In Search of a Breakthrough Therapy for Glioblastoma Multiforme

1
Institute of Living Systems, Immanuel Kant Baltic Federal University, Universitetskaya str, 2, Kaliningrad 236041, Russia
2
School of Physiology, Pharmacology and Neuroscience, University of Bristol, University Walk, Bristol BS8 1TD, UK
*
Author to whom correspondence should be addressed.
†
Those authors make equal contributions to this work.
Neuroglia 2018, 1(2), 292-310; https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020020
Submission received: 21 August 2018
/
Revised: 14 September 2018
/
Accepted: 20 September 2018
/
Published: 26 September 2018
Abstract
:Glioblastoma multiforme (GBM) is an extremely malignant type of brain cancer which originates from astrocytes or their precursors. Glioblastoma multiforme cells share some features with astrocytes but are characterized by highly unstable genomes with multiple driver mutations and aberrations. Effective therapies for GBM are lacking and hardly any progress has been made in the last 15 years in terms of improving the outcomes for patients. The lack of new especially targeted anti-GBM medications has prompted scientists in academia around the world to test whether any of the currently approved drugs might be used to fight this devastating disease. This approach is known as repurposing. Dozens of drugs have been reported to have anti-GBM properties in vitro but there is no solid evidence for the clinical efficacy of any of them. Perhaps the most interesting group of those repurposed are tricyclic antidepressants but the mechanism of their action on GBM cells remains obscure. In this brief review we consider various approaches to repurpose drugs for therapy of GBM and highlight their limitations. We also pay special attention to the mitochondria, which appear to be intimately involved in the process of apoptosis and could be a focus of future developments in search of a better treatment for patients suffering from GBM.
1. Introduction
Glial cells are an essential component of the mammalian brain and are primarily responsible for its homeostasis. In contrast to neurons, which are terminally differentiated cells where genes responsible for cell division are methylated and essentially inactive, glial cells are much more plastic and even in the human brain they probably can divide, albeit seldomly. This can be seen particularly clearly in case of a mechanical brain trauma, leading to the formation of the so-called glial scars, areas densely populated by astrocytes which show signs of “reactivity”. This implies that the parts of the genome required for cell division are accessible in astrocytes, which makes oncogenic transformation of these cells fundamentally possible. Stochastic mutations in genes which control replication eventually trigger uncontrollable division of these cells and formation of a tumor. These tumors can be more or less malignant. The least malignant are low-grade astrocytomas, which in many cases can be treated surgically. Unfortunately, in many patients, multiple mutations and chromosomal aberrations combine, creating increasingly aggressive pools of cells and transformation into glioblastoma multiforme (GBM).
Glioblastoma multiforme is the most common central nervous system (CNS) tumor [1]. There are around 2200 people diagnosed with GBM every year in England, accounting for 55% of malignant brain tumors [2]. According to World Health Organization (WHO) Classification of Tumors of the Central Nervous System, GBM is categorized as grade IV glioma, the most malignant one [1]. Glioblastoma multiforme either appears as de novo (primary GBM), or through progression of a lower grade glioma, leading to a secondary GBM [1]. Despite the dramatic advances in understanding the molecular basis of malignant glioma, this type of cancer is very aggressive and still incurable. The median survival after diagnosis is 10–11 months with standard treatment [3], and the overall 5-year survival is less than 5% [4]. Burnet et al. reported that GBM is the cause of the greatest average loss of life-years among all cancers [5].
Standard treatment of GBM involves surgical resection with radiotherapy and chemotherapy. However, recurrence seems to be inevitable [6]. Complete surgical resection of GMB is hardly ever possible because the boundaries of GBM are diffuse. The tumor sends “streaks” along nervous tracts and blood vessels, and often surgeons have no choice but to leave certain areas untouched because of the risk of causing severe disabilities in the patients [7].
In 2005, protocols consisting of surgery followed by radiotherapy alone were supplemented by a lipophilic alkylating agent, temozolomide (TMZ), approved by the Food and Drug Administration (FDA). Concurrent and adjuvant chemotherapy with TMZ was found to improve median survival by 2.5 months compared to radiotherapy alone in a large 5-year phase III randomized trial [8]. This so-called “Stupp Protocol” is, to this day, the universally accepted standard of care.
Strikingly, despite the desperate need for new treatments, there have been no other major advances for many years now. This is partially due to the complexity of the problem but also reflects the lack of interest of the pharmaceutical companies in this relatively rare form of cancer. From 2005 to 2015, 216 phase-II or III clinical trials on glioblastoma treatment were registered at clinicaltrial.gov database, some of which are still ongoing [9]. Clinical trials are testing different therapeutic approaches including molecular targeted drugs, immunotherapy, viral vector-based gene therapy, electrotherapy and novel strategies to increase tumor sensitivity to radiotherapy [10]. In addition to the traditional strategies based on the research into the mechanisms of oncogenesis, cell division, and tumor resistance, multiple attempts have been made to improve the outcomes in GBM patients by “repurposing” drugs which are already available in clinics. Quite a few various drugs have been claimed to have anti-GBM effects. The problem with most of these studies is that they were carried out on either in vitro or, at best, on mouse models with transplanted GBM and there is very little solid evidence for any of these strategies to be beneficial clinically.
In this review, we summarize some of the available information on repurposed drugs suggested for therapy of GBM. We specifically focus on two key issues. First, how strong is the evidence that any suggested drug is actually more harmful to GBM than to the healthy cells, are the concentrations used to demonstrate the anti-GBM effects physiologically and clinically relevant? Second, is there any common potential cellular mechanism or target for such drugs, something what might be a point of convergence for the action for at least some of them. We believe that GBM mitochondria could be such a “weak spot” of GBM.
2. Repurposing of Drugs for Glioblastoma Multiforme
Fairly low output of new and effective therapies stimulates efforts directed towards finding any possible new treatments among existent medicines. Table 1 illustrates the plethora of drugs suggested for repurposing against GBM, but the list of such studies is actually significantly longer. Specific anticancer drugs developed for other types of tumors and tested against GBM are not included.
Already a quick look at Table 1 suggests that there is very little commonality between the proposed drugs or their suggested mechanisms of action. Moreover, for all these drugs, evidence for their clinical anti-GBM efficacy is weak or lacking altogether. Often anti-GBM effects are reported based on in vitro tests on cultured GBM cells, which in many cases are commercially available lines, which have been in vitro for decades and therefore perhaps are hardly representative of the real biology and genetics of the GBM. It is also noticeable that many of the drugs proposed for therapy of GBM have not been shown to cross the blood-brain barrier (GBM core might have leaky barrier but it the periphery it is probably still sufficiently tight). Importantly, many studies used drugs in vitro without much regard to what is known about the biologically relevant concentrations in humans, or the effect of these chemicals on normal cells at the same concentrations at which they had a negative effect on the GBM. An example of these issues is the reported antiproliferative effect of quinidine which was demonstrated using C6 cell line and a high concentration of the drug, half maximal effective concentration (EC50) = 112 μM [31]. For reference, an average therapeutic plasma concentration of quinidine is 1.68 μg/mL [37], which equals to 2.5 μM when converted to molar concentration. This is just one of many studies with the same limitation. It seems logic that even if an anti-GBM effect of a drug can be demonstrated, one would expect the malignant cells to be more sensitive to such an effect, than the healthy ones. However, studies where accurate comparisons have been made are extremely rare.
Some scientists believe that combination of many repurposed drugs can be advantageous. Kast et al. [38] developed the Coordinated Undermining of Survival Paths protocol, known as CUSP9, based on a combination of nine repurposed drugs which are to be combined with continuous low dose TMZ administration. It was expected to augment the clinical efficacy and tolerability of TMZ. Patients in Belgium are currently being recruited to take part in a phase I clinical trial of this CUSP9 protocol. The primary completion date of this study is March 2019. In theory, combinations of drugs could improve their efficacy but equally their side effects could combine. It remains to be seen whether CUSP9 will be any more successful that previous attempts.
Below we further discuss some commonly used drugs in clinical practice that have been tested on GBM for possible repurposing and try to illustrate some of the limitations of this research.
3. Biguanides: Metformin and Phenformin
Metformin is one of the most commonly prescribed drugs in clinical practice. It is used to treat type II diabetes, polycystic ovary disease, and metabolic syndrome [39]. The use of phenformin has been discontinued because of its side effects.
Biguanides are known to inhibit gluconeogenesis in the liver and stimulate glycolysis by altering the activity of different enzymes involved in these pathways [39]. They also improve the sensitivity of insulin receptors in skeletal muscle cells and enhance insulin-mediated glucose uptake through enhanced activity and translocation of glucose transporters, such as glucose transporter type-4 [39]. Moreover, biguanides increase circulating levels of glucagon-like peptide-1 (GLP-1) and stimulate expression of GLP-1 receptor in the pancreas. GLP-1 increases insulin secretion and decreases glucagon secretion [39]. All these effects are either directly or indirectly related to biguanides’ inhibitory effect on complex I of the mitochondrial electron transport chain, reducing ATP and increasing adenosine monophosphate (AMP) production and AMP-activated kinase activity [39]. This effect is stronger with phenformin, thus the higher incidence of side effects, i.e., lactic acidosis [39].
Anti-cancer properties of biguanides were first demonstrated with metformin on pancreatic, breast, and lung cancer [40,41,42,43]. Metformin was also found to inhibit proliferation, induce apoptosis, and reduce cell adhesion and invasion of GBM [44,45,46,47,48]. Likewise, phenformin was found to inhibit proliferation of glioma stem cells (GSCs), impair sphere formation, decrease stemness, and induce apoptosis [49].
Some of the proposed mechanisms for the antitumor effect of biguanides include: (1) activation of AMPK which leads to blockade of Rheb (Ras homolog enriched in brain)-mTOR (mammalian target of rapamycin) pathways of protein synthesis and cellular growth and activation of tumor suppressor gene p53 [39], (2) reduction of available insulin which reduces the activity of insulin-like growth factor-1 (IGF-1) anabolic pathway [39], (3) suppression of Febulin-3 and Matrix Metalloproteinase-2 expression [44], (4) stimulation of the expression of tumor suppressor micro RNA (miRNA) Lethal-7 [49].
Anticancer effects of metformin described above were achieved using millimolar concentrations of the drug. Lower concentrations either failed to show statistical significance or did only affect a few (1 of 5) glioma cell lines tested [46,47]. The commonly used concentrations are much higher than average plasma concentration for diabetes treatment; 0.86 mg/L (6.6 μM) [50]. In fact, plasma concentrations exceeding 2.5 mg/L (20 μM) are associated with the risk of lactic acidosis [50].
4. Statins: Atorvastatin, Lovastatin, Simvastatin, and Pravastatin
Statins are 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMG-CoA-R) inhibitors, which are prescribed for their lipid-lowering effect. They inhibit the rate limiting step in the mevalonate pathway in hepatocytes, leading to decreased de novo cholesterol synthesis, intracellular lipid stores, and circulating low-density lipoproteins [51].
The reduction in availability of downstream products of the mevalonate pathway is thought to be a key mechanism for the observed growth inhibiting effect of different statins on different cancer cell types including glioma cells [51,52,53,54,55,56,57,58]. Downstream products of the mevalonate pathway are important for prenylation (activation) of cellular proteins Ras, Rho, and Rac, which are small GTPases critical for regulation of cell growth and survival [51]. Other proposed mechanisms include induction of apoptosis and inhibition of cell migration: Apoptosis may be induced by altering the cellular response to stress through the Jun N-terminal kinase (JNK)-dependent cell death pathway [59], by indirectly activating Caspase-3 [56,57], or by decreasing the expression of antiapoptotic proteins such as Bcl-2 and upregulating the expression of proapoptotic proteins such as Bax and Bim [52]. Cellular migration and invasion may be inhibited through inactivation of focal adhesion kinase (FAK) [60], or decreasing the amount of extracellular matrix-degrading enzymes and matrix metalloproteinases released from microglia into the glioma environment [61]. Atorvastatin was also suggested to decrease the expression of proinflammatory proteins and interleukins (IL) [57].
Therapeutic lipid-lowering doses of statins range between 5–80 mg/day and produce plasma concentrations that range from approximately 2 to 15 nM [62]. However, statin concentrations employed in the above-mentioned in vitro experiments commonly ranged between 1–10 μM. The lower end of this range is already 100-fold higher than the average therapeutic plasma concentration of statins in human. One may also wonder why, if these drugs under realistic in vivo conditions inhibit Ras, Rho and Rac signaling, they do not lead to general toxicity, which would have prevented their wide-spread use.
5. Antimicrobial Agents: Dapsone and Nitroxoline
Dapsone is one of three antibiotics used as first line treatment for leprosy [63]. It is also used to treat dermatitis herpetiformis, malaria, and as a disease-modifying anti-rheumatoid drug [64]. Dapsone is bactericidal and bacteriostatic. It works by inhibiting folic acid synthesis in bacteria [63]. For noninfectious indications, dapsone is used for its ability to inhibit synthesis or function of immune chemotactic factors which impairs functions of neutrophils and limits neutrophil-induced tissue destruction [63].
A recent study has shown that dapsone and dapsone derivatives inhibit glioma cells’ anchorage-independent growth (colony formation) and impair glioma cell migration [65]. The authors hypothesized that the antineoplastic effect of dapsone is mediated by inhibition of IL-8. Interleukin-8 is well recognized as growth-promoting and pro-angiogenic factor in many cancer types [64]. By inhibiting IL-8, dapsone impairs neutrophil chemotaxis and migration, and interferes with neutrophil-dependent delivery of vascular endothelial growth factor to glioma cells [65]. In vitro antineoplastic effects of dapsone were achieved using concentrations ranging from 10 to 50 μM, which are slightly higher than average therapeutic molar plasma concentration of dapsone (2–20 μM, calculated from the reported concentration range of 0.5 to 5 mg/L) [66].
Nitroxoline (5-nitro-8-hydroxy-quinoline) is a quinoline-based antibiotic that is FDA approved for treatment of urinary tract infection [67]. Nitroxoline is bactericidal and/or bacteriostatic depending on the type of microorganism [68]. Generally, its mode of action depends on its ability to chelate divalent cations and disrupt the organization of the bacterial cell wall [69]. Nitroxoline has been tested against different types of neoplasia, including bladder, gastrointestinal, lung and breast cancers [67,70,71].
Nitroxoline has been shown to have anti-angiogenic properties. A screen of a library of 175,000 compounds has identified nitroxoline as an inhibitor of methionine aminopeptidase 2 (MetAP-2), which it inhibited in a dose-dependent fashion, with half maximal inhibitory concentration (IC50) = 54.8 nM [71]. Inhibiting MetAP-2 suppresses endothelial cell proliferation. Nitroxoline was also found to inhibit non-cancerous Human Umbilical Vein Endothelial Cells proliferation dose-dependently with IC50 = 1.9 μM [71]. In vivo, 60 mg/(kg∙day) of nitroxoline was able to inhibit neovascularization in breast cancer [71] and bladder cancer xenografts [67]. This dose for mice is equivalent to the common antimicrobial dose used in human (750 mg/day) [67].
Another mechanistic theory explains the anti-cancer effect of nitroxoline through its inhibitory effect on cathepsin B. Cathepsin B is an enzyme which degrades extracellular matrix enabling invasion, migration, and metastasis of tumor cells. Cathepsin B is found in higher concentrations in invading edges of tumors including glioma tumors [72]. Nitroxoline at concentrations ranging from 0.1–100 μM was shown to reversibly inhibit cathepsin B [73].
In relation to GBM, nitroxoline inhibited growth of U251 and U87 glioma cell lines, induced cell cycle arrest at G0/G1, induced apoptosis and decreased invasion in vitro in a dose-dependent manner [74]. Toxic concentrations ranged from 5 to 100 μg/mL (≈26 to 520 μM) [74]. In vivo, a specific strain of genetically engineered mouse (PTEN/KRAS mouse, where PTEN is deleted in astrocytes and human Kirsten rat sarcoma viral oncogene homolog KRAS is overexpressed) which spontaneously develops grade III glioblastomas were injected intraperitonially with 80 mg/(kg∙day) nitroxoline. Magnetic Resonance Imaging taken on days 0, 7, and 14 after treatment showed a significant decrease in tumor sizes in treatment group compared to controls [74]. Immunohistochemical staining of brain slices for TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling assay of apoptosis), revealed significantly more apoptotic cells in treated mice than control mice [74]. It is important to remember that this study used “long term” GBM cell lines which are in many ways different to the cellular populations found in human GBM patients. There is also no good evidence for nitroxoline to accumulate in the brain.
6. Quinolines: Chloroquine and Quinidine
Since 1947, chloroquine has been used to treat malaria infection. It is also used to treat symptoms of some connective tissue disorders, such as systemic lupus erythematosus and rheumatoid arthritis [75].
Quinolines are widely investigated as adjuvant therapy in cancer treatment [76]. They are believed to inhibit lysosome-dependent autophagy and improve chemo and radio sensitivity [76]. Some of the most recent publications on this matter included breast cancer [77], pancreatic cancer [78,79], lung cancer [80], colon cancer [81], and bladder cancer [82].
Chloroquine was found to increase GBM cells’ sensitivity to TMZ by inhibiting autophagy and increasing the production of reactive oxygen species induced by TMZ [83,84,85,86]. Golden et al. have demonstrated this effect in vivo in mice bearing human glioma xenografts, with concentrations similar to therapeutic concentrations used for the original indication of the drug (10 mg/kg of chloroquine) [83]. Chloroquine was also reported to potentiate radiation induced apoptosis in GBM cells [87,88,89]. It seems that the anti-GBM effect of chloroquine deserves more focus, especially as a supplement to TMZ.
7. Antidepressants
Antidepressants often receive special attention as putative anti-GBM agents. Serendipity played a part in their appearance in this field because many cancer and GBM patients are prescribed antidepressants against depression which is a common comorbidity. Depression is not only a result of the psychological burden of the diagnosis but is a consequence of the standard treatment procedures. Interest to the anti-GBM potential has been motivated by findings from retrospective studies such as the large-scale epidemiology study conducted by Walker et al. [90], who found an inverse association between treatment with tricyclic antidepressants (TCA) and the incidence of GBM. Antidepressants have several features which are expected for drugs which could be potentially retargeted towards GBM. They are small and lipid soluble molecules which cross the blood-brain barrier and sequester in the brain in relatively high concentrations. In addition, these drugs are relatively nontoxic and induce few serious side effects.
Many studies investigated the possibility of repurposing antidepressants for GBM treatment. Levkovitz et al. [91] studied the effect of several different antidepressants on apoptotic markers in both glioma C6 and neuroblastoma SH-SY5Y cell lines. They reported that paroxetine and fluoxetine, two serotonin selective reuptake inhibitors (SSRIs), and clomipramine, a TCA, caused apoptosis in both cell lines. Interestingly, the toxic effect of clomipramine on C6 cells developed in almost all-or-nothing manner. At 12 μM there was hardly any toxicity while at 25 μM the effect was already maximal. Similarly, with fluoxetine there was little toxicity at 25 μM but 50 μM had a strong negative effect on viability. This is consistent with the results from a much earlier study in the C6 cell line, showing that fluoxetine caused DNA fragmentation, which is a major known step in apoptosis [92]. Similarly, Liu et al. [93] reported that fluoxetine suppressed the growth of GMB cell lines. The effective concentration of fluoxetine in that study was 25–30 μM in vitro. The authors explained this effect by activation of the intrinsic apoptotic pathway (see below). In vivo, fluoxetine strongly suppressed growth of tumors from U87 implants in the brains of Nu/Nu mice when administered daily at 10 mg/kg orally. Its effect was comparable to that of TMZ at 5 mg/kg intraperitonially. This study illustrates a stark contrast between the available models of GBM and clinic, where fluoxetine has never shown such potency against GMB. Fluvoxamine, another SSRI, at 40 μM was able to suppress migration and invasion of human GBM cell lines (A172, U87-MG, and U251-MG) [94]. This effect was accompanied by inhibition of FAK/Akt mTOR pathway activity. Regarding the feasibility of the concentrations of fluoxetine and other TCA used in anti-GBM studies, human data suggest that they do accumulate in the brain, reaching remarkably high concentrations, up to 10 μg/ml, which converts to approximately 20–30 μM [95,96]. Bielecka-Wajdman et al. [97] examined the influence of six different antidepressants on the phenotypic signature and viability of GSCs isolated from a human GBM cell line. In that study only imipramine and amitriptyline significantly altered cell viability. Imipramine and amitriptyline were most effective in reducing quantity and expression of various stem cell markers, thus silencing the GSC profile. Jeon et al. [98] also used two different GBM cell lines (U87 and C6) and reported that 40 and 60 μM of imipramine-induced cell death in GBM models but, remarkably, not normal primary rat astrocytes. The authors explain the effects of imipramine by activation of autophagy and implicate protein Beclin-1 in this process, because short hairpin RNA (sh-RNA) mediated knock-down of this protein conferred resistance to imipramine-induced cell death. Again, limitations of this study are the use of very old and hypermutated cell lines, and the use of very high concentrations of the antidepressant [98]. In yet another study, Shchors et al. [99] reported that imipramine treatment prolonged the overall survival of glioma-bearing mice by 18 days compared to that of a control cohort. These authors also concluded that TCAs induce autophagic cell death. Their explanation for this effect, however, was different to the previous two studies. The authors proposed that TCAs activate the G-protein αs subunit which, in turn, activates adenylyl cyclase resulting in an elevation of cellular cyclic adenosine monophosphate (cAMP). This was thought to induce autophagy associated cell death in glioma cells via the EPAC branch of the cAMP signaling cascade. This hypothesis was supported by an additional finding that inhibition of the purinergic receptor P2Y12, activation of which inhibits adenylyl cyclase, potentiated the effects of imipramine, making the combination of drugs particularly effective [99]. The problem with this explanation is that it relies on the monoamine theory for the mechanism of action of TCA which is the canonical explanation of the antidepressant effect of TCA. It poses that TCA act by inhibiting reuptake of noradrenaline and serotonin into the monoaminergic terminals from which they are released in the brain. Meanwhile in the in vitro experiments on GBM cultures there are neither monoamines, nor the terminals which could release and then reuptake them and therefore the very substrate for the “classic” monoamine-dependent action is lacking. In addition, TCAs block re-uptake of monoamines in nanomolar concentrations, which is orders of magnitude lower than what is commonly used in GBM experiments. Therefore, the effects reported in that paper require a different explanation.
The studies listed above illustrate the issues common to the literature on anti-GBM effects of TCAs (and, in fact, other repurposed drugs). These issues include: (a) use of the GBM cell lines such as C6, which have been in vitro for decades and accumulated mutations and acquired qualities which make them very different to the real tumors in human brain, (b) the use of unrealistically high concentrations of antidepressants, and (c) lack of coherency in terms of the proposed molecular targets for these drugs between different studies. Another major general limitation is the lack of an adequate model for studying toxic effects of these drugs on healthy human cells. Typically, researchers use either primary rodent astrocytes or human embryonic astrocytes. Neither of these are a close replica of mature human astrocytes or a good match for the GBM cells found in the human brain in the second half of life. Therefore, we do not know whether high concentrations of antidepressants used in GMB studies can be tolerated by healthy adult human brain cells or we are dealing with some un-specific cellular toxicity.
8. Are Mitochondria a Possible “Weak Spot” of Glioblastoma Multiforme?
Of many different explanations for the anti-GBM effects of repurposed drugs, one mechanism stands out. Quite a few studies by unconnected groups of researchers eventually implicate mitochondria in anti-tumor and pro-apoptotic effects, registered under different conditions (Figure 1). Abnormalities in mitochondrial gene regulation and metabolism in GBM are well known and have been reviewed elsewhere [100]. From the analysis presented in that review, it appears that multiple mutations of mitochondrial genes reported in various studies do not directly drive onco-transformation into the GBM but may significantly affect the properties of the individual lines or subclones of GBM cells within the same tumor. It is also important to remember that most mitochondrial proteins are encoded by nuclear DNA and it is the changes in the nuclear DNA which result in onco-transformation. Chaotization of gene expression caused by genomic instability may have an impact on the fine tuning of the reactions mediated by these nucleus-encoded proteins in the mitochondria, which depend on the supply of these proteins, possibly making GBM mitochondria more vulnerable. On the other hand, the principle of clonal selection which takes place in tumors, can lead to elimination of the GBM cells with severely dysregulated mitochondrial function. It is worth notice, that there is a paucity of information, concerning differences between mitochondria in “regular” GBM cells and GSCs [100].
Although mitochondria have been suggested to be the key organelle to target in GBM, different studies approach this idea from completely different angles. Some authors believe that it is possible to use the ability of mitochondria to initiate apoptosis by acting on the GBM mitochondria directly with some of the repurposed drugs [101,102]. It has also been proposed that mitochondrially induced apoptosis can be induced indirectly, via Ca2+ overload [93] or proteasome inhibition [103]. Finally, there is evidence that mitochondrial biogenesis induced by activation of cAMP-mediated signaling can reduce malignancy of GBM cells [104].
In normal glial cells, cellular energy is mainly produced in mitochondria through aerobic respiration [105]. Yet, metabolism starts with glycolysis, whereby glucose is converted into pyruvate with production of ATP in the cytosol. Pyruvate is then transported into the mitochondria where it is oxidized to acetyl-CoA and then used in the citric acid cycle. Unlike normal cells, GBM has a lower number of mitochondria, indicating high mitochondrial degradation activity [106]. Glioblastoma multiforme, as well as other cancer cells, are known to have active aerobic glycolysis despite the presence of normal oxygen concentrations, and rely on it, rather than on oxidative phosphorylation as the main source of energy [107,108]. Early studies have found that this metabolic shift, known as Warburg effect, is due to mitochondrial dysfunction in many tumor cell types including glioma [109,110]. The Warburg effect seems to be an essential feature of GBM, but to this day the exact reason for high glycolytic activity of tumor cells is unknown [111]. Possibly, cancer cells cannot fully use pyruvate due to decreased pyruvate transporter activity [106], which transports pyruvate inside the mitochondrial matrix. Pyruvate transporters isolated from mitochondria of tumor cells are slower and have lower affinity to pyruvate than transporters isolated from normal cells’ mitochondria [112], limiting pyruvate uptake. In addition, pyruvate in mitochondria of tumor cells undergoes decarboxylation into acetaldehyde instead of oxidation [113]. Two acetaldehydes condense to form acetoin which inhibits pyruvate dehydrogenase complex so that pyruvate cannot be converted into acetyl-CoA [113]. In any case, active glycolysis is a landmark of tumors including GBM and seems to confer to them some important survival advantages, possibly by supplying actively dividing cells with new building blocks for lipids, nucleotides and proteins [111].
9. Mitochondria Are the Central Hub of the “Intrinsic” Apoptotic Pathway
Apoptosis is a cascade of events that leads to programmed cell death which can be triggered by both extrinsic and intrinsic pathways (Figure 1). The extrinsic pathway, also known as the death receptor pathway, is initiated when specific ligands bind and stimulate death receptors on the cell surface, initiating a signaling pathway eventually leading to activation of proteases, called caspases [114]. First, procaspases 8 and 10 are cleaved and activated. Next, they cleave and activate the executioner caspases 3 and 7, which start the apoptotic cascade [114]. The intrinsic pathway of apoptosis is activated by direct damage to the cell, such as metabolic failures, hypoxia, radiotherapy, and chemotherapy. In case of direct damage to DNA, upregulation of proapoptotic and downregulation of prosurvival proteins trigger the opening of the mitochondrial permeability transition pore [114]. This pore allows the release of cytochrome C. Cytochrome C binds to apoptotic peptidase activating factor-1 (Apaf-1) eventually leading to the activation of caspase 9 which, similarly to what was described above for the extrinsic pathway, leads to cleavage of the procaspases into the executioner caspases 3, 6 and 7 and triggers fatal apoptotic events [114,115,116]. Another factor released by mitochondria into the cytoplasm is Smac, which blocks the function of inhibitor of apoptosis proteins, thus facilitating activation of the executioner caspases [114].
Abnormalities in both apoptotic pathways are usually found in GBM. The extrinsic pathway is inhibited by developing resistance to TRAIL apoptosis cascade (apoptosis triggered by Tumor necrosis factor-Related Apoptosis-Inducing Ligands) in glioma [117]. This apoptotic resistance may result from suppression by mammalian target of rapamycin (mTOR), which is closely associated with cell proliferation and growth [118].
In the intrinsic pathway, inhibitors of apoptosis proteins are overexpressed in human malignant glioma cells [119]. Immunostaining of the mitochondria in human glioma cell lines showed that the prosurvival Bcl-2 protein is upregulated. This pathway may suppress apoptosis in GBM cells after DNA damage [120]. It has been also reported that more than 90% of human GBM samples exhibit elevated levels of pro-survival Bcl-2 such as 12 (BCL2L12) protein which suppresses the executioner caspases 3 and 7 directly [121]. Other studies show elevated levels of prosurvival Bcl-2 family members but also proapoptotic proteins in GBM compared to normal astrocytes [122,123] However, a significant upregulation of prosurvival Bcl-2 and Bcl-XL and downregulation of proapoptotic Bcl-2 associated X protein (Bax) are shown in GBM recurrences after treatment [124]. Therefore, GBM cells appear to actively counteract the proapoptotic events initiated via mitochondria. Nevertheless, several studies indicate that GBM mitochondria can be affected by drugs leading to the anti-tumor effects.
10. Antidepressants and Glioblastoma Multiforme Mitochondria
Antidepressants appear to be one group where the involvement of mitochondria has been considered by many studies (Figure 1). Daley et al. [101] demonstrated that the TCA clomipramine can cause cell death of human glioma cells without affecting human fetal astrocytes. A toxic effect of clomipramine on GBM cells was evident within 2 hours of exposure, by which time fetal astrocytes exhibited no clear signs of toxicity. However, this effect only became significant with 114 μM of clomipramine, which is improbable in vivo. In that study, clomipramine concentration-dependently decreased oxygen consumption of glioma cells but, again, the lowest concentration used was 140 μM. This was accompanied by a decrease in mitochondrial membrane potential, which is a direct indicator of the activity of oxidative phosphorylation mechanisms. To explain these effects, the authors measured the effect of clomipramine on the activity of mitochondrial complexes I, II, III and IV, isolated from the mitochondria from various organs. The most consistent effect was the inhibition by 25 μM of clomipramine of complex III activity which was approximately the same in mitochondria from different organs (Figure 1). Importantly, mitochondria were not from GBM cells but were isolated from normal rat tissues. The study also reported activation of caspases which was explained by the insult to the mitochondria caused by clomipramine and the consequent recruitment of the intrinsic pathway mentioned above. On balance, while the study highlights the mitochondria as the direct target for clomipramine, the effective concentrations of the drug appear to be very high and human fetal astrocytes cannot be seen as an adequate model of postnatal human astrocytes, raising the possibility that such high concentrations of clomipramine could be equally toxic to malignant and healthy cells in living brain. At least the study offers no answer as to why mitochondria in GBM could be more sensitive to clomipramine. It is also not entirely clear why the focus was on the short-term effects (1–3 h) while it could be more relevant to look for the effects of lower concentrations developing over longer time scale. For further discussion of this topic see [102].
Mitochondria appear to be the ultimate target for the effect of fluoxetine in the study mentioned previously [93]. That study used glioma cell lines C6 (rat) and U87, GBM8401, Hs683 (human). Fluoxetine had a highly non-linear effect on glioma cell lines, decreasing their viability at concentrations 25–30 μM while at 15–20 μM the effect was hardly visible. Fluoxetine evoked strong elevations in intracellular Ca2+ in GBM cell lines, which was attributed to its ability to directly bind to the R1 subunit of glutamate receptors (GluR1) and activate the receptor. Remarkably, normal rat primary astrocytes in that study were fairly resistant to fluoxetine, which the authors explain by high expression of GluR1 on the cell membrane in GBM but not normal astrocytes. It is unclear, though, why GluR1 expression should lead to a strong Ca2+ influx because normally Ca2+ permeability of AMPA receptors which are formed with GluR1 is low. Nevertheless, the study concludes that Ca2+ overload eventually led to mitochondrial damage and activation of the intrinsic apoptotic cascade.
Interestingly, in another study where the effect of fluoxetine was studied on non-GBM cancer cell lines, mitochondrial calcium overload and cell death were explained by a completely different mechanism [127]. The effects were observed after exposure to 100 μM of fluoxetine, a clearly supra-pharmacologic concentration. Fluoxetine is known to enter and accumulate in the mitochondria and seems to be able to inhibit the respiratory chain directly at these concentrations. This could reduce ATP production, which is required for maintenance of the low intracellular Ca2+ concentration. Eventually the increased Ca2+ load was causing direct damage to the mitochondria and release of pro-apoptotic molecules [127].
Overall, it seems that antidepressants can affect mitochondria in GBM when administered at high concentrations but the explanations for this effect put forward by different groups are inconsistent. It is also worth noting that cytochromes residing in mitochondria are involved in oxidation of numerous molecules and drugs, including antidepressants. One might then ask whether this additional chemical activity, caused by extensive oxidation of xenobiotics, is not the reason these molecules become cytotoxic at sufficiently high concentrations, especially if GBM mitochondria are, indeed, somewhat vulnerable.
11. Can Differences in Glioblastoma Multiforme Mitochondria Be Used for Targeted Therapy?
Typically, GBM mitochondria produce rather high quantities of reactive oxygen species (ROS) which is a consequence of inefficient coupling and oxidative phosphorylation. As mentioned above, one possible reason for this is the loss of fine tuning between mitochondrial and nuclear genomes which is required for perfect functioning of these semi-autonomous organelles. Temozolomide is a typical alkylating agent which primarily disrupts nuclear DNA making cell vulnerable to all kinds of damaging factors including ROS. Interestingly, however, in TMZ-resistant lines mitochondrial coupling is improved compared to the susceptible lines and ROS production is reduced, which is probably a result of the clonal selection mentioned above. It is likely that any treatment targeted at mitochondria in GBM mitochondria can make tumors more susceptible to TMZ chemotherapy [128]. An example of realization of this concept is the development of cytochrome C oxidase inhibitors with tropism to chemo-resistant GBM cells [129,130].
Another interesting idea is based on the high activity of one of the isoforms of monoamine oxidases (MAO), MAO-B in GBM [131]. Monoamine oxidase-B is located on the outer mitochondrial membrane, it is normally highly expressed by astrocytes and oxidizes various amines and other molecules. Activity of MAO-B is also particularly high in glial tumors. The authors generated a pro-drug called “MP-MUS” which can be activated by MAO-B and found that this new molecule was selective to primary human glioma cells but, remarkably, had very little toxicity against normal human astrocytes for which the study used commercially available embryonic cells. Encouraging as they are, these results suffer from the same limitation as many other studies mentioned above, because embryonic astrocytes may not be a close match to the astrocytes and other brain cells which populate the brain in the second half of life.
12. Summary
In this brief review we have illustrated some of the current ideas for possible re-targeting of currently available drugs to improve the outcomes for the patients suffering from GBM. Glioblastoma Multiforme represents one of the most difficult cancers to attack not only because of its location and the issues of drug penetration through the blood-brain barrier, but also because of the specific molecular and cellular features of this tumor. Unfortunately, being a relatively small market, GBM does not attract enough interest from the pharmaceutical industry. This puts additional pressure on basic researchers to find new, possibly unconventional, approaches which could help offer a better prognosis to the patients. We have also noted, that within the plethora of suggested mechanisms of action for re-targeted drugs and new developments, mitochondria seem to occupy a particularly prominent place. Potentially mitochondria are the weak spot of GBM which could be exploited to find new therapeutic opportunities.
Author Contributions
Conceptualization, S.K. and A.G.T., Writing—Original Draft, A.V., R.S., L.T., S.K., Writing—Review & Editing, S.K., A.G.T.
Funding
A.V. was supported by 5/100 programme (Russian Federation). R.S. is in receipt of fellowship from King Abdulaziz University (Kingdom of Saudi Arabia). S.K. and A.G.T. were supported by M.R.C. (MR/L020661/1) and BBSRC (BB/L019396/1).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Brodbelt, A.; Greenberg, D.; Winters, T.; Williams, M.; Vernon, S.; Collins, V.P. Glioblastoma in England: 2007–2011. Eur. J. Cancer 2015, 51, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Rick, J.; Chandra, A.; Aghi, M.K. Tumor treating fields: A new approach to glioblastoma therapy. J. Neurooncol. 2018, 137, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.J.; Cai, J.Q.; Liu, C.Y. Evolving Molecular Genetics of Glioblastoma. Chin. Med. J. 2016, 129, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Burnet, N.G.; Jefferies, S.J.; Benson, R.J.; Hunt, D.P.; Treasure, F.P. Years of life lost (YLL) from cancer is an important measure of population burden—and should be considered when allocating research funds. Br. J. Cancer 2005, 92, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Krivosheya, D.; Prabhu, S.S.; Weinberg, J.S.; Sawaya, R. Technical principles in glioma surgery and preoperative considerations. J. Neurooncol. 2016, 130, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Cihoric, N.; Tsikkinis, A.; Minniti, G.; Lagerwaard, F.J.; Herrlinger, U.; Mathier, E.; Soldatovic, I.; Jeremic, B.; Ghadjar, P.; Elicin, O.; et al. Current status and perspectives of interventional clinical trials for glioblastoma—analysis of ClinicalTrials.gov. Radiat. Oncol. 2017, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, F. Glioblastoma update: Molecular biology, diagnosis, treatment, response assessment, and translational clinical trials. F1000Research 2017, 6, 1892. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Basanta, M.; Fang, P.; Maity, A.; Hahn, S.M.; Lustig, R.A.; Dorsey, J.F. A phase I study of nelfinavir concurrent with temozolomide and radiotherapy in patients with glioblastoma multiforme. J. Neurooncol. 2014, 116, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, F.; Yeaton, P.; Brotchi, J.; Kiss, R. Cimetidine, an unexpected anti-tumor agent, and its potential for the treatment of glioblastoma (review). Int. J. Oncol. 2006, 28, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-diclofenac as an anti-cancer agent. Ecancermedicalscience 2016, 10, 610. [Google Scholar] [CrossRef] [PubMed]
- Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P.; Pantziarka, P. Repurposing Drugs in Oncology (ReDO)-nitroglycerin as an anti-cancer agent. Ecancermedicalscience 2015, 9, 568. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.W.; Liang, Y.H.; Kuo, Y.L.; Chuu, C.P.; Lin, C.Y.; Lee, M.H.; Wu, A.T.; Yeh, C.T.; Chen, E.I.; Whang-Peng, J.; et al. Identification of thioridazine, an antipsychotic drug, as an antiglioblastoma and anticancer stem cell agent using public gene expression data. Cell Death Dis. 2015, 6, e1753. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E. Glioblastoma chemotherapy adjunct via potent serotonin receptor-7 inhibition using currently marketed high-affinity antipsychotic medicines. Br. J. Pharmacol. 2010, 161, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kang, S.; Kim, W. Drug Repositioning for Cancer Therapy Based on Large-Scale Drug-Induced Transcriptional Signatures. PLoS ONE 2016, 11, e0150460. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Nam, D.H.; Lee, J. Repurposing antipsychotics as glioblastoma therapeutics: Potentials and challenges. Oncol. Lett. 2016, 11, 1281–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-mebendazole as an anti-cancer agent. Ecancermedicalscience 2014, 8, 443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hothi, P.; Martins, T.J.; Chen, L.; Deleyrolle, L.; Yoon, J.G.; Reynolds, B.; Foltz, G. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget 2012, 3, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Krauze, A.V.; Myrehaug, S.D.; Chang, M.G.; Holdford, D.J.; Smith, S.; Shih, J.; Tofilon, P.J.; Fine, H.A.; Camphausen, K. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 986–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Kim, T.; Joo, J.D.; Han, J.H.; Kim, Y.J.; Kim, I.A.; Yun, C.H.; Kim, C.Y. Survival benefit of levetiracetam in patients treated with concomitant chemoradiotherapy and adjuvant chemotherapy with temozolomide for glioblastoma multiforme. Cancer 2015, 121, 2926–2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesen, C.; Hormann, I.; Roscher, M.; Fichtner, I.; Alt, A.; Hilger, R.; Debatin, K.M.; Miltner, E. Opioid receptor activation triggering downregulation of cAMP improves effectiveness of anti-cancer drugs in treatment of glioblastoma. Cell Cycle 2014, 13, 1560–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robe, P.A.; Bentires-Alj, M.; Bonif, M.; Rogister, B.; Deprez, M.; Haddada, H.; Khac, M.T.; Jolois, O.; Erkmen, K.; Merville, M.P.; et al. In vitro and in vivo activity of the nuclear factor-κB inhibitor sulfasalazine in human glioblastomas. Clin Cancer Res. 2004, 10, 5595–5603. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Guevara, P.; Escobar, E.; García-Navarrete, R.; Pineda, B.; Sotelo, J. Blockage of angiotensin II type I receptor decreases the synthesis of growth factors and induces apoptosis in C6 cultured cells and C6 rat glioma. Br. J. Cancer 2005, 92, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hu, S.; Chen, B. Growth inhibition of epidermal growth factor-stimulated human glioblastoma cells by nicardipine in vitro. J. Neurosurg. Sci. 2001, 45, 151–155. [Google Scholar] [PubMed]
- Zhang, Y.; Cruickshanks, N.; Yuan, F.; Wang, B.; Pahuski, M.; Wulfkuhle, J.; Gallagher, I.; Koeppel, A.F.; Hatef, S.; Papanicolas, C.; et al. Targetable T-type Calcium Channels Drive Glioblastoma. Cancer Res. 2017, 77, 3479–3490. [Google Scholar] [CrossRef] [PubMed]
- Assad Kahn, S.; Costa, S.L.; Gholamin, S.; Nitta, R.T.; Dubois, L.G.; Fève, M.; Zeniou, M.; Coelho, P.L.; El-Habr, E.; Cadusseau, J.; et al. The anti-hypertensive drug prazosin inhibits glioblastoma growth via the PKCδ-dependent inhibition of the AKT pathway. EMBO Mol. Med. 2016, 8, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Durmaz, R.; Deliorman, S.; Uyar, R.; Işiksoy, S.; Erol, K.; Tel, E. The effects of anticancer drugs in combination with nimodipine and verapamil on cultured cells of glioblastoma multiforme. Clin. Neurol. Neurosurg. 1999, 101, 238–244. [Google Scholar] [CrossRef]
- Liu, W.T.; Huang, C.Y.; Lu, I.C.; Gean, P.W. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro-Oncoloy 2013, 15, 1127–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiger, T.M.; Colombatto, S.; Kainz, V.; Heidegger, W.; Grillo, M.A.; Hermann, A. Potassium channel blockers quinidine and caesium halt cell proliferation in C6 glioma cells via a polyamine-dependent mechanism. Biochem. Soc. Trans. 2007, 35, 391–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, W.K.; Kyritsis, A.P.; Gleason, M.J.; Levin, V.A. Treatment of recurrent malignant gliomas with high-dose 13-cis-retinoic acid. Clin. Cancer Res. 1996, 2, 1931–1935. [Google Scholar] [PubMed]
- Baumann, F.; Bjeljac, M.; Kollias, S.S.; Baumert, B.G.; Brandner, S.; Rousson, V.; Yonekawa, Y.; Bernays, R.L. Combined thalidomide and temozolomide treatment in patients with glioblastoma multiforme. J. Neurooncol. 2004, 67, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.R.; Ye, X.; Supko, J.G.; Desideri, S.; Grossman, S.A.; Brem, S.; Mikkelson, T.; Wang, D.; Chang, Y.C.; Hu, J.; et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy 2014, 10, 1359–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toler, S.M.; Noe, D.; Sharma, A. Selective enhancement of cellular oxidative stress by chloroquine: Implications for the treatment of glioblastoma multiforme. Neurosurg. Focus 2006, 21, E10. [Google Scholar] [CrossRef] [PubMed]
- Gey, G.O.; Levy, R.H.; Pettet, G.; Fisher, L. Quinidine plasma concentration and exertional arrhythmia. Am. Heart J. 1975, 90, 19–24. [Google Scholar] [CrossRef]
- Kast, R.E.; Boockvar, J.A.; Brüning, A.; Cappello, F.; Chang, W.W.; Cvek, B.; Dou, Q.P.; Duenas-Gonzalez, A.; Efferth, T.; Focosi, D.; et al. A conceptually new treatment approach for relapsed glioblastoma: Coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget 2013, 4, 502–530. [Google Scholar] [CrossRef] [PubMed]
- Pernicova, I.; Korbonits, M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Antonoff, M.B.; D’Cunha, J. Teaching an old drug new tricks: Metformin as a targeted therapy for lung cancer. Semin. Thorac. Cardiovasc. Surg. 2010, 22, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Memmott, R.M.; Mercado, J.R.; Maier, C.R.; Kawabata, S.; Fox, S.D.; Dennis, P.A. Metformin prevents tobacco carcinogen—induced lung tumorigenesis. Cancer Prev. Res. 2010, 3, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.B.; Matsuzaki, H.; Haorah, J.; Ulrich, A.; Standop, J.; Ding, X.Z.; Adrian, T.E.; Pour, P.M. Prevention of pancreatic cancer induction in hamsters by metformin. Gastroenterology 2001, 120, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.B.; Tian, S.; Gao, H.H.; Xu, Y.Y. Metformin inhibits glioma cell U251 invasion by downregulation of fibulin-3. Neuroreport 2013, 24, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Gritti, M.; Würth, R.; Angelini, M.; Barbieri, F.; Peretti, M.; Pizzi, E.; Pattarozzi, A.; Carra, E.; Sirito, R.; Daga, A.; et al. Metformin repositioning as antitumoral agent: Selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1-mediated ion current. Oncotarget 2014, 5, 11252–11268. [Google Scholar] [CrossRef] [PubMed]
- Isakovic, A.; Harhaji, L.; Stevanovic, D.; Markovic, Z.; Sumarac-Dumanovic, M.; Starcevic, V.; Micic, D.; Trajkovic, V. Dual antiglioma action of metformin: Cell cycle arrest and mitochondria-dependent apoptosis. Cell. Mol. Life Sci. 2007, 64, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Seliger, C.; Meyer, A.L.; Renner, K.; Leidgens, V.; Moeckel, S.; Jachnik, B.; Dettmer, K.; Tischler, U.; Gerthofer, V.; Rauer, L.; et al. Metformin inhibits proliferation and migration of glioblastoma cells independently of TGF-β2. Cell Cycle 2016, 15, 1755–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Zhao, G.; Xie, G.; Zhao, L.; Chen, Y.; Yu, H.; Zhang, Z.; Li, C.; Li, Y. Metformin and temozolomide act synergistically to inhibit growth of glioma cells and glioma stem cells in vitro and in vivo. Oncotarget 2015, 6, 32930–32943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, Z.; Mikkelsen, T.; Poisson, L.; Shackelford, D.B.; Brodie, C. Repurposing phenformin for the targeting of glioma stem cells and the treatment of glioblastoma. Oncotarget 2016, 7, 56456–56470. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.T.; Delfino, S.F.; Feinberg, C.E.; Johnson, M.F.; Nappi, V.L.; Olinger, J.T.; Schwab, A.P.; Swanson, H.I. Current and Emerging Uses of Statins in Clinical Therapeutics: A Review. Lipid Insights 2016, 9, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Bayat, N.; Ebrahimi-Barough, S.; Norouzi-Javidan, A.; Saberi, H.; Tajerian, R.; Ardakan, M.M.M.; Shirian, S.; Ai, A.; Ai, J. Apoptotic effect of atorvastatin in glioblastoma spheroids tumor cultured in fibrin gel. Biomed. Pharmacother. 2016, 84, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- May, M.B.; Glode, A. Novel Uses for Lipid-Lowering Agents. J. Adv. Pract. Oncol. 2016, 7, 181–187. [Google Scholar] [PubMed]
- Oliveira, K.A.; Dal-Cim, T.; Lopes, F.G.; Ludka, F.K.; Nedel, C.B.; Tasca, C.I. Atorvastatin Promotes Cytotoxicity and Reduces Migration and Proliferation of Human A172 Glioma Cells. Mol. Neurobiol. 2018, 55, 1509–1523. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Perez, J.H.; Kirches, E.; Mawrin, C.; Firsching, R.; Schneider, T. Cytotoxic effect of different statins and thiazolidinediones on malignant glioma cells. Cancer Chemother. Pharmacol. 2011, 67, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Yanae, M.; Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yamazoe, Y.; Nishida, S. Statin-induced apoptosis via the suppression of ERK1/2 and Akt activation by inhibition of the geranylgeranyl-pyrophosphate biosynthesis in glioblastoma. J. Exp. Clin. Cancer Res. 2011, 30, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayat, N.; Ebrahimi-Barough, S.; Norouzi-Javidan, A.; Saberi, H.; Ardakan, M.M.M.; Ai, A.; Soleimannejad, M.; Ai, J. Anti-inflammatory Effects of Atorvastatin by Suppressing TRAF3IP2 and IL-17RA in Human Glioblastoma Spheroids Cultured in a Three-dimensional Model: Possible Relevance to Glioblastoma Treatment. Mol. Neurobiol. 2018, 55, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Girgert, R.; Vogt, Y.; Becke, D.; Bruchelt, G.; Schweizer, P. Growth inhibition of neuroblastoma cells by lovastatin and L-ascorbic acid is based on different mechanisms. Cancer Lett. 1999, 137, 167–172. [Google Scholar] [CrossRef]
- Koyuturk, M.; Ersoz, M.; Altiok, N. Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via c-jun N-terminal kinase. Neurosci. Lett. 2004, 370, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Obara, S.; Nakata, M.; Takeshima, H.; Kuratsu, J.; Maruyama, I.; Kitajima, I. Inhibition of migration of human glioblastoma cells by cerivastatin in association with focal adhesion kinase (FAK). Cancer Lett. 2002, 185, 153–161. [Google Scholar] [CrossRef]
- Yongjun, Y.; Shuyun, H.; Lei, C.; Xiangrong, C.; Zhilin, Y.; Yiquan, K. Atorvastatin suppresses glioma invasion and migration by reducing microglial MT1-MMP expression. J. Neuroimmunol. 2013, 260, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Björkhem-Bergman, L.; Lindh, J.D.; Bergman, P. What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br. J. Clin. Pharmacol. 2011, 72, 164–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurien, G.; Pellegrini, M.V. Dapsone. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2018. [Google Scholar]
- Kast, R.E.; Lefranc, F.; Karpel-Massler, G.; Halatsch, M.E. Why dapsone stops seizures and may stop neutrophils’ delivery of VEGF to glioblastoma. Br. J. Neurosurg. 2012, 26, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Karpel-Massler, G.; Kast, R.E.; Siegelin, M.D.; Dwucet, A.; Schneider, E.; Westhoff, M.A.; Wirtz, C.R.; Chen, X.Y.; Halatsch, M.E.; Bolm, C. Anti-glioma Activity of Dapsone and Its Enhancement by Synthetic Chemical Modification. Neurochem. Res. 2017, 42, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Zuidema, J.; Hilbers-Modderman, E.S.; Merkus, F.W. Clinical pharmacokinetics of dapsone. Clin. Pharmacokinet. 1986, 11, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.I.; Wang, S.; Yang, D.; Pan, K.; Li, L.; Yuan, S. Preclinical pharmacodynamic evaluation of antibiotic nitroxoline for anticancer drug repurposing. Oncol. Lett. 2016, 11, 3265–3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelletier, C.; Prognon, P.; Bourlioux, P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob. Agents Chemother. 1995, 39, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Wagenlehner, F.M.E.; Münch, F.; Pilatz, A.; Bärmann, B.; Weidner, W.; Wagenlehner, C.M.; Straubinger, M.; Blenk, H.; Pfister, W.; Kresken, M.; et al. Urinary Concentrations and Antibacterial Activities of Nitroxoline at 250 Milligrams versus Trimethoprim at 200 Milligrams against Uropathogens in Healthy Volunteers. Antimicrob. Agents Chemother. 2014, 58, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Chan-on, W.; Huyen, N.T.B.; Songtawee, N.; Suwanjang, W.; Prachayasittikul, S.; Prachayasittikul, V. Quinoline-based clioquinol and nitroxoline exhibit anticancer activity inducing FoxM1 inhibition in cholangiocarcinoma cells. Drug Des. Dev. Ther. 2015, 9, 2033–2047. [Google Scholar]
- Shim, J.S.; Matsui, Y.; Bhat, S.; Nacev, B.A.; Xu, J.; Bhang, H.E.; Dhara, S.; Han, K.C.; Chong, C.R.; Pomper, M.G.; et al. Effect of nitroxoline on angiogenesis and growth of human bladder cancer. J. Natl. Cancer Inst. 2010, 102, 1855–1873. [Google Scholar] [CrossRef] [PubMed]
- Mai, J.; Sameni, M.; Mikkelsen, T.; Sloane, B.F. Degradation of extracellular matrix protein tenascin-C by cathepsin B: An interaction involved in the progression of gliomas. Biol. Chem. 2002, 383, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, B.; Renko, M.; Turk, S.; Sosic, I.; Jevnikar, Z.; Obermajer, N.; Turk, D.; Gobec, S.; Kos, J. Novel mechanism of cathepsin B inhibition by antibiotic nitroxoline and related compounds. ChemMedChem 2011, 6, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Lazovic, J.; Guo, L.; Nakashima, J.; Mirsadraei, L.; Yong, W.; Kim, H.J.; Ellingson, B.; Wu, H.; Pope, W.B. Nitroxoline induces apoptosis and slows glioma growth in vivo. Neuro-Oncoloy 2015, 17, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Mushtaque, M.; Shahjahan. Reemergence of chloroquine (CQ) analogs as multi-targeting antimalarial agents: A review. Eur. J. Med. Chem. 2015, 90, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Manic, G.; Obrist, F.; Kroemer, G.; Vitale, I.; Galluzzi, L. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell. Oncol. 2014, 1, e29911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Y.D.; Jiang, S.L.; Yu, P.; Wen, M.; Zhang, Y.; Xiao, S.S.; Xu, X.J.; Cheng, Y. Suppression of eEF-2K-mediated autophagy enhances the cytotoxicity of Raddeanin A against human breast cancer cells in vitro. Acta Pharmacol. Sin. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xi, C.; Kuang, J.; Feng, H.; Chen, L.; Liang, J.; Shen, X.; Yuen, S.; Chenghong, P.; Baiyong, S.; et al. CQ sensitizes human pancreatic cancer cells to gemcitabine through the lysosomal apoptotic pathway via reactive oxygen species. Mol. Oncol. 2018, 12, 529–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monma, H.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Tajima, Y.; Harada, M. Chloroquine augments TRAIL-induced apoptosis and induces G2/M phase arrest in human pancreatic cancer cells. PLoS ONE 2018, 13, e0193990. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Cao, L.; Zhang, Q.; Song, Q.; Meng, Z.; Wu, X.; Xu, K. Inhibition of autophagy potentiates the anti-metastasis effect of phenethyl isothiocyanate through JAK2/STAT3 pathway in lung cancer cells. Mol. Carcinog. 2018, 57, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, G.; Xiong, L.; Liu, Z.; Lam, B.; Bui, B.; Ma, L.; Chen, X.; Zhou, P.; Wang, K.; Zhang, Z.; et al. Inhibition of autophagy potentiates the apoptosis-inducing effects of photodynamic therapy on human colon cancer cells. Photodiagn. Photodyn. Ther. 2018, 21, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tang, J.; Li, P.; Si, S.; Yu, H.; Yang, X.; Tao, J.; Lv, Q.; Gu, M.; Yang, H.; et al. Chloroquine Enhances the Radiosensitivity of Bladder Cancer Cells by Inhibiting Autophagy and Activating Apoptosis. Cell. Physiol. Biochem. 2018, 45, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Golden, E.B.; Cho, H.Y.; Jahanian, A.; Hofman, F.M.; Louie, S.G.; Schonthal, A.H.; Chen, T.C. Chloroquine enhances temozolomide cytotoxicity in malignant gliomas by blocking autophagy. Neurosurg. Focus 2014, 37, E12. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Hosoda, R.; Akiyama, Y.; Sebori, R.; Wanibuchi, M.; Mikami, T.; Sugino, T.; Suzuki, K.; Maruyama, M.; Tsukamoto, M.; et al. Chloroquine potentiates temozolomide cytotoxicity by inhibiting mitochondrial autophagy in glioma cells. J. Neurooncol. 2015, 122, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, H.K.; Lee, N.H.; Yi, H.Y.; Kim, H.S.; Hong, S.H.; Hong, Y.K.; Joe, Y.A. The synergistic effect of combination temozolomide and chloroquine treatment is dependent on autophagy formation and p53 status in glioma cells. Cancer Lett. 2015, 360, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Xu, Z.; Dai, S.; Qian, L.; Sun, L.; Gong, Z. Targeting autophagy to sensitive glioma to temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 23. [Google Scholar] [CrossRef] [PubMed]
- Firat, E.; Weyerbrock, A.; Gaedicke, S.; Grosu, A.L.; Niedermann, G. Chloroquine or chloroquine-PI3K/Akt pathway inhibitor combinations strongly promote gamma-irradiation-induced cell death in primary stem-like glioma cells. PLoS ONE 2012, 7, e47357. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Chen, M.; Cao, F.; Huang, H.; Zhan, R.; Zheng, X. Chloroquine, an autophagy inhibitor, potentiates the radiosensitivity of glioma initiating cells by inhibiting autophagy and activating apoptosis. BMC Neurol. 2016, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Qin, Z.; Liang, Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim. Biophys. Sin. 2009, 41, 341–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.J.; Card, T.; Bates, T.E.; Muir, K. Tricyclic antidepressants and the incidence of certain cancers: A study using the GPRD. Br. J. Cancer 2011, 104, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Levkovitz, Y.; Gil-Ad, I.; Zeldich, E.; Dayag, M.; Weizman, A. Differential induction of apoptosis by antidepressants in glioma and neuroblastoma cell lines: Evidence for p-c-Jun, cytochrome c, and caspase-3 involvement. J. Mol. Neurosci. 2005, 27, 29–42. [Google Scholar] [CrossRef]
- Spanová, A.; Kovárů, H.; Lisá, V.; Lukásová, E.; Rittich, B. Estimation of apoptosis in C6 glioma cells treated with antidepressants. Physiol. Res. 1997, 46, 161–164. [Google Scholar] [PubMed]
- Liu, K.H.; Yang, S.T.; Lin, Y.K.; Lin, J.W.; Lee, Y.H.; Wang, J.Y.; Hu, C.J.; Lin, E.Y.; Chen, S.M.; Then, C.K.; et al. Fluoxetine, an antidepressant, suppresses glioblastoma by evoking AMPAR-mediated calcium-dependent apoptosis. Oncotarget 2015, 6, 5088–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, K.; Michiue, H.; Yamada, H.; Takata, K.; Nakayama, H.; Wei, F.Y.; Fujimura, A.; Tazawa, H.; Asai, A.; Ogo, N.; et al. Fluvoxamine, an anti-depressant, inhibits human glioblastoma invasion by disrupting actin polymerization. Sci. Rep. 2016, 6, 23372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.D.; Lewis, R.J.; Angier, M.K. The distribution of fluoxetine in human fluids and tissues. J. Anal. Toxicol. 2007, 31, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Karson, C.N.; Newton, J.E.; Livingston, R.; Jolly, J.B.; Cooper, T.B.; Sprigg, J.; Komoroski, R.A. Human brain fluoxetine concentrations. J. Neuropsychiatry Clin. Neurosci. 1993, 5, 322–329. [Google Scholar] [PubMed]
- Bielecka-Wajdman, A.M.; Lesiak, M.; Ludyga, T.; Sieron, A.; Obuchowicz, E. Reversing glioma malignancy: A new look at the role of antidepressant drugs as adjuvant therapy for glioblastoma multiforme. Cancer Chemother. Pharmacol. 2017, 79, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.H.; Kim, S.H.; Kim, Y.; Kim, Y.S.; Lim, Y.; Lee, Y.H.; Shin, S.Y. The tricyclic antidepressant imipramine induces autophagic cell death in U-87MG glioma cells. Biochem. Biophys. Res. Commun. 2011, 413, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Shchors, K.; Massaras, A.; Hanahan, D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 2015, 28, 456–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, A.; Eder, K.; Selak, M.A.; Kalman, B. Mitochondrial energy metabolism and apoptosis regulation in glioblastoma. Brain Res. 2015, 1595, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Daley, E.; Wilkie, D.; Loesch, A.; Hargreaves, I.P.; Kendall, D.A.; Pilkington, G.J.; Bates, T.E. Chlorimipramine: A novel anticancer agent with a mitochondrial target. Biochem. Biophys. Res. Commun. 2005, 328, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Pilkington, G.J.; Parker, K.; Murray, S.A. Approaches to mitochondrially mediated cancer therapy. Semin. Cancer Biol. 2008, 18, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Zanotto-Filho, A.; Braganhol, E.; Battastini, A.M.; Moreira, J.C. Proteasome inhibitor MG132 induces selective apoptosis in glioblastoma cells through inhibition of PI3K/Akt and NFκB pathways, mitochondrial dysfunction, and activation of p38-JNK1/2 signaling. Investig. New Drugs 2012, 30, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow MeTable 2007, 27, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sanchez, R.; Rodriguez-Enriquez, S.; Marin-Hernandez, A.; Saavedra, E. Energy metabolism in tumor cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordys, B.B.; Launay, S.; Deighton, R.F.; McCulloch, J.; Whittle, I.R. The role of mitochondria in glioma pathophysiology. Mol. Neurobiol. 2010, 42, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Marie, S.K.N.; Shinjo, S.M.O. Metabolism and Brain Cancer. Clinics 2011, 66, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Galarraga, J.; Loreck, D.J.; Graham, J.F.; DeLaPaz, R.L.; Smith, B.H.; Hallgren, D.; Cummins, C.J. Glucose metabolism in human gliomas: Correspondence of in situ and in vitro metabolic rates and altered energy metabolism. Metab. Brain Dis. 1986, 1, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Capuano, F.; Palombini, G.; Galeotti, T.; Papa, S. Transport of Pyruvate in Mitochondria from Different Tumor Cells. Cancer Res. 1983, 43, 5068–5071. [Google Scholar] [PubMed]
- Baggetto, L.G.; Lehninger, A.L. Formation and utilization of acetoin, an unusual product of pyruvate metabolism by Ehrlich and AS30-D tumor mitochondria. J. Biol. Chem. 1987, 262, 9535–9541. [Google Scholar] [PubMed]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.; McCormick, F.; Levitzki, A. Killing time for cancer cells. Nat. Rev. Cancer 2005, 5, 573–580. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J.; Manion, M.; Schwartz, P.; Hockenbery, D.M. Promises and challenges of targeting Bcl-2 anti-apoptotic proteins for cancer therapy. Biochim. Biophys. Acta 2004, 1705, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Kohler, U.; Schackert, G.; Schackert, H.K. Expression of TRAIL and its receptors in human brain tumors. Biochem. Biophys. Res. Commun. 1999, 257, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Vignot, S.; Faivre, S.; Aguirre, D.; Raymond, E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann. Oncol. 2005, 16, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenknecht, B.; Glaser, T.; Naumann, U.; Kugler, S.; Isenmann, S.; Bahr, M.; Korneluk, R.; Liston, P.; Weller, M. Expression and biological activity of X-linked inhibitor of apoptosis (XIAP) in human malignant glioma. Cell Death Differ. 1999, 6, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Malipiero, U.; Aguzzi, A.; Reed, J.C.; Fontana, A. Protooncogene bcl-2 gene transfer abrogates Fas/APO-1 antibody-mediated apoptosis of human malignant glioma cells and confers resistance to chemotherapeutic drugs and therapeutic irradiation. J. Clin. Investig. 1995, 95, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Stegh, A.H.; Kim, H.; Bachoo, R.M.; Forloney, K.L.; Zhang, J.; Schulze, H.; Park, K.; Hannon, G.J.; Yuan, J.; Louis, D.N.; et al. Bcl2L12 inhibits post-mitochondrial apoptosis signaling in glioblastoma. Genes Dev. 2007, 21, 98–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajewska, M.; Krajewski, S.; Epstein, J.I.; Shabaik, A.; Sauvageot, J.; Song, K.; Kitada, S.; Reed, J.C. Immunohistochemical analysis of bcl-2, bax, bcl-X, and mcl-1 expression in prostate cancers. Am. J. Pathol. 1996, 148, 1567–1576. [Google Scholar] [PubMed]
- Nakasu, S.; Nakasu, Y.; Nioka, H.; Nakajima, M.; Handa, J. bcl-2 protein expression in tumors of the central nervous system. Acta Neuropathol. 1994, 88, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushal, S.; Wang, W.; Vaikari, V.P.; Kota, R.; Chen, K.; Yeh, T.S.; Jhaveri, N.; Groshen, S.L.; Olenyuk, B.Z.; Chen, T.C.; et al. Monoamine oxidase A (MAO A) inhibitors decrease glioma progression. Oncotarget 2016, 7, 13842–13853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, E.; Hammadi, M.; Kischel, P.; Delcroix, V.; Demaurex, N.; Castelbou, C.; Vacher, A.M.; Devin, A.; Ducret, T.; Nunes, P.; et al. The antidepressant fluoxetine induces necrosis by energy depletion and mitochondrial calcium overload. Oncotarget 2017, 8, 3181–3196. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Moellering, D.R.; Gillespie, G.Y.; Griguer, C.E. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. PLoS ONE 2011, 6, e24665. [Google Scholar] [CrossRef] [PubMed]
- Oliva, C.R.; Markert, T.; Gillespie, G.Y.; Griguer, C.E. Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas. Oncotarget 2015, 6, 4330–4344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, C.R.; Markert, T.; Ross, L.J.; White, E.L.; Rasmussen, L.; Zhang, W.; Everts, M.; Moellering, D.R.; Bailey, S.M.; Suto, M.J.; et al. Identification of Small Molecule Inhibitors of Human Cytochrome C Oxidase That Target Chemoresistant Glioma Cells. J. Biol. Chem. 2016, 291, 24188–24199. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.A.; Han, J.; Baskin, A.M.; Baskin, D.S. Design and synthesis of a MAO-B-selectively activated prodrug based on MPTP: A mitochondria-targeting chemotherapeutic agent for treatment of human malignant gliomas. ChemMedChem 2015, 10, 621–628. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Some of the drugs which could be acting via mitochondria in GBM [107]. Involvement of mitochondria in the mechanism of action of some of the drugs suggested for therapy of GBM. (1) Effect of imipramine and P2Y12 purinergic receptor blocker ticlopidine (TIC) on GBM cells, as described by Shchors et al. [99]. Imipramine and TIC together act synergistically. Imipramine activates Gsα protein-coupled monoamine receptors, which in turn activate adenylate cyclase. Ticlopidine blocks P2Y12 receptor, a Gi protein-coupled purinergic receptor that normally inhibits adenylyl cyclase. eventually, activity of adenylate cyclase increases and cAMP level rises leading to -via EPAC pathway- autophagy and cell death. (2) Dibutyryl-cAMP (dbcAMP) activates phosphoprotein kinase A and across cAMP response element-binding protein (CREB protein) activates the synthesis of Peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) protein. It finally leads to mitochondrial biogenesis, metabolic reprogramming, and tumor cell differentiation [104]. (3) Сlorgyline alone or with TMZ acts as monoamine oxidase A inhibitor and reduces tumor growth [125]. (4) TCA clomipramine and SSRIs directly interact with complex III of respiratory chain, decreasing O2 consumption and stimulating reactive oxygen species generation. Mitochondrial membrane potential is reduced finally resulting in apoptosis [101]. (5) Chlorpromazine interacts with cytochrome c oxidase and reduces its activity, this leads to cell cycle arrest and inhibition of proliferation of GBM cells [126]. (6) Fluoxetine interacts with GluR1 subunit of AMPA receptors, leading to an increase in intracellular Ca2+, mitochondrial calcium overload and activation of the intrinsic apoptotic pathway [93].
Figure 1.
Some of the drugs which could be acting via mitochondria in GBM [107]. Involvement of mitochondria in the mechanism of action of some of the drugs suggested for therapy of GBM. (1) Effect of imipramine and P2Y12 purinergic receptor blocker ticlopidine (TIC) on GBM cells, as described by Shchors et al. [99]. Imipramine and TIC together act synergistically. Imipramine activates Gsα protein-coupled monoamine receptors, which in turn activate adenylate cyclase. Ticlopidine blocks P2Y12 receptor, a Gi protein-coupled purinergic receptor that normally inhibits adenylyl cyclase. eventually, activity of adenylate cyclase increases and cAMP level rises leading to -via EPAC pathway- autophagy and cell death. (2) Dibutyryl-cAMP (dbcAMP) activates phosphoprotein kinase A and across cAMP response element-binding protein (CREB protein) activates the synthesis of Peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) protein. It finally leads to mitochondrial biogenesis, metabolic reprogramming, and tumor cell differentiation [104]. (3) Сlorgyline alone or with TMZ acts as monoamine oxidase A inhibitor and reduces tumor growth [125]. (4) TCA clomipramine and SSRIs directly interact with complex III of respiratory chain, decreasing O2 consumption and stimulating reactive oxygen species generation. Mitochondrial membrane potential is reduced finally resulting in apoptosis [101]. (5) Chlorpromazine interacts with cytochrome c oxidase and reduces its activity, this leads to cell cycle arrest and inhibition of proliferation of GBM cells [126]. (6) Fluoxetine interacts with GluR1 subunit of AMPA receptors, leading to an increase in intracellular Ca2+, mitochondrial calcium overload and activation of the intrinsic apoptotic pathway [93].


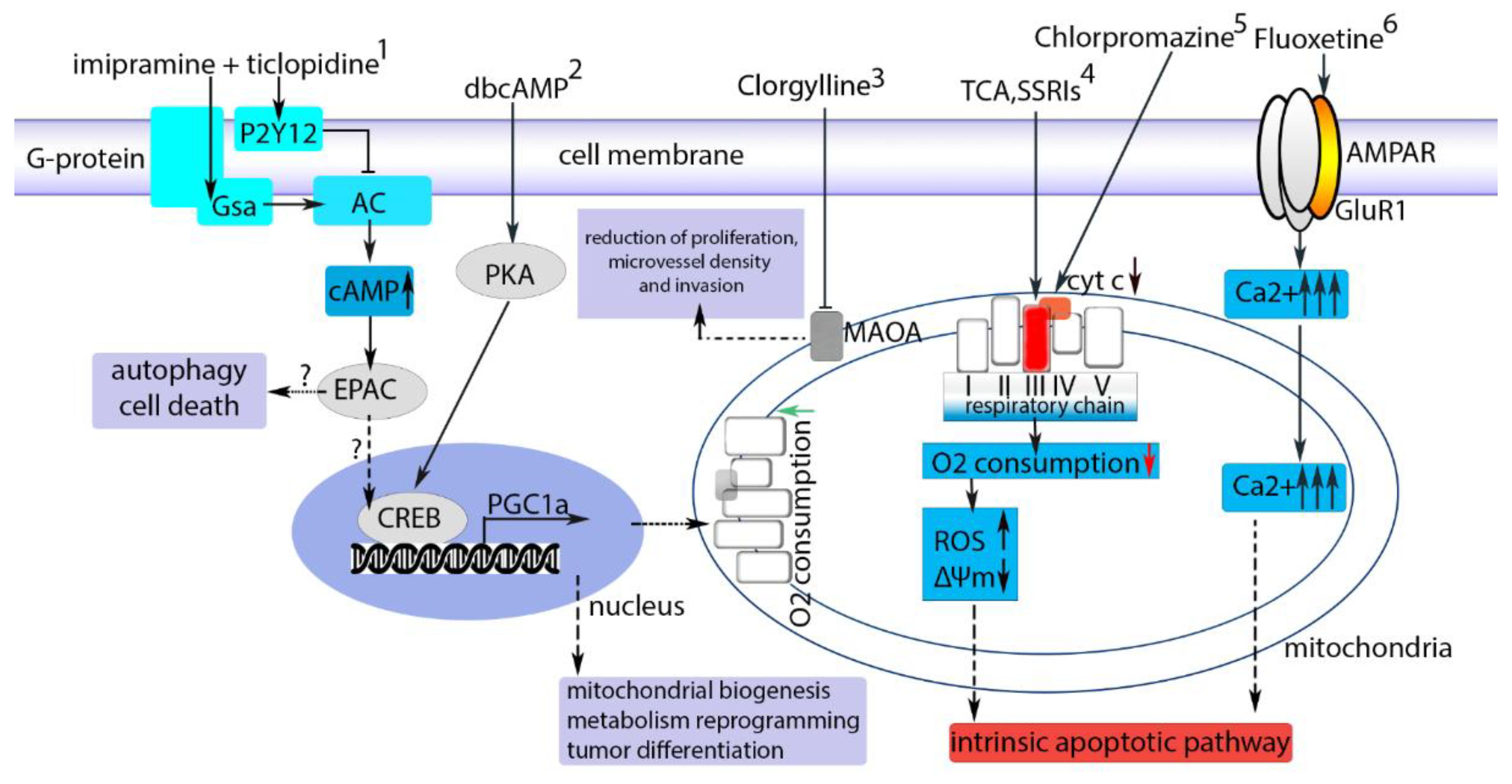{kind=link}
Table 1.
Some of the drugs suggested for repurposing as glioblastoma multiforme (GBM) therapeutics and their proposed mechanisms of action.
Table 1.
Some of the drugs suggested for repurposing as glioblastoma multiforme (GBM) therapeutics and their proposed mechanisms of action.
| Agent | Proposed Anti-GBM Mechanism | Existing Indication and Main Mechanism (If Known) | Reference |
|---|---|---|---|
| Nelfinavir | PI3K-Akt signaling inhibition | HIV protease inhibitor | [11] |
| Cimetidine | Immunomodulation | Peptic ulcers (Histamine H2 blocker) | [12] |
| Diclofenac | Prostaglandin synthesis inhibition | Inflammation and pain (COX-2 inhibitor) | [13] |
| Nitroglycerin | Nitric oxide donor | Angina | [14] |
| Thioridazine | Induces autophagy and upregulates AMPK activity | Antipsychotic psychosis (blocks D2, 5-HT2A and other receptors) | [15] |
| Pimozide | Serotonin receptor-7 inhibition | Antipsychotic (blocks D2, 5-HT2A receptors, has relatively high affinity to 5-HT7 receptors) | [16] |
| Risperidone | Serotonin receptor-7 inhibition | Schizophrenia, bipolar disorder, and irritability | [16] |
| Paliperidone | Serotonin receptor-7 inhibition | Antipsychotic (blocks D2, 5-HT2A receptors, has relatively high affinity to 5-HT7 receptors) | [16] |
| Apomorphine | Mitochondrial metabolic gene downregulation | Emetic, sometimes used in Parkinson disease. Agonist of DA2, DA1, 5-HT2 and α-AR | [17] |
| Flupenthixol | Dopamine receptor modulation | Antipsychotic (typical anti-D2-agent) | [18] |
| Mebendazole | Tubulin polymerization inhibition | Nematode infestations | [19] |
| Disulfiram | Proteasome and alcohol dehydrogenase inhibition | Alcoholism | [20] |
| Valproic acid | Histone deacetylase inhibition | Epilepsy | [21] |
| Levetiracetam | MGMT activity inhibition | Epilepsy | [22] |
| Methadone | cAMP reduction | Severe pain, opioid agonist | [23] |
| Sulfasalazine | NF-κB activity suppression | Inflammatory bowel disease | [24] |
| Captopril | Angiotensin-converting enzyme inhibitor | Hypertension | [25] |
| Nicardipine | EGF and calcium channel antagonism | Hypertension and angina | [26] |
| Mibefradil | T-type calcium channel inhibition | Hypertension and angina | [27] |
| Prazosin | AKT pathway inhibition | Hypertension | [28] |
| Nimodipine | Calcium channel antagonism | Hypertension and angina | [29] |
| Minocycline | Apoptosis and autophagy | Antibiotic has multiple known central side effects | [30] |
| Quinidine | Ornithine decarboxylase activity inhibition | Heart arrhythmia | [31] |
| Accutane | Reduction of EGFR activity | Acne (13-cis-retinoic acid. Has known central side effects) | [32] |
| Thalidomide | Angiogenesis inhibition | Multiple myeloma, leprosy. | [33] |
| Dichloroacetate | Inhibition of anaerobic metabolism | Topically: warts removal. Congenital lactic acidosis. Inhibits pyruvate dehydrogenase kinase, which increase mitochondrial consumption of pyruvate. | [34] |
| Hydroxy-chloroquine | Autophagy inhibition | Malaria | [35] |
| Chloroquine | Oxidative stress enhancement | Malaria | [36] |
PI3K-Akt: phosphoinositide 3-kinase-protein kinase B, HIV: human immunodeficiency virus, COX-2: cyclooxygenase 2, AMPK: adenosine monophosphate activated protein kinase, EGFR: epidermal growth factor receptor, DA: Dopamine, 5-HT: Serotonin, cAMP: Cyclic adenosine monophosphate, NFκB: Nuclear factor kappa-light-chain-enhancer of activated B cells. Table 1 has been compiled based on the literature searches at the time of writing using standard keywords, data from clinical trials database and recent reviews. Conventional anti-cancer therapies are not included, since cancer is their main indication.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vasilev, A.; Sofi, R.; Tong, L.; Teschemacher, A.G.; Kasparov, S. In Search of a Breakthrough Therapy for Glioblastoma Multiforme. Neuroglia 2018, 1, 292-310. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020020
AMA Style
Vasilev A, Sofi R, Tong L, Teschemacher AG, Kasparov S. In Search of a Breakthrough Therapy for Glioblastoma Multiforme. Neuroglia. 2018; 1(2):292-310. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020020
Chicago/Turabian StyleVasilev, Alex, Roba Sofi, Li Tong, Anja G. Teschemacher, and Sergey Kasparov. 2018. "In Search of a Breakthrough Therapy for Glioblastoma Multiforme" Neuroglia 1, no. 2: 292-310. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020020