
Neurovascular Coupling in Seizures

1
Department of Cell Biology and Anatomy, Hotchkiss Brain Institute, Cumming School of Medicine, University of Calgary, Calgary, AB T2N 1N4, Canada
2
Department of Physiology and Cell Biology, University of Nevada, Reno School of Medicine, Reno, NV 89557, USA
*
Author to whom correspondence should be addressed.
Neuroglia 2021, 2(1), 36-47; https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia2010005
Submission received: 31 August 2021
/
Revised: 1 October 2021
/
Accepted: 5 October 2021
/
Published: 11 October 2021
(This article belongs to the Special Issue Dynamic Vascular-Glial-Neuronal Interactions in Health and Disease)
{kind=link}
Abstract
:Neurovascular coupling is a key control mechanism in cerebral blood flow (CBF) regulation. Importantly, this process was demonstrated to be affected in several neurological disorders, including epilepsy. Neurovascular coupling (NVC) is the basis for functional brain imaging, such as PET, SPECT, fMRI, and fNIRS, to assess and map neuronal activity, thus understanding NVC is critical to properly interpret functional imaging signals. However, hemodynamics, as assessed by these functional imaging techniques, continue to be used as a surrogate to map seizure activity; studies of NVC and cerebral blood flow control during and following seizures are rare. Recent studies have provided conflicting results, with some studies showing focal increases in CBF at the onset of a seizure while others show decreases. In this brief review article, we provide an overview of the current knowledge state of neurovascular coupling and discuss seizure-related alterations in neurovascular coupling and CBF control.
1. Introduction
The brain is a high-energy-consuming organ that lacks significant energy reserves. As such, it requires a constant and continuous supply of oxygen and glucose via the blood for its normal computational functions, not only during heightened activity but also at rest. An inadequate blood supply to a certain brain region can injure or kill brain tissue [1,2,3]. Such vascular impairments have slowly emerged as both a cause and consequence of many neural pathological conditions, such as vascular dementia and epilepsy [1,2,3]. To ensure both a constant blood supply throughout the brain and the matching of blood delivery with metabolic needs in different brain regions, the brain possesses two major control mechanisms: cerebrovascular autoregulation and neurovascular coupling (NVC). Cerebrovascular autoregulation is an endogenous vascular-controlled process that is intrinsic to vascular myocytes that typically manifests as pressure-dependent constriction (increased pressure) or dilation (decreased pressure) of blood vessels known as the myogenic response [4]. This mechanism ensures a constant blood supply to the brain despite changes in systemic pressure. In contrast, NVC mechanisms involve multiple cell types, including neurons, glia (i.e., astrocytes and microglia), and vascular cells (i.e., vascular smooth muscle cells, pericytes, and endothelial cells), which collectively form the neurovascular unit. Individual components of the neurovascular unit operate in an integrated manner to increase the local blood flow in response to increases in neuronal activity, which is a process that is termed functional hyperemia [5,6].
We know a fair amount about cerebrovascular autoregulation and its critical role in cerebral blood flow (CBF), as well as the players involved in the process, but the underlying mechanistic details remain incompletely understood [7]. Even less is known about cerebral autoregulation in pathological brain states, such as seizures. Similarly, much remains to be learned regarding NVC mechanisms, but it is generally accepted that, under normal physiological conditions, increases in neuronal activity trigger a release of synaptic glutamate that can ultimately induce an increase in CBF via two major pathways: (1) neuron signaling directly to blood vessels and (2) neuron signaling indirectly to blood vessels via astrocytes [5,8]. Importantly, despite a general understanding of cerebral autoregulatory and NVC processes under physiological conditions, NVC during pathological states, such as seizures, remains poorly defined [9,10]. Electrographic seizures are sustained bouts of hypersynchronous and hyperexcited neuronal activity that impose an enormous metabolic demand on the vasculature [11]. Whether NVC during a seizure (ictal) operates as it does under normal physiological conditions remains an outstanding question. Furthermore, it is still not clear how CBF is affected during preictal, ictal, and postictal periods [9,12]. Most reports have tended to focus on the ictal period, with less attention devoted to the postictal period. In this brief review, we provide an overview of the current knowledge state of physiological cerebrovascular autoregulation and NVC and discuss seizure-related alterations in NVC during ictal and postictal periods. Insights into these processes will help to consolidate our understanding of CBF regulation and provide the clarity that is needed to interpret data from functional imaging techniques, such as functional magnetic resonance imaging (fMRI), which are used as proxies for neuronal activity or as diagnostic or prognostic tools in pathologies like epilepsy.
2. Cerebral Blood Flow
Region-specific brain activity is ever-changing; therefore, it requires both a continuous blood supply to maintain baseline activity and on-demand delivery of blood to support metabolically active regions. The unique angioarchitecture of the cerebral circulation is organized into three distinct topological tiers—pial artery and venule, penetrating arteriole and venule, and the subsurface microvascular network [13]—that collectively ensure that every neural cell is adequately nourished. Fluctuations in blood flow, oxygen consumption, blood volume, and glucose utilization that were probed by functional imaging techniques, such as positron emission tomography (PET) and fMRI, were shown to be important surrogates for changes in brain activity [14]. Early work in the 1950s by Mangold and colleagues [15] showed that oxygen consumption is unaltered between awake resting and sleep states, prompting questions about whether the metabolic activity of the brain is constant during baseline activity. It also raises questions about what is truly baseline activity and whether such a baseline can be established in the brain. Gusnard and Raichle [16] defined “baseline” as the absence of activation and showed that the oxygen extraction rate (OEF) was uniform across brain regions, indicating the establishment of an equilibrium between local metabolic requirements to sustain the ongoing activity and blood flow. Does this baseline or resting state of brain function affect stimulation-induced responses? Work in anesthetized rats provides some insight [17,18]. Simultaneous measurement of blood oxygenation level-dependent (BOLD) fMRI and the relative spiking frequency of a neuronal ensemble during forepaw stimulation from two different baselines (i.e., different anesthesia levels) showed that forepaw stimulation induced different increments from each baseline, but that the final values reached were ultimately approximately the same. The authors of these studies concluded that the same total energy was required to support neuronal activity regardless of baseline. Most of the energy consumed by the brain is provided by the ATP produced via the oxidative metabolism of glucose. Using modeling, investigators attributed the high energy consumption of the brain to several specific cellular signaling processes, including restoration of ion movements generated by action potentials, postsynaptic currents, and neurotransmitter release and recycling [19,20].
In a physiological state, total blood flow to the brain is relatively constant, which is due in part to the contributions of large arteries and parenchymal arterioles that control vascular resistance [21,22]. Using PET, Fox and colleagues reported a strong correlation between CBF and cerebral oxygen consumption in the resting state; however, this strong correlation was lost during neuronal activation via somatosensory stimulation [23]. Thus, there is an oversupply of oxygen during activation [24,25], but the rationale for this excess remains poorly understood. There are some caveats to these observations using PET, where the size of the detected area influences the magnitude of the responses detected. For example, metabolic responses are confined to smaller brain region than the hemodynamic responses and that may contribute to the discrepancy observed between CBF and oxygen consumption during neuronal activity. On the other hand, this oversupply of oxygen could be a mechanism to prevent the tissue O2 concentration from dropping when CMRO2 increases [26]. In contrast to this uncoupling between CBF and oxygen consumption, CBF and glucose consumption are closely coupled during activation [27]. The mismatch between excess increases in CBF and oxygen consumption raises questions regarding the relationship between energy supply and demands and cautions against uncritical interpretations of functional imaging data, such as BOLD signals imaged with fMRI and fNIRS, which are based on relative changes in oxygenated versus deoxygenated hemoglobin. In addition to commonly reported increases in BOLD signals and associated increases in neuronal activity [28,29], functional brain imaging studies also present sustained negative BOLD responses [30,31,32]. The relationship between negative BOLD signals and neural activity remains poorly understood. While some studies proposed that a drop in hemodynamic responses correspond to the suppression of neuronal activity [31], others suggest that a drop in hemodynamic response is a passive process and independent of changes in neuronal activity [33]. Simultaneous recordings of electrical signals and fMRI in anesthetized macaque monkeys reported a negative BOLD signal that was associated with decreases in neuronal activity in regions beyond the stimulated site [31]. Moreover, decreases in CBF and the associated negative BOLD signal did not cause a decrease in neuronal activity. However, these findings do not rule out the possibility that other neuromodulators could act directly on the vasculature. CBF is determined by the cerebral perfusion pressure (i.e., the difference between the mean arterial pressure and intracranial pressure), cardiac output, and the vascular tone of the microvasculature. Resistance vessels, such as parenchymal arterioles and capillaries, play an essential role in actively regulating CBF through changes in vascular tone. Arteriole tone is regulated by the contractility of the vascular smooth muscle cells (SMCs) that circumferentially line the vascular wall. The massive capillary network supports blood distribution throughout the brain parenchyma, ensuring that every cell is adequately nourished [34]. Tissue oxygenation is further regulated by red blood cells (RBCs) flux through individual capillaries. Increases or decreases in RBC flux through capillaries depend in part on the dilation or constriction of upstream pial arteries and arterioles. Recent studies have also revealed that pericytes, which are the predominant mural cells within capillary networks, contribute to basal blood flow resistance and modulate blood flow [35]. Because there is not enough blood in the cerebral circulation to adequately supply the entire brain if all regions were activated at the same time, brain blood flow must be modulated such that the needs of regions with high metabolic demand are met while other regions of the brain still receive a sufficient supply of blood. How cerebrovascular autoregulation and NVC are integrated to regulate CBF remains an outstanding question.
3. Cerebrovascular Autoregulation
Cerebrovascular autoregulation maintains CBF despite changes in arterial pressure [36]. Extensive work on autoregulation has established the vital role of this protective mechanism. A key aspect of cerebrovascular autoregulation is the myogenic response, which is an intrinsic vascular-dependent and neuronal-independent process that allows the vasculature to constrict or dilate in response to increases or decreases in intraluminal pressure, respectively, to maintain a relatively constant blood supply. Autoregulation is not unique to the cerebral vasculature, as it also operates in other resistance vessels [36], but it does play a vital role in brain circulation. Although the concept of autoregulation was first proposed in the late 19th century by Bayliss, Hill, and Gulland [37], and notwithstanding subsequent significant advances in our understanding of the process, the mechanistic basis of autoregulation is not yet fully understood. We know that autoregulation only operates within a range of pressures and that outside this range, autoregulation fails, causing the vasculature to passively respond to changes in blood pressure and leaving the brain at risk of either hyperperfusion or hypoperfusion. Original work from Lassen reported that autoregulation manifested as a plateau in pressure-diameter curves in the range of ~60–150 mmHg, which is approximately a 90 mmHg range [38]. However, more recent studies in humans have reported a considerably smaller mean arterial pressure plateau range of ~5–10 mmHg [39], underscoring how vital autoregulation is as a vascular protective mechanism. In vivo and in vitro studies on animals showed that myogenic responses act primarily through a Ca2+-dependent pathway in SMCs [40] such that increases in arterial pressure induce a depolarization of vascular SMCs that leads to Ca2+ entry through voltage-gated Ca2+ channels [41]. Despite evidence showing that increases in pressure induce depolarization, the cause-and-effect relationship between membrane depolarization and the myogenic response remains to be confirmed. It was proposed that mechanosensitive channels are activated in response to increased pressure and allow for Ca2+ entry through voltage-gated Ca2+ channels [42]. Studies also provided evidence that, in addition to electromechanical coupling, Ca2+ sensitization may contribute to the myogenic response. Moreover, some studies have reported a potential role for K+ channels, in particular, the large-conductance Ca2+-dependent K+ (BKCa) channel, in myogenic responses [43], while others pointed to the involvement of hydroxyeicosatetraenoic acid (20-HETE) [44]. It is anticipated that future investigations using advanced experimental techniques, such as Cre-lox technologies, will be able to provide some clarity on the subject.
4. Physiological Neurovascular Coupling
Neurovascular coupling is a collection of mechanisms that regulate CBF in response to increases in neuronal activity. Our understanding of NVC at the cellular level has advanced significantly because of the development of new imaging techniques that allow us to explore the interaction between members of the neurovascular units: glia (e.g., astrocytes), neurons, and cells of the vasculature (endothelial cells, SMCs, pericytes). The recent development of awake in vivo two-photon imaging [45] also facilitates NVC research under near-physiological conditions without the confounding effects of anesthesia. Evidence from in vitro (i.e., brain slices) and in vivo preparations led to the formulation of two NVC models: (1) activation of neurons directly triggers signaling pathways that release vasoactive agents and cause vasodilation and (2) activation of neurons elicits functional hyperemia through the activation of astrocytes.
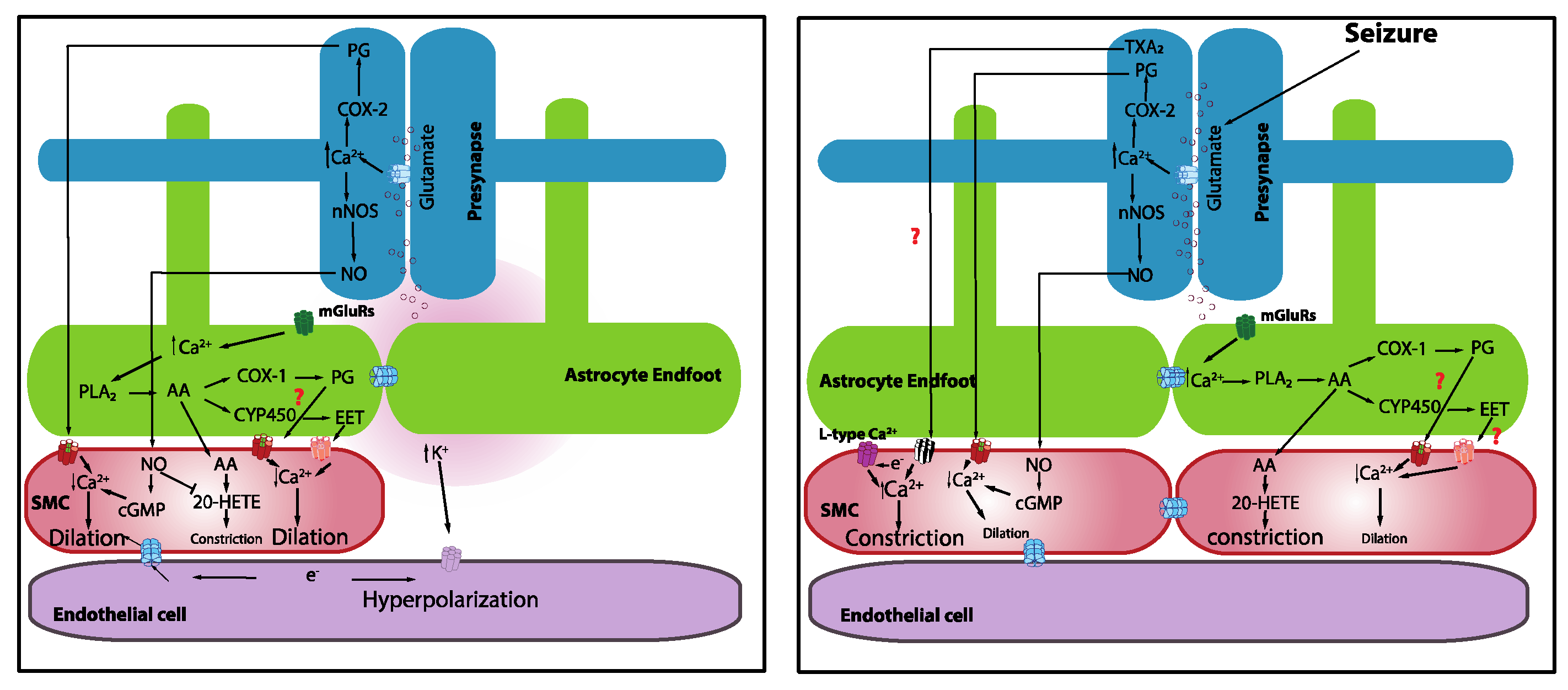
It is known that increases in neuronal activity lead to a synaptic release of glutamate, which acts through N-methyl-D-aspartate receptors (NMDARs) to elevate neuronal Ca2+. Increases in neuronal Ca2+, in turn, activate neuronal nitric oxide synthase (nNOS), triggering the synthesis and release of nitric oxide (NO), which subsequently elicits vasodilation [46]. Topical application of NMDA elicits vasodilation [47]. Moreover, inhibition of nNOS attenuates whisker-stimulation-induced functional hyperemia in anesthetized rats [48] and awake mice [49], implicating a role for NO in NVC. However, the idea that neuronally derived NO acts directly on the vasculature may be more complicated than it appears given that mice lacking nNOS (type I) do not show significant alterations in functional hyperemia [48] and NO donors restored the inhibitory effects of the nNOS blockade [50]. It should also be noted that the role of NO in NVC appears to vary across different regions of the brain [5]. It is possible that neurons directly induce vasodilation through the release of NO, but this may not be the only factor. It was also shown that prostaglandins that are elaborated by the cyclooxygenase (COX)-2 pathway are involved [49] (Figure 1). The contributions of interneurons were also proposed [51].
One proposed NVC mechanism posits that the synaptic release of glutamate during increased neuronal activity activates metabotropic glutamate receptors on astrocytes, leading to an increase in astrocyte intracellular Ca2+. This increase in astrocyte intracellular free Ca2+ activates phospholipase A2, which induces the production of arachidonic acid (AA), which is a substrate of COX-2. The subsequent action of COX-2 on AA produces a series of metabolites, including prostaglandins and epoxyeicosatrienoic acid (EETs), that typically causes vasodilation. Evidence from brain slices revealed that astrocytic Ca2+ elevations that are induced by neuronal afferent stimulation [52] or uncaging Ca2+ using photolysis [53,54] are accompanied by delayed vasodilation. Gordon and colleagues [55] proposed that oxygen availability and, consequently, lactate, as well as adenosine levels, determine the polarity of astrocyte-mediated vascular response. Low oxygen availability facilitates vasodilation rather than vasoconstriction, suggesting that metabolic activity contributes to driving the hemodynamic responses. Girouard and Nelson further reported that astrocytic endfoot Ca2+ levels determine the polarity of the vascular response, such that normal astrocytic endfoot Ca2+ levels (~300 nM) induce vasodilation, whereas high Ca2+ concentrations (>732 nM) switched the response from vasodilation to vasoconstriction [54]. Interestingly, astrocytes were implicated in mediating vasomotor changes independently of neural activity [56].
However, in vivo studies presented conflicting findings, with some studies reporting a rapid onset of astrocytic Ca2+ (i.e., within hundreds of milliseconds) after the onset of sensory stimulation [57,58,59], and others, including our own, showing that functional hyperemia occurs before the onset of astrocytic Ca2+ elevations in different astrocytic cellular compartments [49,60]. Importantly, some groups observed functional hyperemia in response to sensory stimulation in the absence of a rise in astrocytic Ca2+ [61] or the presence of only sporadic astrocytic Ca2+ events [60]. Recent work from Stobart and colleagues [62] demonstrated that a sub-population of astrocyte Ca2+ signals exhibit temporal dynamics that are similar to those of neurons, and these fast signals are detected in both the endfeet and fine processes. These authors inferred that these fast astrocytic Ca2+ signals that are observed in microdomains are the driving force that elicits the release of vasoactive agents that lead to functional hyperemia. However, direct evidence to show that these fast, discrete microdomain events mediate functional hyperemia is still needed. Other studies proposed the existence of Ca2+-independent mechanisms that are capable of driving astrocytes to release vasoactive agents that cause vessel dilation or constriction. Manipulations of astrocytic Ca2+ have suggested a role for inositol 1,4,5-trisphosphate (IP3), but knockout of the type 2 IP3 receptor (IP3R2) that is thought to mediate IP3 effects in these cells does not appear to affect astrocytic Ca2+. However, this conclusion should be interpreted with caution, as astrocytic Ca2+ from the soma rather than the endfoot Ca2+ was measured in these studies. Ca2+ signal dynamics in the endfoot are a key consideration in the study of NVC given that the vast majority of the microcirculation in the brain is ensheathed by this specialized subcellular compartment of astrocytes [63]. Recent work also underscored the heterogeneous nature of astrocytic Ca2+, highlighting the need to study astrocytic Ca2+ in all cellular compartments and different cortical layers. By extension, data obtained from a particular region of a cell should not be generalized [64]. There may be organelles within the cell that can regionally regulate Ca2+ dynamics and thus can either directly affect the onset of functional hyperemia or maintain functional hyperemia in response to increased neuronal activity.
5. Neurovascular Coupling in Seizures
Seizures are intense and highly synchronous neuronal discharges that increase metabolic activity and subsequently increase energy demands [65]. Under physiological conditions, the NVC provides sufficient blood supply in response to increase neuronal activity to match metabolic needs. However, whether the NVC is functional and capable of achieving an adequate blood supply during seizures is an open question. Equally important is whether CBF is affected during the postictal period as behavioral impairments following the termination of a seizure were reported [66]. Very few studies in people with epilepsy have examined blood flow during the postictal period, and those studies that do exist presented inconsistent evidence, with some reporting local hypoperfusion [67] and others reporting hyperperfusion [68,69]. In general, the available evidence relating CBF and epileptic seizures give discordant results, likely reflecting differences in experimental designs and time points at which the measurements were obtained. Additional insights into NVC in seizures are essential to further our understanding of the progression of the chronic epileptic state since this is a prerequisite for a proper scientific and diagnostic interpretation of metabolic imaging data in patients. Furthermore, NVC forms the basis of functional imaging, where fMRI is commonly used to define the epileptogenic zone and guide surgical therapy of untreatable focal epilepsies that are medically refractory [70]. In this section, we focus on CBF during the ictal event and postictal period.
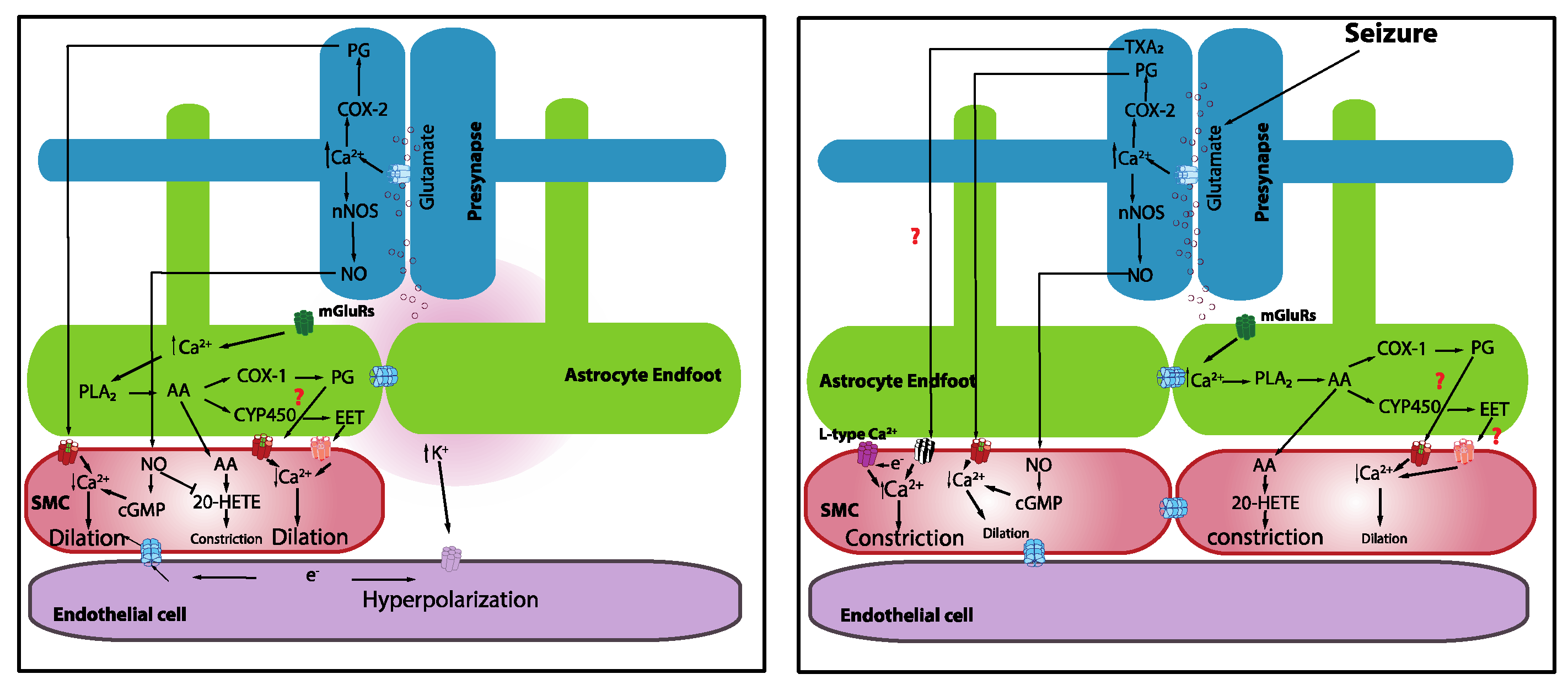
It is well established that there is a dramatic, transient increase in blood flow during seizures [71,72,73], accompanied by a transient “dip” in oxygenation that quickly recovers [71,74,75]. Superficially, this suggests that NVC is functional during a seizure. The transient drop in oxygenation could be due to increased metabolic activity and delayed oxygen extraction. Farrell et al. (2016) showed that self-generated and electrically kindled seizures caused a brief, small drop in hippocampal oxygenation at seizure onset and a subsequent increase in oxygenation during the electrographic seizure. Interestingly, using awake in vivo two-photon imaging, Tran and colleagues [76] reported a dramatic constriction of cortical penetrating arterioles at the onset of an induced maximal electroconvulsive seizure that corresponded with a strong rise in vascular SMC Ca2+. Simultaneous monitoring of astrocyte Ca2+ showed that seizures robustly elevated endfoot Ca2+. This robust rise in astrocyte Ca2+, accompanied by strong vasoconstriction at the onset of a seizure, mirrors previous results that were obtained following uncaging high levels of Ca2+ in astrocytes [54]. Using a 4-amipyridine (4-AP)-induced epilepsy model in anesthetized mice, Zhang and colleagues observed a large increase in astrocyte endfeet Ca2+ during a seizure that was accompanied by the dilation of a nearby arteriole [10]. They further showed that the basal level of astrocyte Ca2+, and consequently basal tone, determined how the diameter of the arteriole changed in response to a seizure. In vitro work that was performed in cortical slices revealed that ictal seizure-like discharges evoked massive homogeneous astrocytic Ca2+ elevations in both endfeet and soma [77,78]. Similar to in vivo observations from Zhang and colleagues, Gomez-Gonzalo et al. demonstrated that elevations in astrocytes Ca2+ were accompanied by vasodilation with a 5.5 s delayed onset after ictal discharge onset [78]. The discordance in these data could be attributable to the different models of seizure that were used and different regions of the brain that were studied, as well as the use of anesthesia in one study and not in the other. It should also be noted that the in vitro condition in brain slices differs substantially from the in vivo condition; the lack of blood flow and physiological pressure in the brain slice preparation is especially notable. One important result from the work of Zhang et al. was the finding of vasodilation at the epileptic focus but vasoconstriction in areas that were remote from the epileptic focus [10]. Interestingly, hemodynamic imaging showed an increase in CBF in the seizure focus but a decrease in the cortical area around the seizure focus [79]. These findings similarly reflect the center–surround phenomena that were observed during sensory stimulation under physiological conditions [51,80]. This implies that the brain may be able to partly manage the distribution of the blood supply to match metabolic demands by “stealing” supply from metabolically low-demand regions and distributing it to highly active regions, i.e., the “steal effect.” It could also be an active shunting process due to vasoconstriction in the surrounding region.
Seizures are often followed by acute impairments in sensory, cognitive, or motor functions that are associated with the affected area [66]. These impairments are not the direct consequence of the seizures themselves but rather are the result of compromised blood perfusion during the postictal period. Most seizure-related hemodynamic studies focused on changes before or during the seizure rather than during the postictal period. Functional imaging data that was obtained from people during the postictal period are conflicting, with some studies reporting hypoperfusion [67,81,82] and others reporting hyperperfusion [68,69]. Leonhardt and colleagues reported both responses. The responses are not only varied between patients but also between regions of the brain. One brain region experiences hypoperfusion while the other has hyperperfusion in the early phase of the postictal period, and the responses appear to reverse in those two brain regions in the late phase of the postictal period [67]. Variance in seizure type and when and where the perfusion measurement was taken appear to contribute to the discordance. Using continuous oxygen detection with high temporal resolution in non-human subjects, Farrell et al. showed that seizures result in severe local brain hypoxia (i.e., <10 mmHg) that remained for more than an hour after the termination of seizures. This severe postictal hypoxia was accompanied by reduced blood flow [83] and corresponded with sustained arteriole constriction in acute hippocampal slice preparations that lasted for 60–90 min post seizure [83]. Tran and colleagues [76] also reported dramatic, prolonged vasoconstriction following an induced seizure that lasted for more than 1 h after the seizure initiation. At the cellular level, this constriction was associated with a strong rise in vascular SMC Ca2+; however, SMC Ca2+ did not remain elevated for the entire period of the vasoconstriction. Such Ca2+-independent constriction could be attributable to Ca2+ sensitization mechanisms that operate within vascular SMCs [84]. Accompanying the changes in vascular tone during the initiation of vasoconstriction was a rapid elevation in astrocyte endfoot Ca2+, which decayed to a plateau for approximately 20 s before returning to baseline [76]. A similar rapid Ca2+ rise was seen in the arbor, but it rapidly returned to baseline without decaying to a plateau level. The inhibition of COX-2 and blocking of L-type Ca2+ channels was shown to inhibit severe postictal hypoxia [83]. Interestingly from a temporal perspective is the observation that pre-seizure administration of COX-2 antagonists effectively inhibits postictal hypoxia, whereas administration of the antagonist immediately following termination of seizures is completely ineffective. On the other hand, L-type Ca2+ channel blockers are effective in abolishing postictal hypoxia whether administered prior to the seizure or immediately following their termination [83]. These findings are among the first to show that seizure-induced functional deficits reflect impaired blood perfusion during the postictal period and highlight the potential utility of L-type Ca2+ channels, as well as the COX-2 signaling pathways as molecular targets for drug discovery and/or drug repurposing efforts [85].
6. Future Directions
Our knowledge about NVC and CBF regulation has advanced significantly over the past few decades. However, there are still many questions that remain to be investigated. These include whether and how cerebrovascular autoregulation and NVC integrate to regulate blood flow, what useful information about neuronal activity that we can reliably obtain from non-invasive functional imaging, and how autoregulation and/or NVC is disturbed in disease. To answer these questions, it is essential for us to understand the mechanisms underlying NVC at the microscopic level. Several findings at the cellular level suggest that the activation of different sub-populations of neurons triggers different vascular responses (i.e., vasoconstriction vs. vasodilation) through the release of vasoactive molecules [51]. In vivo studies showed a correlation between arteriole constriction and neuronal inhibition [86]. The development of optogenetics, chemogenetics, and Cre-lox technologies started a new era of study regarding NVC with cell specificity that has not been possible before. One can manipulate a specific cell type to examine the role of different neuronal populations, astrocytes, or vascular cells and establish their integrated action on CBF regulation. Providing a basic understanding of NVC mechanisms in physiology will pave the way for us to better understand how NVC behaves in diseases such as epilepsy. Multiple vasoactive messengers were proposed to be involved in NVC; none appears to take precedence over others. It is possible that not all of them act at the same time. One specific signaling pathway that gets activated in normal physiology to elicit functional hyperemia may not be activated during pathological conditions such as seizures. Likewise, the signaling pathway that is activated during a pathological condition may tip the balance between vasodilation and vasoconstriction. The rise in astrocyte Ca2+ may trigger the vasoconstriction seen in the ictal period, while the sustained constriction seen during the postictal period could be due to Ca2+ sensitization. Simultaneous recording of the activity of neurons, astrocytes, and the vasculature in awake animals will provide clear information about the integration of the members of the neurovascular unit in physiological and pathological conditions without the confounding effects of anesthesia. Different seizure models, brain regions, and time points of recording are all critical for providing a more defined picture of the cell type and signaling pathway involved and, ultimately, provide insights into possible therapeutics.
7. Conclusions
The more we learn about cerebrovascular autoregulation and NVC in physiology, the more we appreciate the complexity of CBF regulation. While this makes the scientific question more interesting, it also makes the task of studying NVC in disease more challenging. Our increasing understanding of NVC has helped us to better interpret functional imaging data while cautioning us regarding their use as a proxy for neuronal activity. With recent advances in related technologies, we have gradually come to uncover the integrative nature of cells of the neurovascular unit and their contributions to the physiological regulation of CBF. There are still many missing pieces of the puzzle, and extensive future research will be required to understand the mechanistic basis of cerebrovascular autoregulation and NVC and how these processes are impaired in disease states, such as epilepsy.
Author Contributions
Conceptualization, C.H.T.T. and G.C.T.; writing—original draft preparation, C.H.T.T.; writing—review and editing, C.H.T.T. and G.C.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Centers of Biomedical Research Excellence 1P20GM130459 (NIH) to CHT. GCT was supported by a grant from the Canadian Institutes for Health Research (PJT-152956).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Iadecola, C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 2004, 5, 347–360. [Google Scholar] [CrossRef]
- Tarantini, S.; Tran, C.H.T.; Gordon, G.R.; Ungvari, Z.; Csiszar, A. Impaired neurovascular coupling in aging and Alzheimer’s disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 2017, 94, 52–58. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [Green Version]
- McCarron, J.G.; Osol, G.; Halpern, W. Myogenic Responses Are Independent of the Endothelium in Rat Pressurized Posterior Cerebral Arteries. J. Vasc. Res. 1989, 26, 315–319. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.; Charpak, S.; Lauritzen, M.; MacVicar, B.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Nippert, A.R.; Biesecker, K.R.; Newman, E.A. Mechanisms Mediating Functional Hyperemia in the Brain. Neuroscientist 2017, 24, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Longden, T.; Hill-Eubanks, D.C.; Nelson, M.T. Ion channel networks in the control of cerebral blood flow. Br. J. Pharmacol. 2015, 36, 492–512. [Google Scholar] [CrossRef] [Green Version]
- Stackhouse, T.L.; Mishra, A. Neurovascular Coupling in Development and Disease: Focus on Astrocytes. Front. Cell Dev. Biol. 2021, 9, 702832. [Google Scholar] [CrossRef] [PubMed]
- Baruah, J.; Vasudevan, A.; Köhling, R. Vascular Integrity and Signaling Determining Brain Development, Network Excitability, and Epileptogenesis. Front. Physiol. 2020, 10, 1583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Tabatabaei, M.; Bélanger, S.; Girouard, H.; Moeini, M.; Lu, X.; Lesage, F. Astrocytic endfoot Ca2+ correlates with parenchymal vessel responses during 4-AP induced epilepsy: An in vivo two-photon lifetime microscopy study. Br. J. Pharmacol. 2017, 39, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, D.E.; Engel, J.; Phelps, M.E.; Selin, C. Epileptic patterns of local cerebral metabolism and perfusion in humans determined by emission computed tomography of 18FDG and 13NH3. Ann. Neurol. 1980, 8, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, T.; Teaw, S.; Bordey, A. Hypervascularization in mTOR-dependent focal and global cortical malformations displays differential rapamycin sensitivity. Epilepsia 2019, 60, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Shih, A.Y.; Rühlmann, C.; Blinder, P.; Devor, A.; Drew, P.J.; Friedman, B.; Knutsen, P.M.; Lyden, P.D.; Mateo, C.; Mellander, L.; et al. Robust and fragile aspects of cortical blood flow in relation to the underlying angioarchitecture. Microcirculation 2015, 22, 204–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, S.; Lee, T.M.; Kay, A.R.; Tank, D.W. Brain magnetic resonance imaging with contrast dependent on blood oxygenation. Proc. Natl. Acad. Sci. USA 1990, 87, 9868–9872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangold, R.; Sokoloff, L.; Conner, E.; Kleinerman, J.; Therman, P.-O.G.; Kety, S.S. The effects of sleep and lack of sleep on the cerebral circulation and metabolism of normal young men 1. J. Clin. Investig. 1955, 34, 1092–1100. [Google Scholar] [CrossRef] [Green Version]
- Gusnard, D.A.; Raichle, M.E. Searching for a baseline: Functional imaging and the resting human brain. Nat. Rev. Neurosci. 2001, 2, 685–694. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Shulman, R.G. Total neuroenergetics support localized brain activity: Implications for the interpretation of fMRI. Proc. Natl. Acad. Sci. USA 2002, 99, 10771–10776. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.J.; Blumenfeld, H.; Behar, K.; Rothman, D.L.; Shulman, R.G.; Hyder, F. Cerebral energetics and spiking frequency: The neurophysiological basis of fMRI. Proc. Natl. Acad. Sci. USA 2002, 99, 10765–10770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attwell, D.; Laughlin, S. An Energy Budget for Signaling in the Grey Matter of the Brain. Br. J. Pharmacol. 2001, 21, 1133–1145. [Google Scholar] [CrossRef]
- Howarth, C.; Gleeson, P.; Attwell, D. Updated Energy Budgets for Neural Computation in the Neocortex and Cerebellum. Br. J. Pharmacol. 2012, 32, 1222–1232. [Google Scholar] [CrossRef]
- Faraci, F.; Heistad, D. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ. Res. 1990, 66, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, M.J.; Smith, J.; Kohlmeyer, M.M.; Godfrey, J.A. SKCa and IKCa Channels, myogenic tone, and vasodilator responses in middle cerebral arteries and parenchymal arterioles: Effect of ischemia and reperfusion. Stroke 2009, 40, 1451–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, P.; Raichle, M.E. Focal physiological uncoupling of cerebral blood flow and oxidative metabolism during somatosensory stimulation in human subjects. Proc. Natl. Acad. Sci. USA 1986, 83, 1140–1144. [Google Scholar] [CrossRef] [Green Version]
- Lecoq, J.; Tiret, P.; Najac, M.; Shepherd, G.M.; Greer, C.A.; Charpak, S. Odor-evoked oxygen consumption by action potential and synaptic transmission in the olfactory bulb. J. Neurosci. 2009, 29, 1424–1433. [Google Scholar] [CrossRef]
- Parpaleix, A.; Houssen, Y.G.; Charpak, S. Imaging local neuronal activity by monitoring PO2 transients in capillaries. Nat. Med. 2013, 19, 241–246. [Google Scholar] [CrossRef]
- Devor, A.; Sakadžić, S.; Saisan, P.A.; Yaseen, M.A.; Roussakis, E.; Srinivasan, V.J.; Vinogradov, S.A.; Rosen, B.R.; Buxton, R.B.; Dale, A.M.; et al. ‘Overshoot’ of O2 is required to maintain baseline tissue oxygenation at locations distal to blood vessels. J. Neurosci. 2011, 31, 13676–13681. [Google Scholar] [CrossRef] [Green Version]
- Paulson, O.B.; Hasselbalch, S.G.; Rostrup, E.; Knudsen, G.M.; Pelligrino, D. Cerebral Blood Flow Response to Functional Activation. Br. J. Pharmacol. 2009, 30, 2–14. [Google Scholar] [CrossRef]
- Logothetis, N.K.; Pauls, J.; Augath, M.; Trinath, T.; Oeltermann, A. Neurophysiological investigation of the basis of the fMRI signal. Nature 2001, 412, 150–157. [Google Scholar] [CrossRef]
- Devor, A.; Ulbert, I.; Dunn, A.K.; Narayanan, S.N.; Jones, S.; Andermann, M.L.; Boas, D.A.; Dale, A.M. Coupling of the cortical hemodynamic response to cortical and thalamic neuronal activity. Proc. Natl. Acad. Sci. USA 2005, 102, 3822–3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmuel, A.; Yacoub, E.; Pfeuffer, J.; Van de Moortele, P.-F.; Adriany, G.; Hu, X.; Ugurbil, K. Sustained Negative BOLD, Blood Flow and Oxygen Consumption Response and Its Coupling to the Positive Response in the Human Brain. Neuron 2002, 36, 1195–1210. [Google Scholar] [CrossRef] [Green Version]
- Shmuel, A.; Augath, M.; Oeltermann, A.; Logothetis, N.K. Negative functional MRI response correlates with decreases in neuronal activity in monkey visual area V1. Nat. Neurosci. 2006, 9, 569–577. [Google Scholar] [CrossRef]
- Klingner, C.M.; Ebenau, K.; Hasler, C.; Brodoehl, S.; Görlich, Y.; Witte, O.W. Influences of negative BOLD responses on positive BOLD responses. NeuroImage 2011, 55, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Harel, N.; Lee, S.-P.; Nagaoka, T.; Kim, D.-S.; Kim, S.-G. Origin of negative blood oxygenation lev-el-dependent fMRI signals. J. Cereb. Blood Flow Metab. 2002, 22, 908–917. [Google Scholar] [CrossRef]
- Blinder, P.; Tsai, P.S.; Kaufhold, J.; Knutsen, P.M.; Suhl, H.; Kleinfeld, D. The cortical angiome: An interconnected vascular network with noncolumnar patterns of blood flow. Nat. Neurosci. 2013, 16, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.A.; Berthiaume, A.-A.; Grant, R.I.; Harrill, S.A.; Koski, T.; Tieu, T.; McDowell, K.P.; Faino, A.V.; Kelly, A.L.; Shih, A.Y. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 2021, 24, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Hill, M. Signaling Mechanisms Underlying the Vascular Myogenic Response. Physiol. Rev. 1999, 79, 387–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayliss, W.M.; Hill, L.; Gulland, G.L. On Intra-Cranial Pressure and the Cerebral Circulation: Part I. Physiological; Part II. Histological. J. Physiol. 1895, 18, 334–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassen, N.A. Cerebral Blood Flow and Oxygen Consumption in Man. Physiol. Rev. 1959, 39, 183–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.O.; Hamner, J.W.; Taylor, J.A. The role of myogenic mechanisms in human cerebrovascular regulation. J. Physiol. 2013, 591, 5095–5105. [Google Scholar] [CrossRef]
- Brayden, J.E.; Earley, S.; Nelson, M.T.; Reading, S. Transient receptor potential (trp) channels, vascular tone and autoregulation of cerebral blood flow. Clin. Exp. Pharmacol. Physiol. 2008, 35, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Meininger, G.A.; Davis, M.J. Cellular mechanisms involved in the vascular myogenic response. Am. J. Physiol. Circ. Physiol. 1992, 263, H647–H659. [Google Scholar] [CrossRef]
- VanBavel, E.; Wesselman, J.P.M.; Spaan, J.A.E. Myogenic Activation and Calcium Sensitivity of Cannulated Rat Mesenteric Small Arteries. Circ. Res. 1998, 82, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Brayden, J.; Nelson, M. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science 1992, 256, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Harder, D.R.; Lange, A.R.; Gebremedhin, D.; Birks, E.K.; Roman, R.J. Cytochrome P450 metabolites of arachidonic acid as intracellular signaling molecules in vascular tissue. J. Vasc. Res. 1997, 34, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.H.; Gordon, G.R. Acute two-photon imaging of the neurovascular unit in the cortex of active mice. Front. Cell. Neurosci. 2015, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busija, D.W.; Bari, F.; Domoki, F.; Louis, T. Mechanisms involved in the cerebrovascular dilator effects of N-methyl-d-aspartate in cerebral cortex. Brain Res. Rev. 2007, 56, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domoki, F.; Perciaccante, J.V.; Shimizu, K.; Puskar, M.; Busija, D.W.; Bari, F. N-methyl-d-aspartate-induced vasodilation is mediated by endothelium-independent nitric oxide release in piglets. Am. J. Physiol. Circ. Physiol. 2002, 282, H1404–H1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Meng, W.; Ayata, C.; Huang, P.L.; Fishman, M.C.; Moskowitz, M.A. L-NNA-sensitive regional cerebral blood flow augmentation during hypercapnia in type III NOS mutant mice. Am. J. Physiol. Circ. Physiol. 1996, 271, H1717–H1719. [Google Scholar] [CrossRef]
- Tran, C.H.; Peringod, G.; Gordon, G.R. Astrocytes Integrate Behavioral State and Vascular Signals during Functional Hyperemia. Neuron 2018, 100, 1133–1148.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindauer, U.; Megow, D.; Matsuda, H.; Dirnagl, U. Nitric oxide: A modulator, but not a mediator, of neuro-vascular coupling in rat somatosensory cortex. Am. J. Physiol. 1999, 277, H799–H811. [Google Scholar] [CrossRef]
- Uhlirova, H.; Kılıç, K.; Tian, P.; Thunemann, M.; Desjardins, M.; Saisan, P.A.; Sakadžić, S.; Ness, T.V.; Mateo, C.; Cheng, Q.; et al. Cell type specificity of neurovascular coupling in cerebral cortex. eLife 2016, 5, e14315. [Google Scholar] [CrossRef] [PubMed]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.-A.; Pozzan, T.; Carmignoto, P. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2002, 6, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, S.J.; MacVicar, B. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 2004, 431, 195–199. [Google Scholar] [CrossRef]
- Girouard, H.; Bonev, A.D.; Hannah, R.M.; Meredith, A.; Aldrich, R.W.; Nelson, M.T. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc. Natl. Acad. Sci. USA 2010, 107, 3811–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, G.R.J.; Choi, H.B.; Rungta, R.; Ellis-Davies, G.C.R.; MacVicar, B.A. Brain metabolism dictates the polarity of astrocyte control over arterioles. Nature 2008, 456, 745–749. [Google Scholar] [CrossRef] [Green Version]
- Nett, W.J.; Oloff, S.H.; McCarthy, K.D. Hippocampal Astrocytes In Situ Exhibit Calcium Oscillations That Occur Independent of Neuronal Activity. J. Neurophysiol. 2002, 87, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Lind, B.L.; Brazhe, A.R.; Jessen, S.B.; Tan, F.C.C.; Lauritzen, M. Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E4678–E4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, B.L.; Jessen, S.B.; Lønstrup, M.; Joséphine, C.; Bonvento, G.; Lauritzen, M. Fast Ca2+ responses in astrocyte end-feet and neurovascular coupling in mice. Glia 2017, 66, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Winship, I.R.; Plaa, N.; Murphy, T.H. Rapid Astrocyte Calcium Signals Correlate with Neuronal Activity and Onset of the Hemodynamic Response In Vivo. J. Neurosci. 2007, 27, 6268–6272. [Google Scholar] [CrossRef] [Green Version]
- Nizar, K.; Uhlirova, H.; Tian, P.; Saisan, P.A.; Cheng, Q.; Reznichenko, L.; Weldy, K.L.; Steed, T.; Sridhar, V.B.; Macdonald, C.L.; et al. In vivo Stimulus-Induced Vasodilation Occurs without IP3 Receptor Activation and May Precede Astrocytic Calcium Increase. J. Neurosci. 2013, 33, 8411–8422. [Google Scholar] [CrossRef]
- Bonder, D.E.; McCarthy, K.D. Astrocytic Gq-GPCR-Linked IP3R-Dependent Ca2+ Signaling Does Not Mediate Neurovascular Coupling in Mouse Visual Cortex In Vivo. J. Neurosci. 2014, 34, 13139–13150. [Google Scholar] [CrossRef]
- Stobart, J.; Ferrari, K.D.; Barrett, M.J.; Glück, C.; Stobart, M.J.; Zuend, M.; Weber, B. Cortical Circuit Activity Evokes Rapid Astrocyte Calcium Signals on a Similar Timescale to Neurons. Neuron 2018, 98, 726–735.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Gordon, G.R.J.; Tran, C.H.T. Heterogeneity of Sensory-Induced Astrocytic Ca2+ Dynamics During Functional Hyperemia. Front. Physiol. 2020, 11, 587-10. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, T.H. Neurovascular Coupling and Epilepsy: Hemodynamic Markers for Localizing and Predicting Seizure Onset. Epilepsy Curr. 2007, 7, 91–94. [Google Scholar] [CrossRef]
- Farrell, J.S.; Wolff, M.D.; Teskey, G.C. Neurodegeneration and Pathology in Epilepsy: Clinical and Basic Perspectives. Neurodegener. Dis. 2017, 15, 317–334. [Google Scholar] [CrossRef]
- Leonhardt, G.; De Greiff, A.; Weber, J.; Ludwig, T.; Wiedemayer, H.; Forsting, M.; Hufnagel, A. Brain Perfusion Following Single Seizures. Epilepsia 2005, 46, 1943–1949. [Google Scholar] [CrossRef]
- Fong, G.C.Y.; Fong, K.Y.; Mak, W.; Tsang, K.L.; Chan, K.H.; Cheung, R.T.F.; Ho, S.L. Postictal psychosis related regional cerebral hyperfusion. J. Neurol. Neurosurg. Psychiatr. 2000, 68, 100–101. [Google Scholar] [CrossRef] [Green Version]
- Tatlidil, R. Persistent postictal hyperperfusion demonstrated with PET. Epilepsy Res. 2000, 42, 83–88. [Google Scholar] [CrossRef]
- Gotman, J.; Kobayashi, E.; Bagshaw, A.; Bénar, C.-G.; Dubeau, F. Combining EEG and fMRI: A multimodal tool for epilepsy research. J. Magn. Reson. Imaging 2006, 23, 906–920. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, H.; Suh, M.; Schwartz, T.H. Spatiotemporal Dynamics of Perfusion and Oximetry during Ictal Discharges in the Rat Neocortex. J. Neurosci. 2009, 29, 2814–2823. [Google Scholar] [CrossRef] [Green Version]
- Penfield, W.; Santha von, K.; Cipriani, A. Cerebral Blood Flow During Induced Epileptiform Seizures in Animals and Man. J. Neurophysiol. 1939, 2, 257–267. [Google Scholar] [CrossRef]
- Penfield, W. Epilepsy and the cerebral lesions of birth and infancy. Can. Med. Assoc. J. 1939, 41, 527–534. [Google Scholar]
- Bahar, S.; Suh, M.; Zhao, M.; Schwartz, T.H. Intrinsic optical signal imaging of neocortical seizures: The ‘epileptic dip’. NeuroReport 2006, 17, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Suh, M.; Ma, H.; Zhao, M.; Sharif, S.; Schwartz, T.H. Neurovascular Coupling and Oximetry During Epileptic Events. Mol. Neurobiol. 2006, 33, 181–198. [Google Scholar] [CrossRef]
- Tran, C.H.T.; George, A.G.; Teskey, G.C.; Gordon, G.R. Seizures elevate gliovascular unit Ca2+ and cause sustained vasoconstriction. JCI Insight 2020, 5, e136469. [Google Scholar] [CrossRef]
- Gómez-Gonzalo, M.; Losi, G.; Chiavegato, A.; Zonta, M.; Cammarota, M.; Brondi, M.; Vetri, F.; Uva, L.; Pozzan, T.; de Curtis, M.; et al. An excitatory loop with astrocytes contributes to drive neurons to seizure thresh-old. PLoS Biol. 2010, 8, e1000352. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Gonzalo, M.; Losi, G.; Brondi, M.; Uva, L.; Sato, S.S.; de Curtis, M.; Ratto, G.M.; Carmignoto, G. Ictal but Not Interictal Epileptic Discharges Activate Astrocyte Endfeet and Elicit Cerebral Arteriole Responses. Front. Cell. Neurosci. 2011, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Nguyen, J.; Ma, H.; Nishimura, N.; Schaffer, C.B.; Schwartz, T.H. Preictal and Ictal Neurovascular and Metabolic Coupling Surrounding a Seizure Focus. J. Neurosci. 2011, 31, 13292–13300. [Google Scholar] [CrossRef] [Green Version]
- Devor, A.; Hillman, E.M.C.; Tian, P.; Waeber, C.; Teng, I.C.; Ruvinskaya, L.; Shalinsky, M.H.; Zhu, H.; Haslinger, R.H.; Narayanan, S.N.; et al. Stimulus-Induced Changes in Blood Flow and 2-Deoxyglucose Uptake Dissociate in Ipsilateral Somatosensory Cortex. J. Neurosci. 2008, 28, 14347–14357. [Google Scholar] [CrossRef] [Green Version]
- Rowe, C.C.; Berkovic, S.; Austin, M.C.; Saling, M.; Kalnins, R.M.; McKay, W.J.; Bladin, P.F. Visual and quantitative analysis of interictal SPECT with technetium-99m-HMPAO in temporal lobe epilepsy. J. Nucl. Med. 1991, 32, 1688–1694. [Google Scholar] [PubMed]
- Newton, M.R.; Berkovic, S.F.; Austin, M.C.; Rowe, C.C.; McKay, W.J.; Bladin, P.F. Postictal switch in blood flow distribution and temporal lobe seizures. J. Neurol. Neurosurg. Psychiatry 1992, 55, 891–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, J.S.; Gaxiola-Valdez, I.; Wolff, M.D.; David, L.S.; Dika, H.I.; Geeraert, B.L.; Wang, X.R.; Singh, S.; Spanswick, S.C.; Dunn, J.F.; et al. Postictal behavioural impairments are due to a severe prolonged hypoperfusion/hypoxia event that is COX-2 dependent. Elife 2016, 5, e19352. [Google Scholar] [CrossRef] [PubMed]
- Gokina, N.I.; Osol, G. Temperature and protein kinase C modulate myofilament Ca2+ sensitivity in pressurized rat cerebral arteries. Am. J. Physiol. 1998, 274, H1920–H1927. [Google Scholar] [CrossRef]
- Farrell, J.S.; Colangeli, R.; Dong, A.; George, A.G.; Addo-Osafo, K.; Kingsley, P.J.; Morena, M.; Wolff, M.D.; Dudok, B.; He, K.; et al. In vivo endocannabinoid dynamics at the timescale of physiological and pathological neural activity. Neuron 2021, 109, 2398–2403.e4. [Google Scholar] [CrossRef]
- Devor, A.; Tian, P.; Nishimura, N.; Teng, I.C.; Hillman, E.M.; Narayanan, S.N.; Ulbert, I.; Boas, D.A.; Kleinfeld, D.; Dale, A.M. Suppressed neuronal activity and concurrent arteriolar vasoconstriction may explain negative blood oxygenation level-dependent signal. J. Neurosci. 2007, 27, 4452–4459. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Mechanistic basis of neurovascular coupling in physiology and seizures.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Teskey, G.C.; Tran, C.H.T. Neurovascular Coupling in Seizures. Neuroglia 2021, 2, 36-47. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia2010005
AMA Style
Teskey GC, Tran CHT. Neurovascular Coupling in Seizures. Neuroglia. 2021; 2(1):36-47. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia2010005
Chicago/Turabian StyleTeskey, G. Campbell, and Cam Ha T. Tran. 2021. "Neurovascular Coupling in Seizures" Neuroglia 2, no. 1: 36-47. https://0-doi-org.brum.beds.ac.uk/10.3390/neuroglia2010005