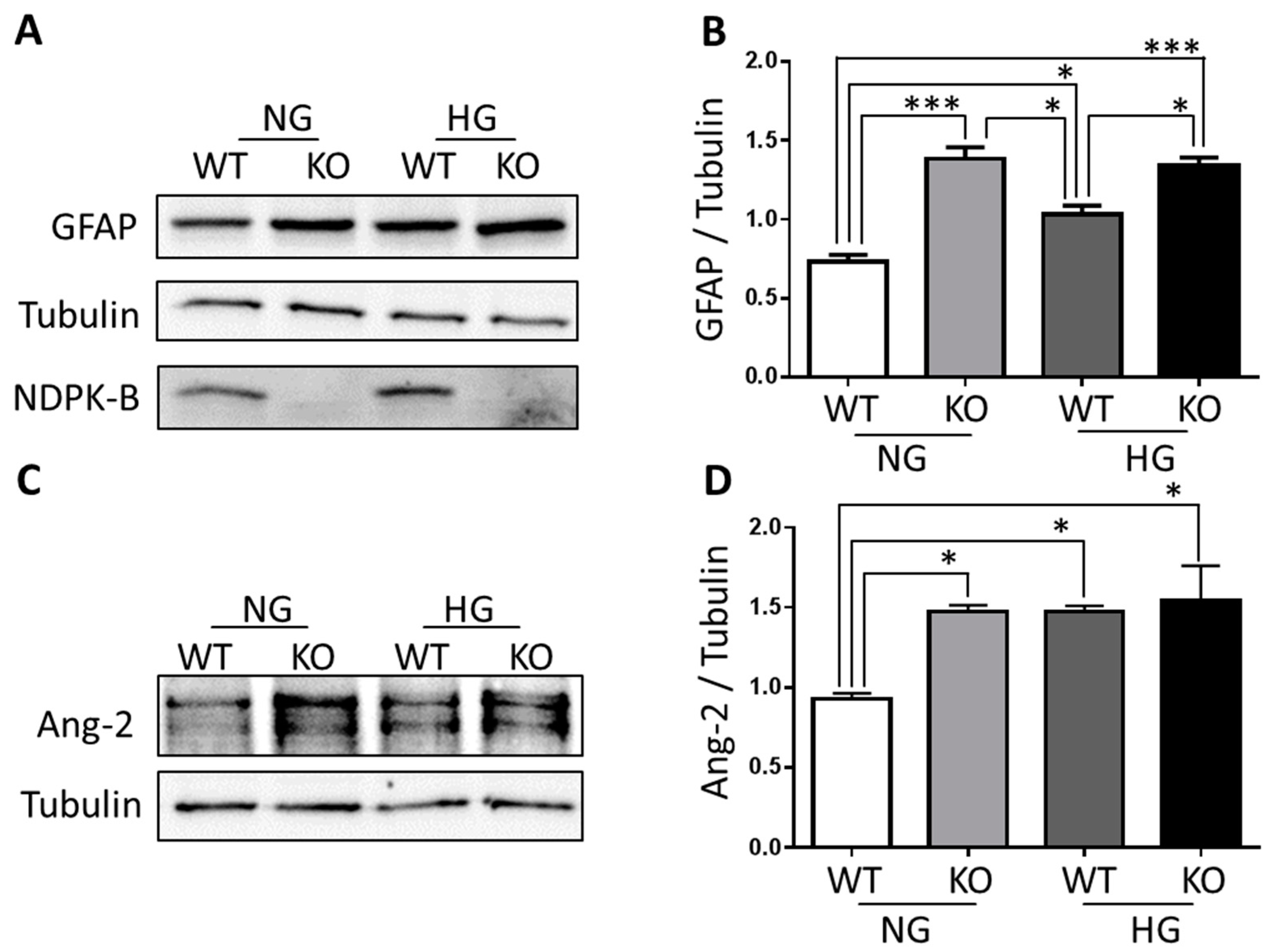
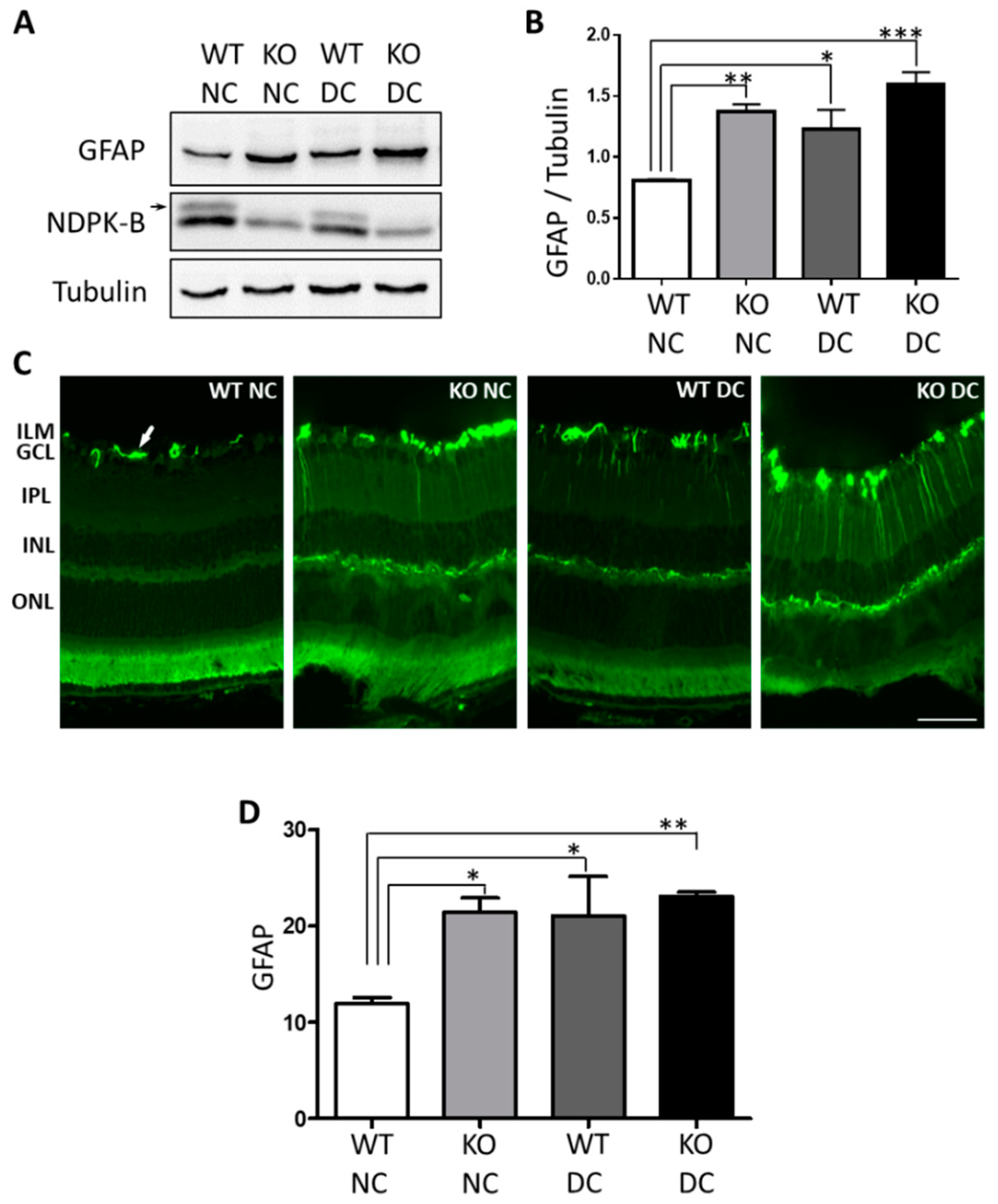
3.1. Müller Cells Are Activated in NDPK-B Deficient Retinas
3.2. Ang-2 Is Preferentially Upregulated in Müller Cells in the Retina during Vascular Degeneration
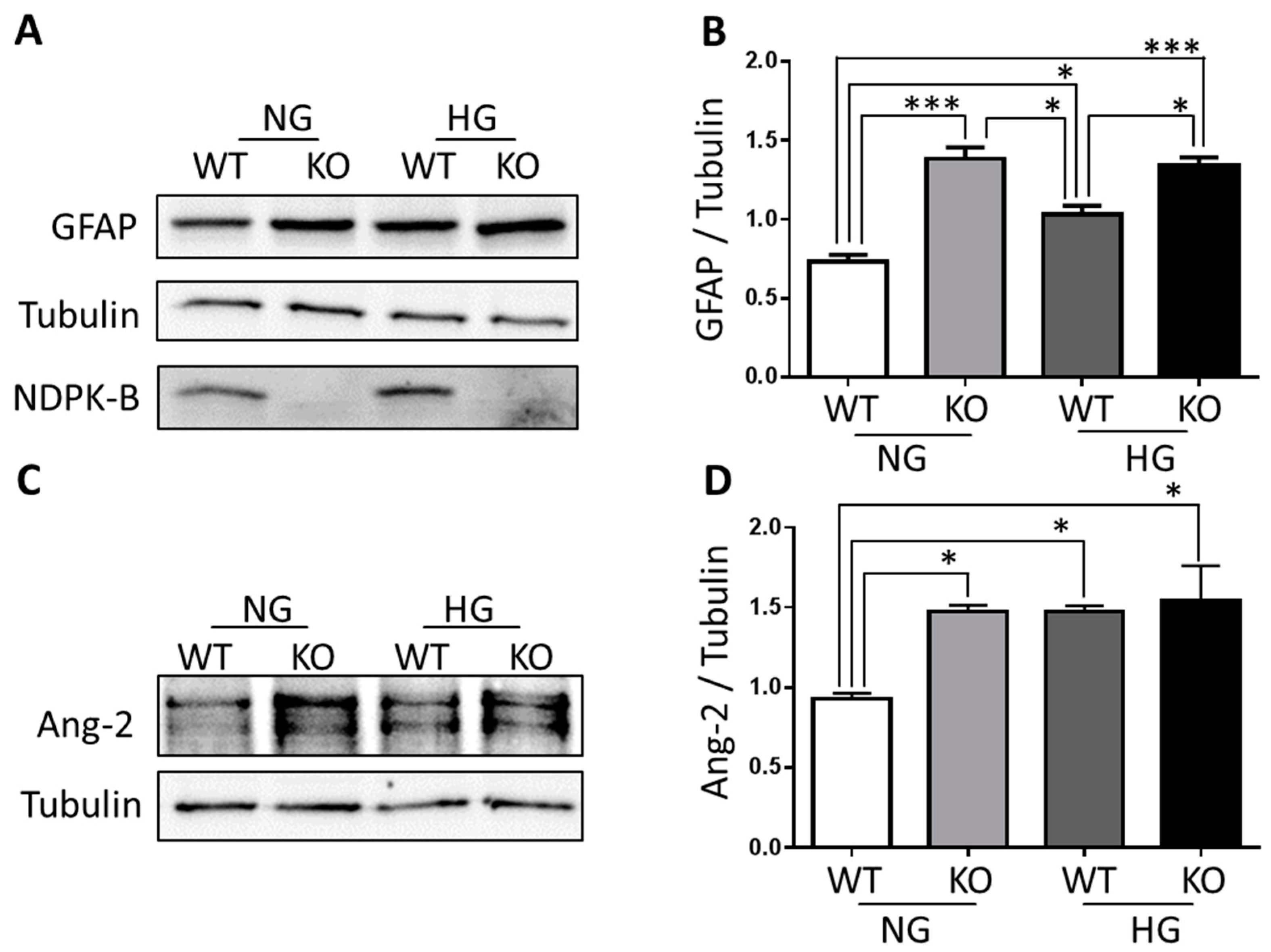
3.3. NDPK-B Deficiency Caused Enhancement of Ang-2 in Isolated Müller Cells
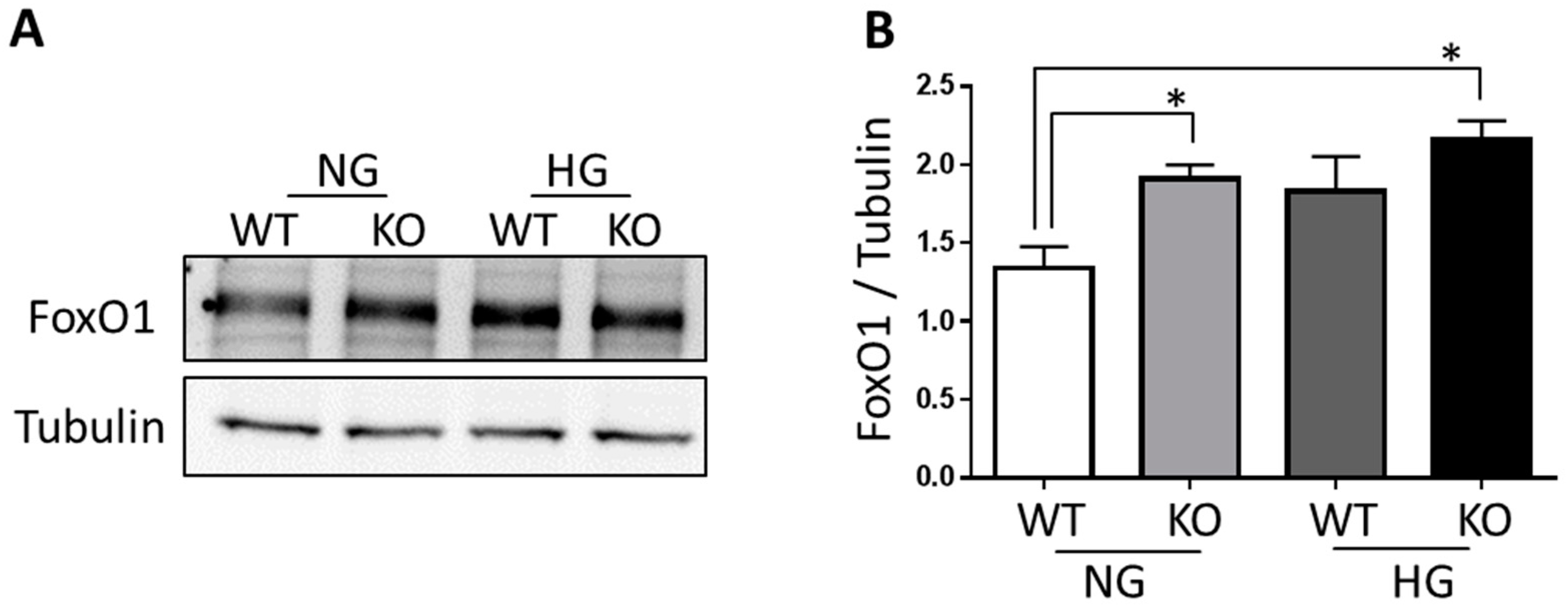
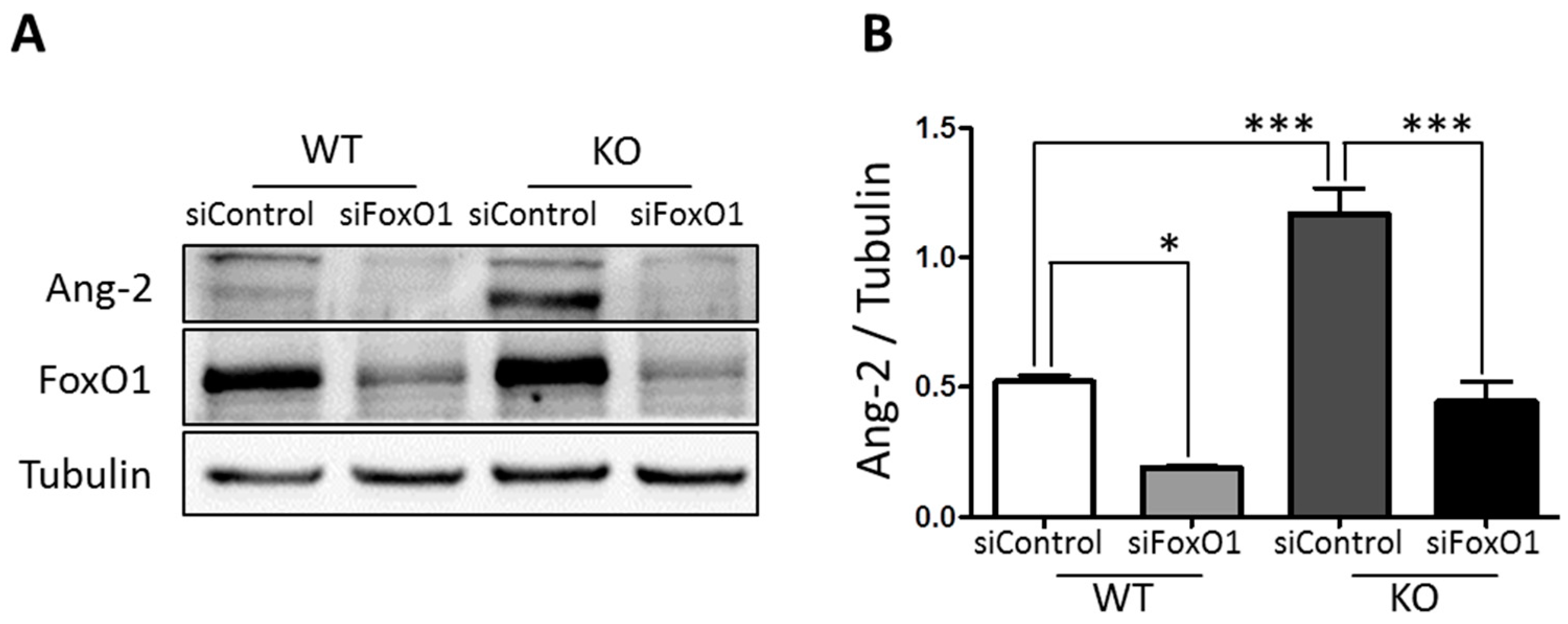
3.4. FoxO1 Is Required for NDPK-B Deficiency Induced Ang-2 Upregulation in Müller Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hammes, H.P.; Feng, Y.; Pfister, F.; Brownlee, M. Diabetic retinopathy: Targeting vasoregression. Diabetes 2011, 60, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Frank, R.N. Diabetic retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Vom Hagen, F.; Wang, Y.; Beck, S.; Schreiter, K.; Pfister, F.; Hoffmann, S.; Wagner, P.; Seeliger, M.; Molema, G. The absence of angiopoietin-2 leads to abnormal vascular maturation and persistent proliferative retinopathy. Thromb. Haemost. 2009, 102, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.-P.; Lin, J.; Wagner, P.; Feng, Y.; Vom Hagen, F.; Krzizok, T.; Renner, O.; Breier, G.; Brownlee, M.; Deutsch, U. Angiopoietin-2 causes pericyte dropout in the normal retina: Evidence for involvement in diabetic retinopathy. Diabetes 2004, 53, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Pfister, F.; Wang, Y.; Schreiter, K.; Vom Hagen, F.; Altvater, K.; Hoffmann, S.; Deutsch, U.; Hammes, H.-P.; Feng, Y. Retinal overexpression of angiopoietin-2 mimics diabetic retinopathy and enhances vascular damages in hyperglycemia. Acta Diabetol. 2010, 47, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Iandiev, I.; Pannicke, T.; Wurm, A.; Hollborn, M.; Wiedemann, P.; Osborne, N.N.; Reichenbach, A. Cellular signaling and factors involved in Müller cell gliosis: Neuroprotective and detrimental effects. Prog. Retin. Eye Res. 2009, 28, 423–451. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Ward, M.; Madden, A.; Yong, P.; Limb, G.; Curtis, T.; Stitt, A. Hyperglycaemia-induced pro-inflammatory responses by retinal Müller glia are regulated by the receptor for advanced glycation end-products (rage). Diabetologia 2010, 53, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, A.; Pannicke, T.; Biedermann, B.; Francke, M.; Iandiev, I.; Grosche, J.; Wiedemann, P.; Albrecht, J.; Reichenbach, A. Role of retinal glial cells in neurotransmitter uptake and metabolism. Neurochem. Int. 2009, 54, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Reichelt, W.; Stabel-Burow, J.; Pannicke, T.; Weichert, H.; Heinemann, U. The glutathione level of retinal Müller glial cells is dependent on the high-affinity sodium-dependent uptake of glutamate. Neuroscience 1997, 77, 1213–1224. [Google Scholar] [CrossRef]
- Bai, Y.; Ma, J.X.; Guo, J.; Wang, J.; Zhu, M.; Chen, Y.; Le, Y.Z. Müller cell-derived VEGF is a significant contributor to retinal neovascularization. J. Pathol. 2009, 219, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.-P.; Porta, M. Subject Index. In Experimental Approaches to Diabetic Retinopathy; Karger Publishers: Basel, Switzerland, 2010; Volume 20, pp. 229–232. [Google Scholar]
- Matsumura, T.; Hammes, H.-P.; Thornalley, P.J.; Edelstein, D.; Brownlee, M. Hyperglycemia increases angiopoietin-2 expression in retinal muller cells through superoxide-induced overproduction of [Alpha]-Oxoaldehyde age precursors. Diabetes 2000, 49, A55. [Google Scholar]
- Gilles, A.-M.; Presecan, E.; Vonica, A.; Lascu, I. Nucleoside diphosphate kinase from human erythrocytes. Structural characterization of the two polypeptide chains responsible for heterogeneity of the hexameric enzyme. J. Biol. Chem. 1991, 266, 8784–8789. [Google Scholar] [PubMed]
- Lascu, I.; Gonin, P. The catalytic mechanism of nucleoside diphosphate kinases. J. Bioenerg. Biomembr. 2000, 32, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Janin, J.; Dumas, C.; Moréra, S.; Xu, Y.; Meyer, P.; Chiadmi, M.; Cherfils, J. Three-dimensional structure of nucleoside diphosphate kinase. J. Bioenerg. Biomembr. 2000, 32, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Fancsalszky, L.; Monostori, E.; Farkas, Z.; Pourkarimi, E.; Masoudi, N.; Hargitai, B.; Bosnar, M.H.; Deželjin, M.; Zsákai, A.; Vellai, T. NDK-1, the homolog of NM23-H1/H2 regulates cell migration and apoptotic engulfment in C. Elegans. PLoS ONE 2014, 9, e92687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, H.-N.; Albigès-Rizo, C.; Block, M.R. New insights into NM23 control of cell adhesion and migration. J. Bioenerg. Biomembr. 2003, 35, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Snider, N.T.; Altshuler, P.J.; Omary, M.B. Modulation of cytoskeletal dynamics by mammalian nucleoside diphosphate kinase (NDPK) proteins. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Hippe, H.-J.; Luedde, M.; Lutz, S.; Koehler, H.; Eschenhagen, T.; Frey, N.; Katus, H.A.; Wieland, T.; Niroomand, F. Regulation of cardiac camp synthesis and contractility by nucleoside diphosphate kinase b/g protein βγ dimer complexes. Circ. Res. 2007, 100, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Hippe, H.-J.; Lutz, S.; Cuello, F.; Knorr, K.; Vogt, A.; Jakobs, K.H.; Wieland, T.; Niroomand, F. Activation of heterotrimeric G proteins by a high energy phosphate transfer via nucleoside diphosphate kinase (NDPK) B and Gβ subunits specific activation of Gsα by an NDPK B·Gβγ complex in H10 cells. J. Biol. Chem. 2003, 278, 7227–7233. [Google Scholar] [CrossRef] [PubMed]
- Hippe, H.-J.; Wolf, N.M.; Abu-Taha, H.I.; Lutz, S.; Le Lay, S.; Just, S.; Rottbauer, W.; Katus, H.A.; Wieland, T. Nucleoside diphosphate kinase B is required for the formation of heterotrimeric G protein containing caveolae. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Gross, S.; Wolf, N.M.; Butenschön, V.M.; Qiu, Y.; Devraj, K.; Liebner, S.; Kroll, J.; Skolnik, E.Y.; Hammes, H.-P. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arterioscler. Throm. Vasc. Biol. 2014, 34, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhao, D.; Butenschön, V.-M.; Bauer, A.T.; Schneider, S.W.; Skolnik, E.Y.; Hammes, H.-P.; Wieland, T.; Feng, Y. Nucleoside diphosphate kinase B deficiency causes a diabetes-like vascular pathology via up-regulation of endothelial angiopoietin-2 in the retina. Acta Diabetol. 2016, 53, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanova, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the k+ channel KCa3. 1, resulting in defective T cell activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.; Courtois, Y. The growth and behaviour of rat retinal Müller cells in vitro 1. An improved method for isolation and culture. Exp. Eye Res. 1990, 51, 119–129. [Google Scholar] [CrossRef]
- Hu, J.; Popp, R.; Frömel, T.; Ehling, M.; Awwad, K.; Adams, R.H.; Hammes, H.-P.; Fleming, I. Müller glia cells regulate Notch signaling and retinal angiogenesis via the generation of 19,20-dihydroxydocosapentaenoic acid. J. Exp. Med. 2014, 211, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.J.; Sarthy, V.P. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Wong, V.; Burova, E.; Wei, Y.; Zabski, S.; Griffiths, J.; Lai, K.-M.; Lin, H.C.; Ioffe, E.; Yancopoulos, G.D. Angiopoietin-1 modulates endothelial cell function and gene expression via the transcription factor FKHR (FOXO1). Genes Dev. 2004, 18, 1060–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potente, M.; Urbich, C.; Sasaki, K.-I.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; DePinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005, 115, 2382–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Signaling vascular morphogenesis and maintenance. Science 1997, 277, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Vom, H.F.; Pfister, F.; Djokic, S.; Hoffmann, S.; Back, W.; Wagner, P.; Lin, J.; Deutsch, U.; Hammes, H.P. Impaired pericyte recruitment and abnormal retinal angiogenesis as a result of angiopoietin-2 overexpression. Thromb. Haemost. 2007, 97, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Berberich, S.; Postel, E. PuF/NM23-H2/NDPK-B transactivates a human c-myc promoter-CAT gene via a functional nuclease hypersensitive element. Oncogene 1995, 10, 2343–2347. [Google Scholar] [PubMed]
- Eijkelenboom, A.; Burgering, B.M.T. Foxos: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Nakae, J.; Kitamura, T.; Silver, D.L.; Accili, D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J. Clin. Investig. 2001, 108, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alikhani, M.; Roy, S.; Graves, D.T. FoxO1 plays an essential role in apoptosis of retinal pericytes. Mol. Vis. 2010, 16, 408–415. [Google Scholar] [PubMed]
- Behl, Y.; Krothapalli, P.; Desta, T.; Roy, S.; Graves, D.T. FoxO1 plays an important role in enhanced microvascular cell apoptosis and microvascular cell loss in type 1 and type 2 diabetic rats. Diabetes 2009, 58, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.; Zilberfarb, V.; Gangneux, N.; Christeff, N.; Issad, T. O-glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6-phosphatase gene. FEBS Lett. 2008, 582, 829–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, Y.; Huang, H.; Chatterjee, A.; Teuma, L.D.; Baumann, F.S.; Hammes, H.-P.; Wieland, T.; Feng, Y. Mediation of FoxO1 in Activated Neuroglia Deficient for Nucleoside Diphosphate Kinase B during Vascular Degeneration. Neuroglia 2018, 1, 280-291. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1010019
Qiu Y, Huang H, Chatterjee A, Teuma LD, Baumann FS, Hammes H-P, Wieland T, Feng Y. Mediation of FoxO1 in Activated Neuroglia Deficient for Nucleoside Diphosphate Kinase B during Vascular Degeneration. Neuroglia. 2018; 1(1):280-291. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1010019
Chicago/Turabian StyleQiu, Yi, Hongpeng Huang, Anupriya Chatterjee, Loïc Dongmo Teuma, Fabienne Suzanne Baumann, Hans-Peter Hammes, Thomas Wieland, and Yuxi Feng. 2018. "Mediation of FoxO1 in Activated Neuroglia Deficient for Nucleoside Diphosphate Kinase B during Vascular Degeneration" Neuroglia 1, no. 1: 280-291. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1010019