Putative Receptors Underpinning l-Lactate Signalling in Locus Coeruleus

 ,
,
Abstract
:1. Introduction
1.1. Targets and Mechanisms of l-Lactate Signalling in the Brain
1.2. Hydroxy-carboxylic acid receptor (HCA1)
1.3. Olfactory l-Lactate Receptors
1.4. Orphan G-protein-coupled receptor 4 (GPR4)
1.5. The l-Lactate Receptor Responsible for Central noradrenaline Release
2. Materials and Methods
2.1. Chemicals
2.2. Organotypic Brain Slice Preparation
2.3. Fluorescence-Activated Cell Sorting and RNA Sequencing of Locus Coeruleus Neurones and Astrocytes
2.4. Measurement of Noradrenaline Release
2.5. Patch Clamp of Noradrenergic Neurones
2.6. GloSensor Assay of Intracellular cAMP Accumulation
2.7. Statistical Analysis
3. Results
3.1. l-Lactate Releases Noradrenaline from Locus Coeruleus Neurones
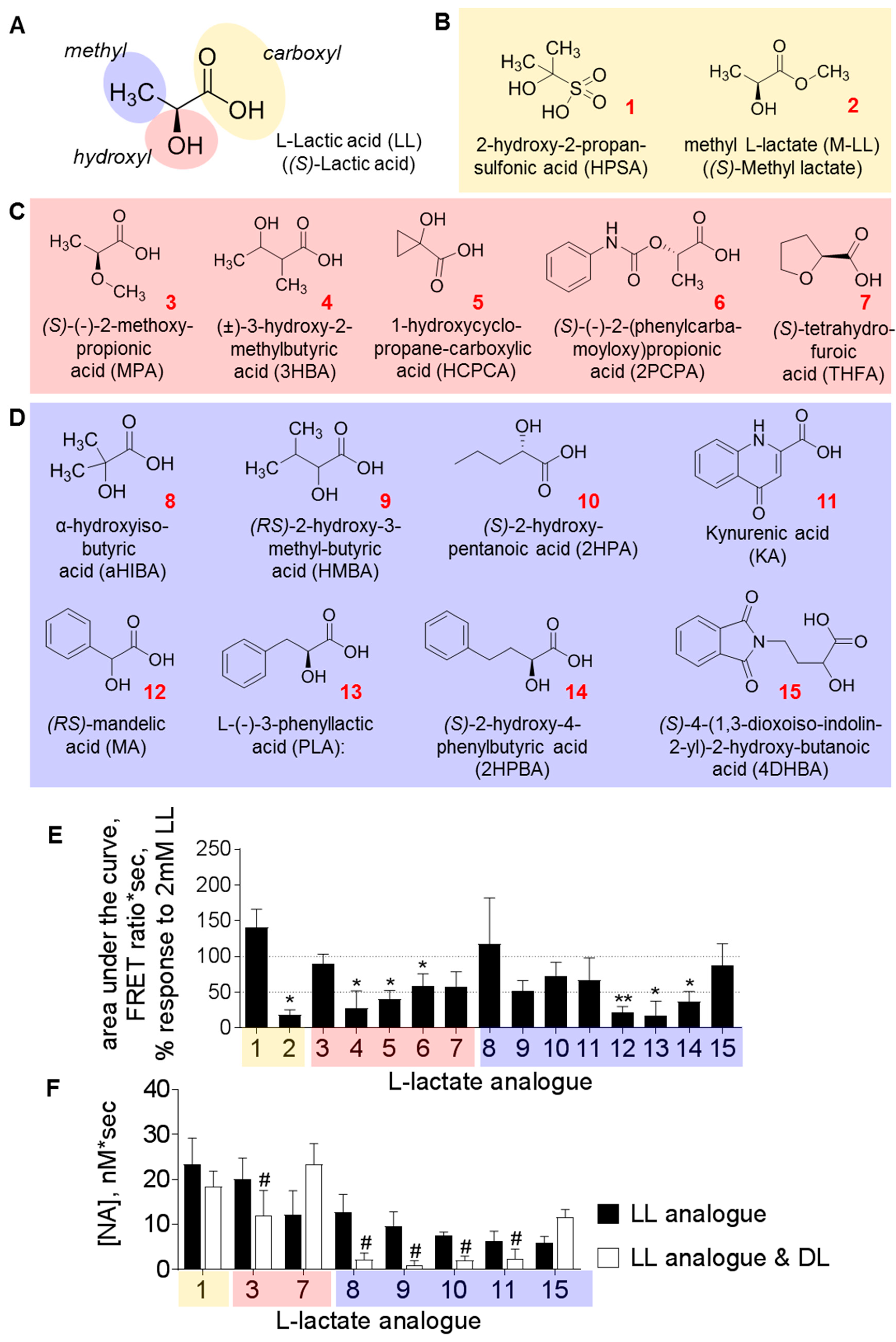
3.2. Structural Requirements for the Agonistic Action of l-Lactate on Noradrenaline Release
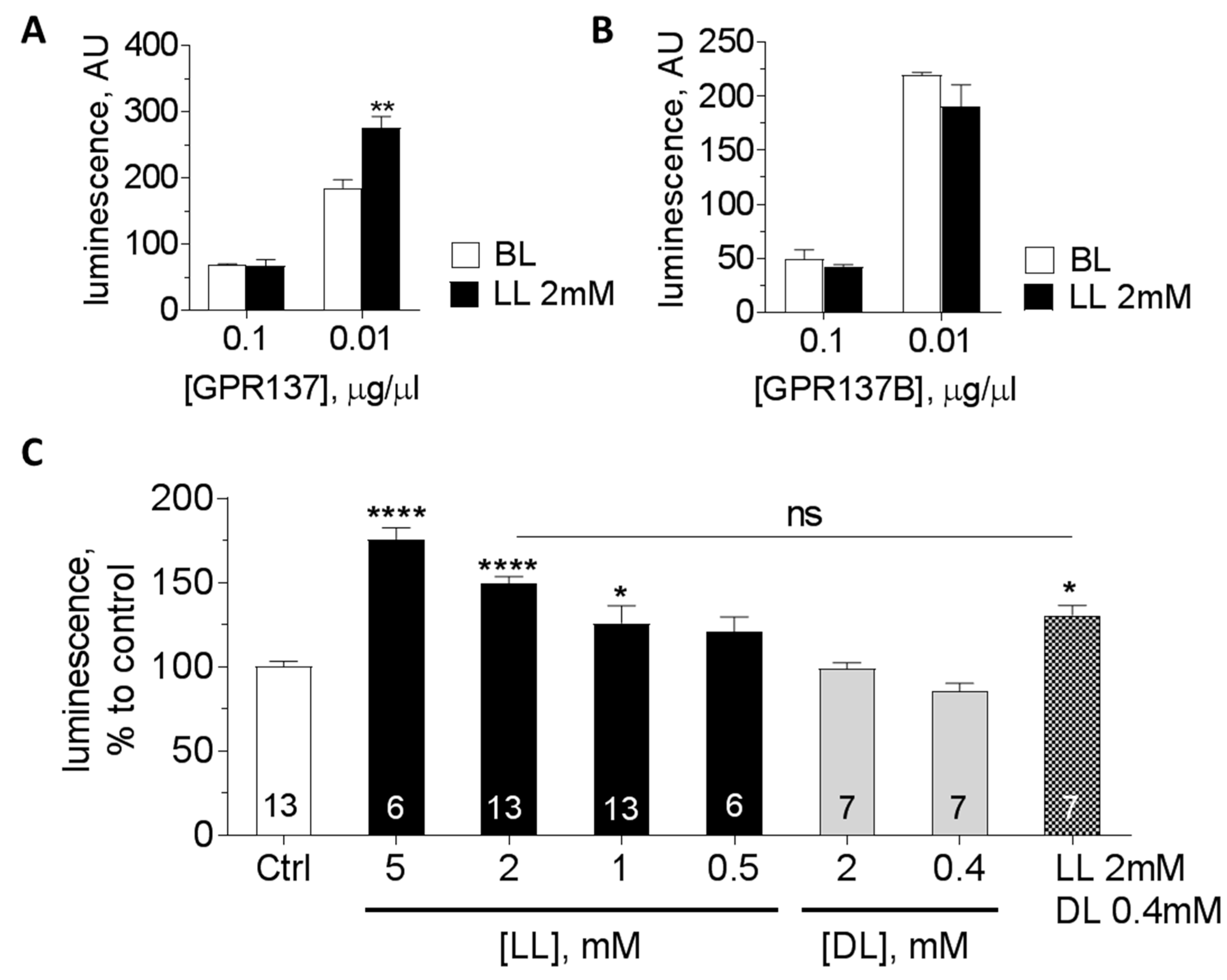
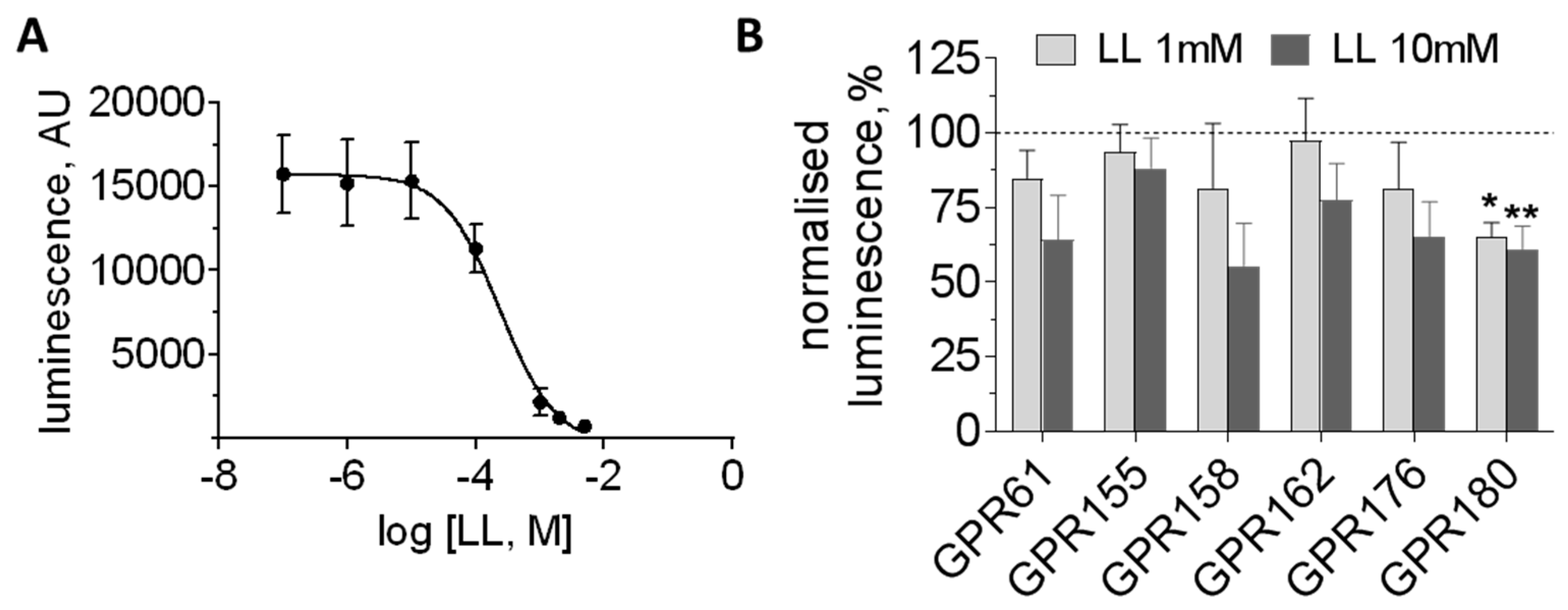
3.3. Orphan GPCRs as Putative Targets of l-Lactate
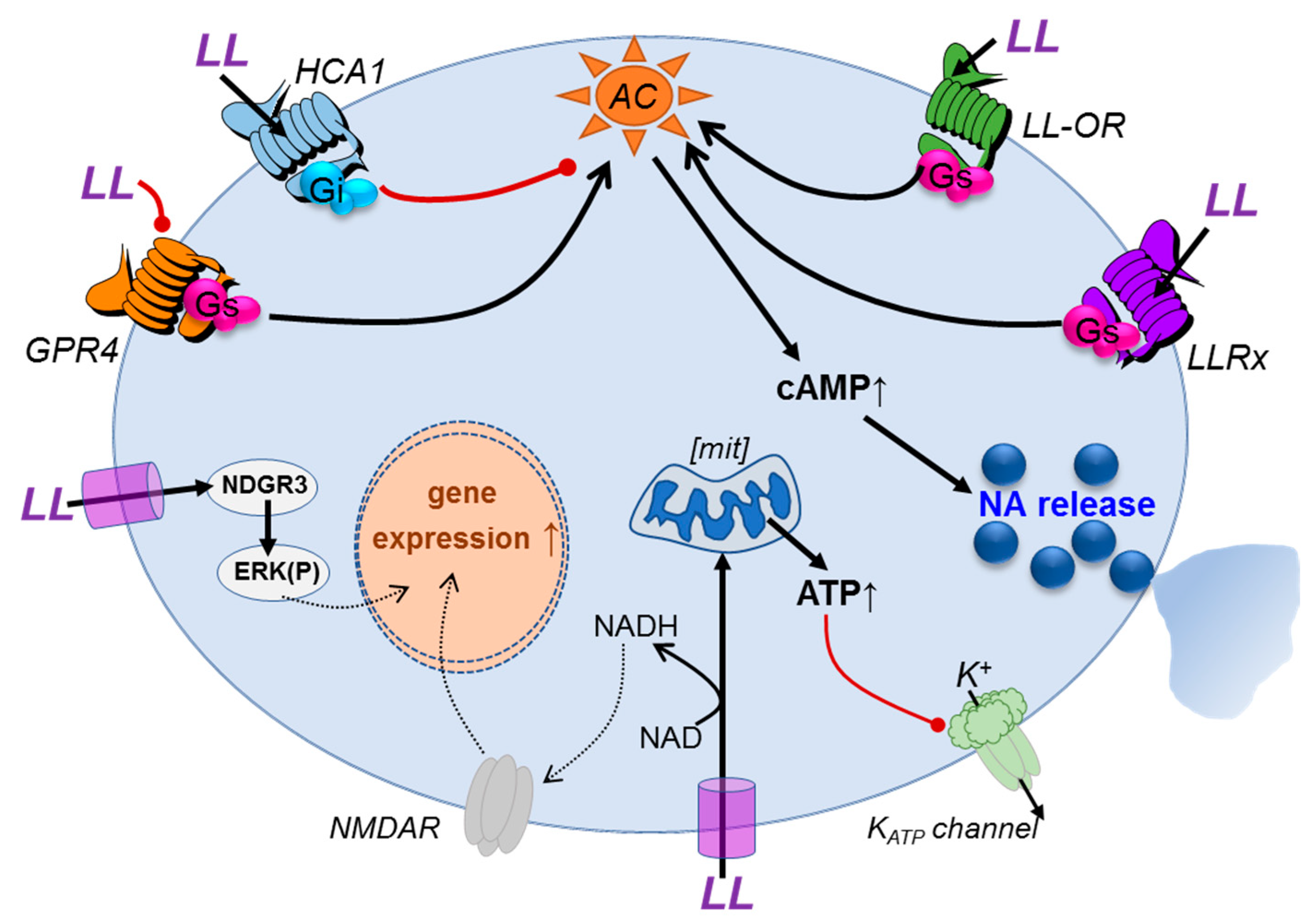
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oe, Y.; Baba, O.; Ashida, H.; Nakamura, K.C.; Hirase, H. Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. GLIA 2016, 64, 1532–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell. Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi-Saab, W.M.; Maggs, D.G.; Jones, T.; Jacob, R.; Srihari, V.; Thompson, J.; Kerr, D.; Leone, P.; Krystal, J.H.; Spencer, D.D.; et al. Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: Effects of hyperglycemia and hypoglycemia. J. Cereb. Blood Flow Metab. 2002, 22, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Mosienko, V.; Teschemacher, A.G.; Kasparov, S. Is l-lactate a novel signaling molecule in the brain? J. Cereb. Blood Flow Metab. 2015, 35, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Machler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martin, A.; Romero-Gomez, I.; et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Supplie, L.M.; Duking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Gotz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.A. Respiration-Deficient Astrocytes Survive as Glycolytic Cells In Vivo. J. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Moncada, I.; Ruminot, I.; Robles-Maldonado, D.; Alegria, K.; Deitmer, J.W.; Barros, L.F. Neuronal control of astrocytic respiration through a variant of the Crabtree effect. Proc. Natl. Acad. Sci. USA 2018, 115, 1623–1628. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2011, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Barros, L.F. The Astrocyte: Powerhouse and Recycling Center. Cold Spring Harb. Perspect. Biol. 2015, 7, a020396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [PubMed]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef] [PubMed]
- Alberini, C.M.; Cruz, E.; Descalzi, G.; Bessieres, B.; Gao, V. Astrocyte glycogen and lactate: New insights into learning and memory mechanisms. GLIA 2017, 66, 1244–1262. [Google Scholar] [CrossRef] [PubMed]
- Clasadonte, J.; Scemes, E.; Wang, Z.; Boison, D.; Haydon, P.G. Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron 2017, 95, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Naylor, E.; Aillon, D.V.; Barrett, B.S.; Wilson, G.S.; Johnson, D.A.; Johnson, D.A.; Harmon, H.P.; Gabbert, S.; Petillo, P.A. Lactate as a biomarker for sleep. Sleep 2012, 35, 1209–1222. [Google Scholar] [CrossRef] [PubMed]
- Rempe, M.J.; Wisor, J.P. Cerebral lactate dynamics across sleep/wake cycles. Front Comput. Neurosci. 2014, 8, 174. [Google Scholar] [CrossRef] [PubMed]
- Carrard, A.; Elsayed, M.; Margineanu, M.; Boury-Jamot, B.; Fragniere, L.; Meylan, E.M.; Petit, J.M.; Fiumelli, H.; Magistretti, P.J.; Martin, J.L. Peripheral administration of lactate produces antidepressant-like effects. Mol. Psychiatry 2018, 23, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Reiach, J.S.; Li, P.P.; Warsh, J.J.; Kish, S.J.; Young, L.T. Reduced adenylyl cyclase immunolabeling and activity in postmortem temporal cortex of depressed suicide victims. J. Affect Disord. 1999, 56, 141–151. [Google Scholar] [CrossRef]
- Vardjan, N.; Chowdhury, H.H.; Horvat, A.; Velebit, J.; Malnar, M.; Muhic, M.; Kreft, M.; Krivec, S.G.; Bobnar, S.T.; Mis, K.; et al. Enhancement of Astroglial Aerobic Glycolysis by Extracellular Lactate-Mediated Increase in cAMP. Front Mol. Neurosci. 2018, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Lane, S.; Korsak, A.; Paton, J.F.; Gourine, A.V.; Kasparov, S.; Teschemacher, A.G. Lactate-mediated glia-neuronal signaling in the mammalian brain. Nat. Commun. 2014, 5, 3284. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ruchti, E.; Petit, J.M.; Jourdain, P.; Grenningloh, G.; Allaman, I.; Magistretti, P.J. Lactate promotes plasticity gene expression by potentiating NMDA signaling in neurons. Proc. Natl. Acad. Sci. USA 2014, 111, 12228–12233. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.C.; Sohn, H.A.; Park, Z.Y.; Oh, S.; Kang, Y.K.; Lee, K.M.; Kang, M.; Jang, Y.J.; Yang, S.J.; Hong, Y.K.; et al. A lactate-induced response to hypoxia. Cell 2015, 161, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.P.; Hirasawa, M. ATP-sensitive potassium channel-mediated lactate effect on orexin neurons: Implications for brain energetics during arousal. J. Neurosci. 2010, 30, 8061–8070. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.Q.; Ren, N.; Jin, L.; Cheng, K.; Kash, S.; Chen, R.; Wright, S.D.; Taggart, A.K.; Waters, M.G. Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem. Biophys. Res. Commun. 2008, 377, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, J.; Zhu, J.; Kuei, C.; Yu, J.; Shelton, J.; Sutton, S.W.; Li, X.; Yun, S.J.; Mirzadegan, T.; et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J. Biol. Chem. 2009, 284, 2811–2822. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Lauritzen, K.H.; Puchades, M.; Holm-Hansen, S.; Andersson, K.; Gjedde, A.; Attramadal, H.; Storm-Mathisen, J.; Bergersen, L.H. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1, Expression and action in brain. J. Neurosci. Res. 2015, 93, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, L.; Puyal, J.; Chatton, J.Y. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE 2013, 8, e71721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.J.; Ortega, F.E.; Riegler, J.; Madison, D.V.; Krasnow, M.A. Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 2015, 527, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosford, P.S.; Mosienko, V.; Kishi, K.; Jurisic, G.; Seuwen, K.; Kinzel, B.; Ludwig, M.G.; Wells, J.A.; Christie, I.N.; Koolen, L.; et al. CNS distribution, signalling properties and central effects of G-protein coupled receptor 4. Neuropharmacology 2018, 138, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Conzelmann, S.; Levai, O.; Bode, B.; Eisel, U.; Raming, K.; Breer, H.; Strotmann, J. A novel brain receptor is expressed in a distinct population of olfactory sensory neurons. Eur. J. Neurosci. 2000, 12, 3926–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Torrelo, H.; Ortega-Saenz, P.; Macias, D.; Omura, M.; Zhou, T.; Matsunami, H.; Johnson, R.S.; Mombaerts, P.; Lopez-Barneo, J. The role of Olfr78 in the breathing circuit of mice. Nature 2018, 561, E33–E40. [Google Scholar] [CrossRef] [PubMed]
- McBryde, F.D.; Abdala, A.P.; Hendy, E.B.; Pijacka, W.; Marvar, P.; Moraes, D.J.; Sobotka, P.A.; Paton, J.F. The carotid body as a putative therapeutic target for the treatment of neurogenic hypertension. Nat. Commun. 2013, 4, 2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marina, N.; Tang, F.; Figueiredo, M.; Mastitskaya, S.; Kasimov, V.; Mohamed-Ali, V.; Roloff, E.; Teschemacher, A.G.; Gourine, A.V.; Kasparov, S. Purinergic signalling in the rostral ventro-lateral medulla controls sympathetic drive and contributes to the progression of heart failure following myocardial infarction in rats. Basic Res. Cardiol. 2013, 108, 317. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.T.; Toy, P.; McClary, J.A.; Lin, R.J.; Miyamoto, N.G.; Kretschmer, P.J. Cloning and genetic characterization of an evolutionarily conserved human olfactory receptor that is differentially expressed across species. Gene 2001, 278, 41–51. [Google Scholar] [CrossRef]
- Weber, M.; Pehl, U.; Breer, H.; Strotmann, J. Olfactory receptor expressed in ganglia of the autonomic nervous system. J. Neurosci. Res. 2002, 68, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Mosienko, V.; Chang, A.J.; Alenina, N.; Teschemacher, A.G.; Kasparov, S. Rodents and humans are able to detect the odour of l-Lactate. PLoS ONE 2017, 12, e0178478. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.G.; Vanek, M.; Guerini, D.; Gasser, J.A.; Jones, C.E.; Junker, U.; Hofstetter, H.; Wolf, R.M.; Seuwen, K. Proton-sensing G-protein-coupled receptors. Nature 2013, 425, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.N.; Velic, A.; Soliz, J.; Shi, Y.; Li, K.; Wang, S.; Weaver, J.L.; Sen, J.; Abbott, S.B.; Lazarenko, R.M.; et al. PHYSIOLOGY. Regulation of breathing by CO2 requires the proton-activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 2015, 348, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Teschemacher, A.G.; Wang, S.; Lonergan, T.; Duale, H.; Waki, H.; Paton, J.F.; Kasparov, S. Targeting specific neuronal populations in the rat brainstem using adeno- and lentiviral vectors: Applications for imaging and studies of cell function. Exp. Physiol. 2005, 90, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.; Lane, S.; Stout, R.F., Jr.; Liu, B.; Parpura, V.; Teschemacher, A.G.; Kasparov, S. Comparative analysis of optogenetic actuators in cultured astrocytes. Cell Calcium 2014, 56, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Lobo, M.K.; Karsten, S.L.; Gray, M.; Geschwind, D.H.; Yang, X.W. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat. Neurosci. 2006, 9, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Joseph, V.; Slesinger, P.A.; Kleinfeld, D. Cell-based reporters reveal in vivo dynamics of dopamine and norepinephrine release in murine cortex. Nat. Methods 2014, 11, 1245–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Mosienko, V.; Vaccari, C.B.; Prokudina, D.; Huentelman, M.; Teschemacher, A.G.; Kasparov, S. Glio- and neuro-protection by prosaposin is mediated by orphan G-protein coupled receptors GPR37L1 and GPR37. GLIA 2018. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Noriega, L.E.; Stevens, C.F. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J. Neurosci. 1994, 14, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.T.; Bobker, D.H.; Harris, G.C. Synaptic potentials in locus coeruleus neurons in brain slices. Prog. Brain Res. 1991, 88, 167–172. [Google Scholar] [PubMed]
- Pepper, C.M.; Henderson, G. Opiates and opioid peptides hyperpolarize locus coeruleus neurons in vitro. Science 1980, 209, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, G.K.; Vandermaelen, C.P. α 2-adrenoceptor-mediated hyperpolarization of locus coeruleus neurons: Intracellular studies in vivo. Science 1982, 215, 1394–1396. [Google Scholar] [CrossRef] [PubMed]
- Cedarbaum, J.M.; Aghajanian, G.K. Activation of locus coeruleus neurons by peripheral stimuli: Modulation by a collateral inhibitory mechanism. Life Sci. 1978, 23, 1383–1392. [Google Scholar] [CrossRef]
- Foote, S.L.; Aston-Jones, G.; Bloom, F.E. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc. Natl. Acad. Sci. USA 1980, 77, 3033–3037. [Google Scholar] [CrossRef] [PubMed]
- Alt, A.; Weiss, B.; Ogden, A.M.; Knauss, J.L.; Oler, J.; Ho, K.; Large, T.H.; Bleakman, D. Pharmacological characterization of glutamatergic agonists and antagonists at recombinant human homomeric and heteromeric kainate receptors in vitro. Neuropharmacology 2004, 46, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Zong, G.; Wang, H.; Li, J.; Xie, Y.; Bian, E.; Zhao, B. Inhibition of GPR137 expression reduces the proliferation and colony formation of malignant glioma cells. Neurol. Sci. 2014, 35, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Koelzer, V.H.; Stuber, R.; Thoo, L.; Keller, I.; Koeck, I.; Langenegger, M.; Simillion, C.; Pfister, S.P.; Faderl, M.; et al. The ESRP1-GPR137 axis contributes to intestinal pathogenesis. Elife 2017, 6, e28366. [Google Scholar] [CrossRef] [PubMed]

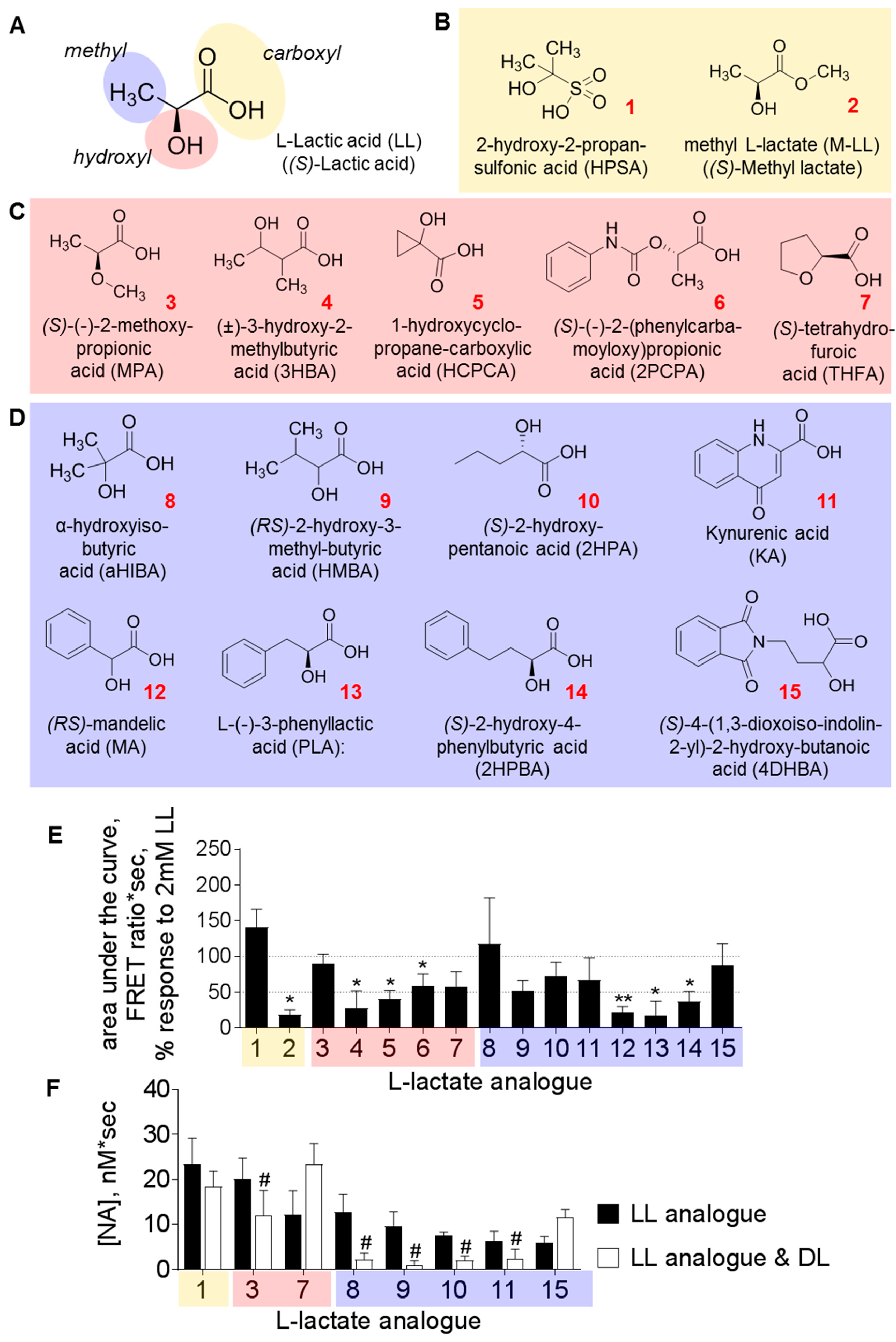


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | LL Potency (Range) | Intracellular Signalling | Other Agonists | Antagonists | References |
|---|---|---|---|---|---|
| HCA1 | 5–30 mM | Gi, cAMP↓ | 3,5-DHBA; α-HBA; glycolate; γ-HBA | nd | [25,26] |
| OR51E2 | ~4 mM | Gs, cAMP↑ | Acetate; propionate | nd | [29] |
| GPR4 | 1–10 mM | allosteric modulation? | H+ | NE 52-QQ57 | [30] |
| LLRx | 0.5 mM | Gs, cAMP↑ | MPA; aHIBA; HMBA; 2HPA; KA (see Figure 2) | d-lactate | [20] |
| Gene Product of Interest | LC (p29) | LC (p7) | LC Neurones | Astrocytes |
|---|---|---|---|---|
| DbH | 43.3 | 527.7 | 3556.2 | 2.5 |
| α2A-AR | 116.8 | 307.0 | 268.4 | 115.9 |
| HCA1 | 0.0 | 0.0 | 0.0 | 0.0 |
| OR51E2 | 7.6 | 21.3 | 0.0 | 0.0 |
| GPR4 | 70.5 | 26.0 | 34.6 | 24.5 |
| LL Analogue (Figure 2) | Cells Analysed (number) | Cells Depolarised (%) | Vm (mV) |
|---|---|---|---|
| LL | 94 | 34 | 7.3 ± 1.1 |
| 3 | 8 | 0 | |
| 8 | 28 | 43 | 6.2 ± 1.3 |
| 9 | 9 | 67 | 5.3 ± 1.0 |
| 10 | 8 | 25 | |
| 11 | 7 | 14 |
| DiscoveRx Activation (%) by LL (mM) | Expression in LC (NGS) | cAMP Assay: Activation by LL (0.5–10 mM) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| GPCR | Coupling? | 2 | 4 | 8 | p29 | p7 | Neuron | Astrocyte | |
| GPR61 | Gs | −2% | 16% | 19% | 97 | 183 | 163 | 0 | No response |
| GPR81 | Gi | 1% | 3% | 3% | 0 | 0 | 0 | 0 | Decrease |
| GPR137 | Gs? | 6% | 4% | 3% | 959 | 628 | 196 | 577 | Increase |
| GPR137B | ? | nd | nd | nd | 226 | 83 | 140 | 125 | No response |
| GPR146 | ? | −3% | 10% | 20% | 129 | 49 | 6 | 312 | nd |
| GPR155 | RGS? | nd | nd | nd | 842 | 412 | 140 | 36 | No response |
| GPR158 | RGS7 | nd | nd | nd | 1692 | 433 | 613 | 1 | No response |
| GPR162 | ? | −9% | 15% | 30% | 1293 | 1012 | 282 | 64 | No response |
| GPR176 | Gz | −8% | −2% | 9% | 193 | 196 | 190 | 9 | No response |
| GPR180 | Gi? | nd | nd | nd | 768 | 542 | 520 | 83 | Decrease |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosienko, V.; Rasooli-Nejad, S.; Kishi, K.; De Both, M.; Jane, D.; Huentelman, M.J.; Kasparov, S.; Teschemacher, A.G. Putative Receptors Underpinning l-Lactate Signalling in Locus Coeruleus. Neuroglia 2018, 1, 365-380. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020025
Mosienko V, Rasooli-Nejad S, Kishi K, De Both M, Jane D, Huentelman MJ, Kasparov S, Teschemacher AG. Putative Receptors Underpinning l-Lactate Signalling in Locus Coeruleus. Neuroglia. 2018; 1(2):365-380. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020025
Chicago/Turabian StyleMosienko, Valentina, Seyed Rasooli-Nejad, Kasumi Kishi, Matt De Both, David Jane, Matt J. Huentelman, Sergey Kasparov, and Anja G. Teschemacher. 2018. "Putative Receptors Underpinning l-Lactate Signalling in Locus Coeruleus" Neuroglia 1, no. 2: 365-380. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia1020025