Removal of Arsenate and Arsenite in Equimolar Ferrous and Ferric Sulfate Solutions through Mineral Coprecipitation: Formation of Sulfate Green Rust, Goethite, and Lepidocrocite



Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Formation of Coprecipitation Solids
2.3. X-Ray Diffraction (XRD) Analysis
2.4. X-ray Absorption Spectroscopy Analysis
2.5. Raman Spectroscopy Analysis
2.6. Aqueous Solution Analysis
2.7. Arsenic Concentration in Solid Phases
3. Results
3.1. Iron and Arsenic Removal
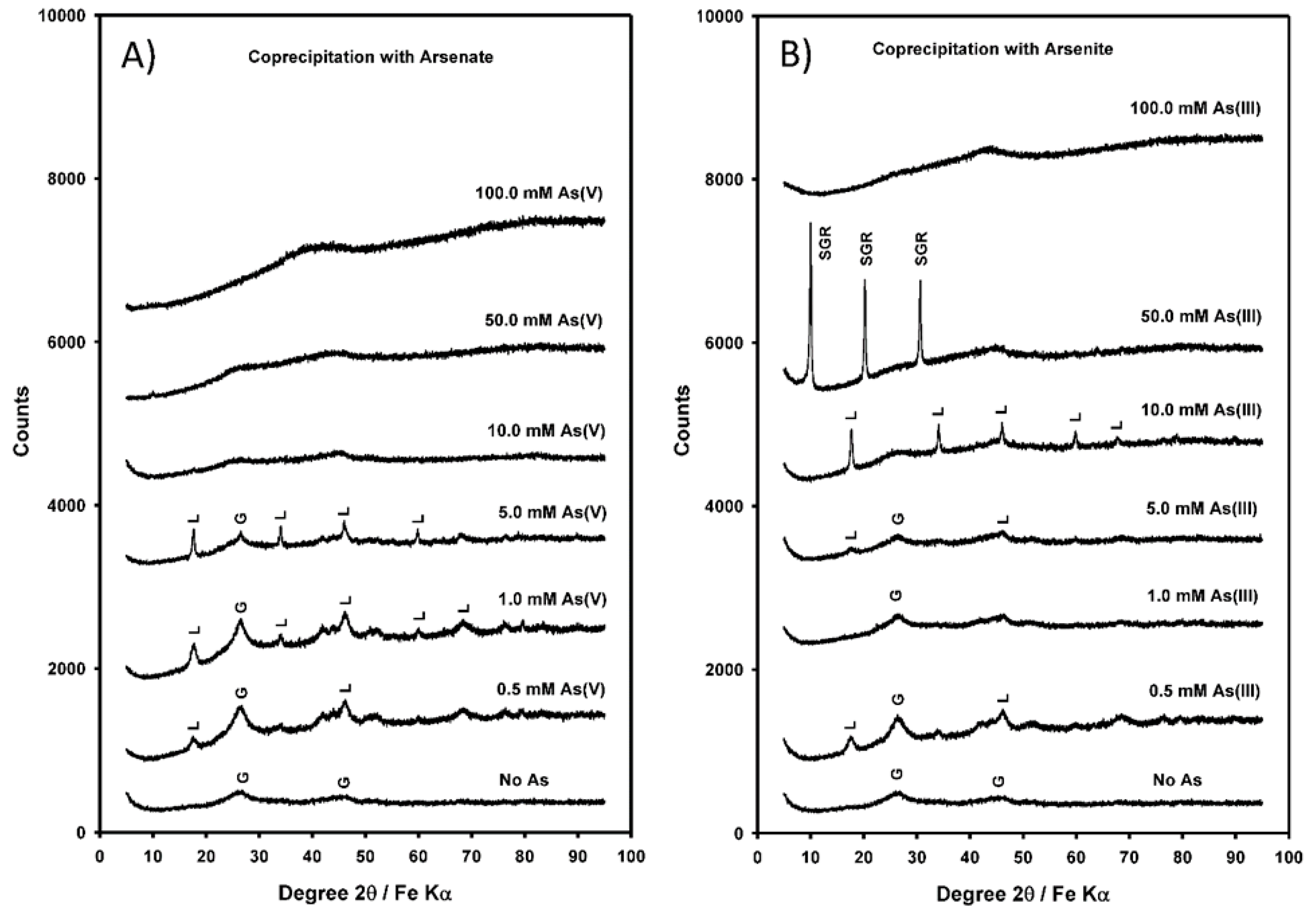
3.2. Mineralogy of Coprecipitated Solids
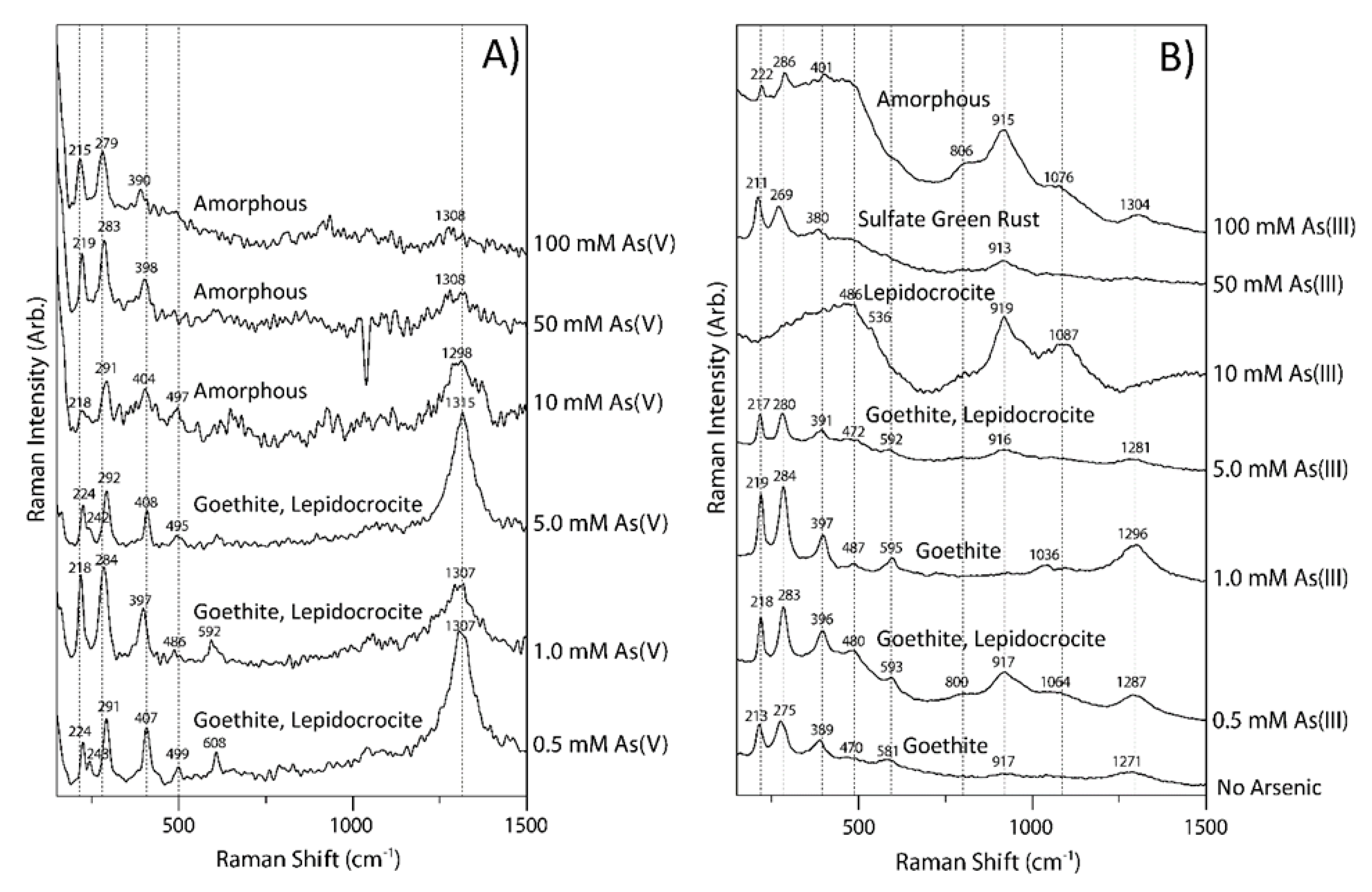
3.3. Raman Spectra of Coprecipitates
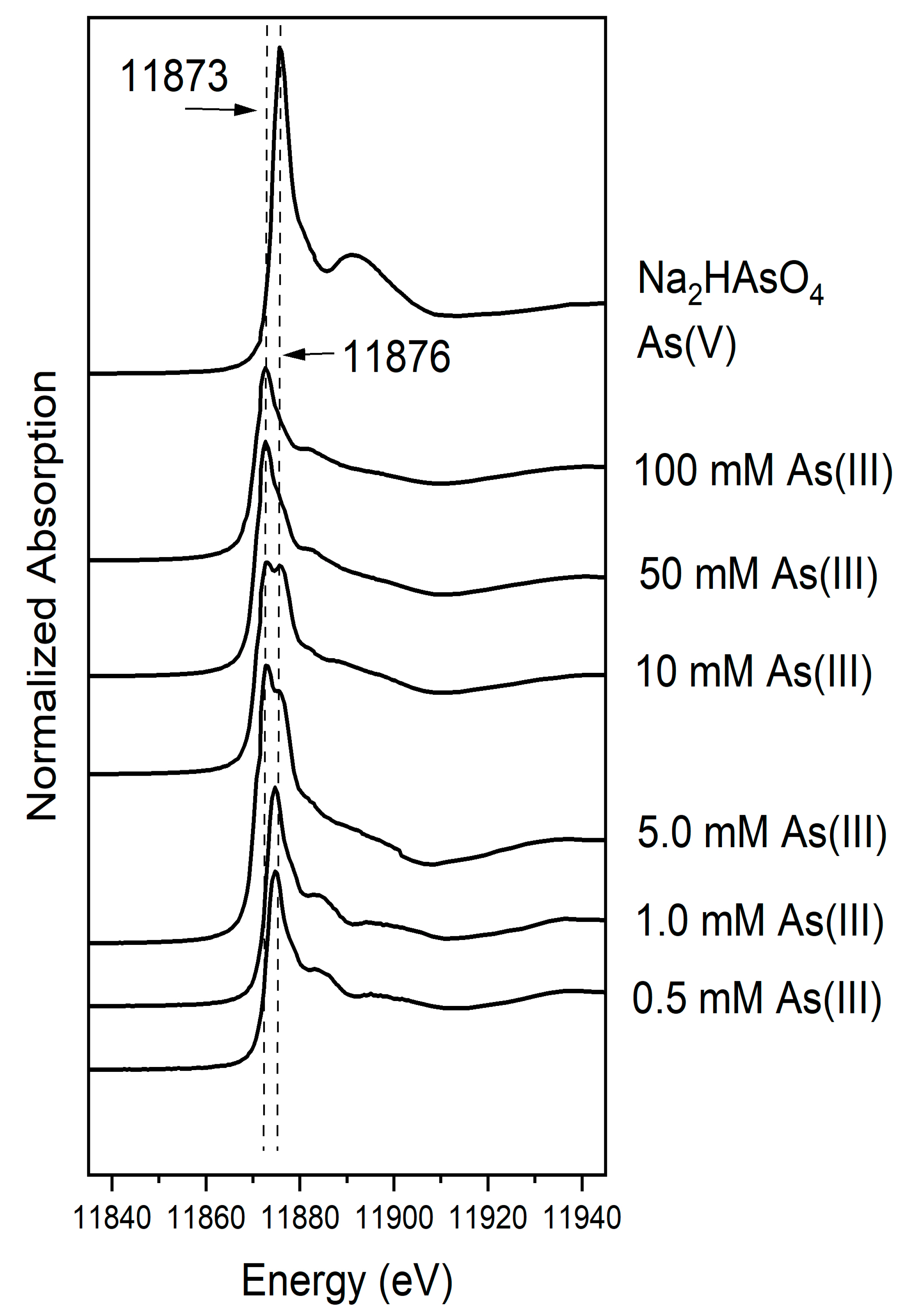
3.4. Extent and Nature of Coprecipitated As(V) and As(III)
4. Discussion
4.1. Stabilization of Sulfate Green Rust by As(III)
4.2. Was Dissolved O2 An Oxidant for As(III) Oxidation?
4.3. Implications of Iron-Based Remediation of Groundwater
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Smedley, P.L.; Kinniburgh, D.G. A review of the source, behavior and distribution of arsenic in natural waters. Appl. Geochem. 2002, 17, 517–568. [Google Scholar]
- Stollenwerk, K.G.; Breit, G.N.; Welch, A.H.; Yount, J.C.; Whitney, J.W.; Foster, A.L.; Uddin, M.N.; Majumder, R.K.; Ahmed, N. Arsenic attenuation by oxidized aquifer sediments in Bangladesh. Sci. Total Environ. 2007, 379, 133–150. [Google Scholar] [PubMed]
- Smith, S.J. Naturally Occurring Arsenic in Ground Water, Norman, Oklahoma, 2004, and Remediation Options for Produced Water; U.S. Geological Survey Fact Sheet 2005–3111; U.S. Geological Survey: Oklahoma City, OK, USA, 2005; p. 6.
- Nickson, R.; McArthur, J.; Ravenscroft, P.; Burgess, W.; Ahmed, K. Mechanism of arsenic release to groundwater, Bangladesh and West Bengal. Appl. Geochem. 2000, 15, 403–413. [Google Scholar]
- Wang, S.; Mulligan, C.N. Effect of natural organic matter on arsenic release from soils and sediments into groundwater. Environ. Geochem. Health 2006, 28, 197–214. [Google Scholar] [PubMed]
- Sadiq, M. Arsenic chemistry in soils: An overview of thermodynamic predictions and field observations. Water Air Soil Pollut. 1997, 93, 117–136. [Google Scholar]
- Cherry, J.A.; Shaikh, A.U.; Tallman, D.E.; Nicholson, R.V. Arsenic species as an indicator of redox conditions in groundwater. J. Hydrol. 1979, 43, 373–392. [Google Scholar]
- Zheng, Y.; Stute, M.; van Geen, A.; Gavrieli, I.; Dhar, R.; Simpson, H.J.; Schlosser, P.; Ahmed, K.M. Redox control of arsenic mobilization in Bangladesh groundwater. Appl. Geochem. 2004, 19, 201–214. [Google Scholar]
- Horneman, A.; Van Geen, A.; Kent, D.V.; Mathe, P.E.; Zheng, Y.; Dhar, R.K.; O’Connell, S.; Hoque, M.A.; Aziz, Z.; Shamsudduha, M.; et al. Decoupling of As and Fe release to Bangladesh groundwater under reducing conditions. Part 1: Evidence from sediment profiles. Geochim. Cosmochim. Acta 2004, 68, 3459–3473. [Google Scholar]
- Johnston, R.B.; Singer, P.C. Solubility of symplesite (ferrous arsenate): Implications for reduced groundwaters and other geochemical environments. Soil Sci. Soc. Am. J. 2007, 71, 101–107. [Google Scholar]
- Jönsson, J.; Sherman, D.M. Sorption of As(III) and As(V) to siderite, green rust (fougerite) and magnetite: Implications for arsenic release in anoxic groundwaters. Chem. Geol. 2008, 255, 173–181. [Google Scholar]
- Hansen, H.C.B. Environmental chemistry of iron(II)-iron(III) LDHs. In Layered Double Hydroxides; Rives, V., Ed.; Nova Science Publishers: Huntington, NY, USA, 2001; pp. 413–434. [Google Scholar]
- Cuttler, A.H.; Man, V.; Cranshaw, T.E.; Longworth, G.A. Mossbauer study of green rust precipitates; I. Preparations from sulphate solutions. Clay Miner. 1990, 25, 289–301. [Google Scholar]
- Kukkadapu, R.K.; Zachara, J.M.; Fredrickson, J.K.; Kennedy, D.W. Biotransformation of two-line silica-ferrihydrite by a dissimilatory Fe(III)-reducing bacterium: Formation of carbonate green rust in the presence of phosphate. Geochim. Cosmochim. Acta 2004, 68, 2799–2814. [Google Scholar]
- Legrand, L.; Abdelmoula, M.; Géhin, A.; Chaussé, A.; Génin, J.M.R. Electrochemical formation of a new Fe(II)-Fe(III) hydroxy-carbonate green rust: Characterization and morphology. Electrochim. Acta 2001, 46, 1815–1822. [Google Scholar]
- Ona-Nguema, G.; Abdelmoula, M.; Jorand, F.; Benali, O.; Géhin, A.; Block, J.C.; Génin, J.-M.R. Iron(II, III) hydrocycarbonate green rust formation and stabilization from lepidocrocite bioreduction. Environ. Sci. Technol. 2002, 36, 16–20. [Google Scholar]
- Ruby, C.; Géhin, A.; Aissa, R.; Ghanbaja, J.; Abdelmoula, M.; Génin, J.M.R. Chemical stability of hydroxysulphate green rust synthetized in the presence of foreign anions: Carbonate, phosphate and silicate. Hyperfine Interact. 2006, 167, 803–807. [Google Scholar]
- Ahmed, I.A.M.; Benning, L.G.; Kakonyi, G.; Sumoondur, A.D.; Terrill, N.J.; Shaw, S. Formation of green rust sulfate: A combined in situ time-resolved X-ray scattering and electrochemical study. Langmuir 2010, 26, 6593–6603. [Google Scholar]
- Hansen, H.C.B.; Koch, C.B.; Nancke-Krogh, H.; Borggaard, O.K.; Sørensen, J. Abiotic nitrate reduction to ammonium: Key role of green rust. Environ. Sci. Technol. 1996, 30, 2053–2056. [Google Scholar]
- Legrand, L.; Figuigui, A.E.; Mercier, F.; Chausse, A. Reduction of aqueous chromate by Fe(II)/Fe(III) carbonate green rust: Kinetics and mechanistic studies. Environ. Sci. Technol. 2004, 38, 4587–4595. [Google Scholar]
- Skovbjerg, L.L.; Stipp, S.L.S.; Utsunomiya, S.; Ewing, R.C. The mechanisms of reduction of hexavalent chromium by green rust sodium sulfate: Formation of Cr-goethite. Geochimica 2006, 70, 3582–3592. [Google Scholar]
- O’Loughlin, E.J.; Kelly, S.D.; Cook, R.E.; Csencsits, R.; Kemner, K.M. Reduction of uranium(VI) by mixed iron(II)/iron(III) hydroxide (green rust): Formation of UO2 nanoparticles. Environ. Sci. Technol. 2003, 37, 721–727. [Google Scholar]
- O’Loughlin, E.J.; Kelly, S.D.; Kemner, K.M.; Csencsits, R.; Cook, R.E. Reduction of AgI, AuIII, CuII, and HgII by FeII/FeIII hydroxysulfate green rust. Chemosphere 2003, 53, 437–446. [Google Scholar] [PubMed]
- Erbs, M.; Hansen, H.C.B.; Olsen, C.E. Reductive dechlorination of carbon tetrachloride using iron(II) iron(III) hydroxide sulfate (green rust). Environ. Sci. Technol. 1999, 33, 307–311. [Google Scholar]
- O’Loughlin, E.J.; Kemner, K.M.; Burris, D.R. Effects of AgI, AuIII, and CuII on the reductive dechlorination of carbon tetrachloride by green rust. Environ. Sci. Technol. 2003, 37, 2905–2912. [Google Scholar]
- Liang, X.; Butler, E. Effects of natural organic matter model compounds on the transformation of carbon tetrachloride by chloride green rust. Water Res. 2010, 44, 2125–2132. [Google Scholar]
- Su, C.; Puls, R.W. Significance of iron(II, III) hydroxycarbonate green rust in arsenic remediation using zerovalent iron in laboratory column tests. Environ. Sci. Technol. 2004, 38, 5224–5231. [Google Scholar] [PubMed]
- Feder, F.; Trolard, F.; Klingelhöfer, G.; Bourrie, G. In situ Mössbauer spectroscopy: Evidence for green rust (fougerite) in a gleysol and its mineralogical transformations with time and depth. Geochim. Cosmochim. Acta 2005, 69, 4463–4483. [Google Scholar]
- Ruby, C.; Upadhyay, C.; Géhin, A.; Ona-Nguema, G.; Génin, J.M.R. In situ redox flexibility of FeII-III oxyhydroxycarbonate green rust and fougerite. Environ. Sci. Technol. 2006, 40, 4696–4702. [Google Scholar]
- Christiansen, B.C.; Balic-Zunic, T.; Dideriksen, K.; Stipp, S.L.S. Identification of green rust in Groundwater. Environ. Sci. Technol. 2009, 43, 3436–3441. [Google Scholar]
- Hansen, H.C.B. Composition, stabilization, and light absorption of Fe(II)Fe(III) hydroxy-carbonate (‘green rust’). Clay Miner. 1989, 24, 663–669. [Google Scholar]
- Génin, J.-M.R.; Aїssa, R.; Géhin, A.; Abdelmoula, M.; Benali, O.; Ernstsen, V.; Ona-Nguema, G.; Upadhyay, C.; Ruby, C. Fougerite and FeII-III hydroxycarbonate green rust; ordering, deprotonation and/or cation substitution; structure of hydrotalcite-like compounds and mythic ferrosic hydroxide Fe(OH)(2+x). Solid State Sci. 2005, 7, 545–572. [Google Scholar]
- Drissi, S.H.; Refait, P.; Génin, J.M.R. The preparation and thermodynamic properties of Fe(II)-Fe(III) hydroxide-carbonate (green rust 1); Pourbaix diagram of iron in carbonate-containing aqueous media. Corros. Sci. 1995, 37, 2025–2041. [Google Scholar]
- Simon, L.; François, M.; Refait, P.; Renaudin, G.; Lelaurain, M.; Génin, J.M.R. Structure of the Fe(II-III) layered double hydroxysulfate green rust two from Rietveld analysis. Solid State Sci. 2003, 5, 327–334. [Google Scholar]
- Randall, S.R.; Sherman, D.M.; Ragnarsdottir, K.V. Sorption of As(V) on green rust (Fe4(II)Fe2(III)(OH)12SO4·3H2O) and lepidocrocite (γ-FeOOH): Surface complexes from EXAFS spectroscopy. Geochim. Cosmochim. Acta 2001, 65, 1015–1023. [Google Scholar]
- Su, C.; Wilkin, R.T. Arsenate and arsenite sorption on and arsenite oxidation by iron (II, III) hydroxycarbonate green rust. Am. Chem. Soc. Symp. Ser. 2005, 915, 25–40. [Google Scholar]
- Sposito, G. The Surface Chemistry of Soils; Oxford University Press: New York, NY, USA, 1984. [Google Scholar]
- Ford, R.G. Rates of hydrous ferric oxide crystallization and the influence on coprecipitated arsenate. Environ. Sci. Technol. 2002, 36, 2459–2463. [Google Scholar]
- Das, S.; Hendry, M.J. Application of Raman spectroscopy to identify iron minerals commonly found in mine wastes. Chem. Geol. 2011, 290, 101–108. [Google Scholar]
- Thoral, S.; Rose, J.; Garnier, J.M.; Van Geen, A.; Refait, P.; Traverse, A.; Fonda, E.; Nahon, D.; Bottero, J.Y. XAS study of iron and arsenic speciation during Fe(II) oxidation in the presence of As(III). Environ. Sci. Technol. 2005, 39, 9478–9485. [Google Scholar]
- Ravel, B.; Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data Analysis for X-Ray Absorption Spectroscopy Using IFEFFIT. J. Synchrotron Rad. 2005, 12, 537–541. [Google Scholar]
- Hansel, C.M.; Benner, S.G.; Fendorf, S. Competing Fe(II)-induced mineralization pathways of ferrihydrite. Environ. Sci. Technol. 2005, 39, 7147–7153. [Google Scholar]
- Song, J.; Jia, S.Y.; Yu, B.; Wu, S.H.; Han, X. Formation of iron(hydr)oxides during the abiotic oxidation of Fe(II) in the presence of arsenate. J. Hazard. Mat. 2015, 294, 70–79. [Google Scholar]
- Han, X.; Song, J.; Li, Y.L.; Jia, S.Y.; Wang, W.H.; Huang, F.G.; Wu, S.H. As(III) removal and speciation of Fe (oxyhydr)oxides during simultaneous oxidation of As(III) and Fe(II). Chemosphere 2016, 147, 337–344. [Google Scholar] [PubMed]
- Wang, Y.; Morin, G.; Ona-Nguema, G.; Menguy, N.; Brown, G.E., Jr. Arsenic(III) and As(V) speciation during transformation of lepidocrocite to magnetite. Environ. Sci. Technol. 2014, 48, 14282–14290. [Google Scholar]
- Masue-Slowey, Y.; Slowey, A.J.; Michel, F.M.; Webb, S.M.; Fendorf, S. Constrains on precipitation of the ferrous arsenite solid H7Fe4(AsO3)5. J. Environ. Qual. 2014, 43, 947–954. [Google Scholar] [PubMed]
- Sumoondur, A.; Shaw, S.; Ahmed, I.; Benning, L.G. Green rust as a precursor for magnetite: An in situ synchrotron based study. Mineralog. Magaz. 2008, 72, 201–204. [Google Scholar]
- Ruby, C.; Géhin, A.; Abdelmoula, M.; Génin, J.M.R.; Jolivet, J.P. Coprecipitation of Fe(II) and Fe(III) cations in sulphated aqueous medium and formation of hydroxysulphate green rust. Solid State Sci. 2003, 5, 1055–1062. [Google Scholar]
- Schwertmann, U.; Fechter, H. The formation of green rust and its transformation to lepidocrocite. Clay Mineral. 1994, 29, 87–92. [Google Scholar]
- Cumplido, J.; Barron, V.; Torrent, J. Effect of phosphate on the formation of nanophase lepidocrocite from Fe(II) sulfate. Clays Clay Mineral. 2000, 48, 503–510. [Google Scholar]
- Dove, P.M.; Rimstidt, J.D. The solubility and stability of scorodite, FeAsO4·2H2O. Am. Mineral. 1985, 70, 838–844. [Google Scholar]
- Rochette, E.A.; Li, G.C.; Fendorf, S. Stability of arsenate minerals in soil under biotically generated reducing conditions. Soil Sci. Soc. Am. J. 1998, 62, 1530–1537. [Google Scholar]
- Paktunc, D.; Dutrizac, J.; Gertsman, V. Synthesis and phase transformations involving scorodite, ferric arsenate and arsenical ferrihydrite: Implications for arsenic mobility. Geochim. Cosmochim. Acta 2008, 72, 2649–2672. [Google Scholar]
- Le Berre, J.F.; Gauvin, R.; Demopoulos, G.P. Characterization of poorly-crystalline ferric arsenate precipitated from equimolar Fe(III)-As(V) solutions in the pH range 2 to 8. Metallurg. Material. Transact. B 2007, 38, 751–762. [Google Scholar]
- Richmond, W.R.; Loan, M.; Morton, J.; Parkinson, G.M. Arsenic removal from aqueous solution via ferrihydrite crystallization control. Environ. Sci. Technol. 2004, 38, 2368–2372. [Google Scholar]
- Refait, P.; Gehin, A.; Abdelmoula, M.; Genin, J.M.R. Coprecipitation thermodynamics of iron(II-III) hydroxysulphate green rust from Fe(II) and Fe(III) salts. Corros. Sci. 2003, 45, 659–676. [Google Scholar]
- Mazeina, L.; Navrotsky, A.; Dyar, D. Enthalpy of formation of sulfate green rusts. Geochim. Cosmochim. Acta 2008, 72, 1143–1153. [Google Scholar]
- Ayala-Luis, K.B.; Koch, C.B.; Hansen, H.C.B. The standard Gibbs energy of formation of Fe(II)Fe(III) hydroxide sulfate green rust. Clays Clay Miner. 2008, 56, 633–644. [Google Scholar]
- Legodi, M.A.; de Waal, D. The preparation of magnetite, goethite, hematite and maghemite of pigment quality from mill scale iron waste. Dyes Pigm. 2007, 74, 161–168. [Google Scholar]
- Legrand, L.; Sagon, G.; Lecomte, S.; Chausse, A.; Messina, R. A Raman and infrared study of a new carbonate green rust obtained by electrochemical way. Corros. Sci. 2001, 43, 1739–1749. [Google Scholar]
- Boucherit, N.; Hugot-Le Goff, A.; Joiret, S. Raman studies of corrosion films grown on Fe and Fe-6Mo in pitting conditions. Corros. Sci. 1991, 32, 497–507. [Google Scholar]
- Perez, J.P.H.; Freeman, H.M.; Brown, A.P.; van Genuchten, C.M.; Dideriksen, K.; S’ari, M.; Tobler, D.J.; Benning, L.G. Direct visualization of arsenic binding on green rust sulfate. Environ. Sci. Technol. 2020, 54, 3297–3305. [Google Scholar]
- Maillot, F.; Morina, G.; Juillot, F.; Bruneel, O.; Casiot, C.; Ona-Nguema, G.; Wang, Y.; Lebrun, S.; Aubry, E.; Vlaic, G.; et al. Structure and reactivity of As(III)- and As(V)-rich schwertmannites and amorphous ferric arsenate sulfate from the Carnoulès acid mine drainage, France: Comparison with biotic and abiotic model compounds and implications for As remediation. Geochim. Cosmochim. Acta 2013, 104, 310–329. [Google Scholar]
- Kim, M.J.; Nriagu, J. Oxidation of arsenite in groundwater using ozone and oxygen. Sci. Total Environ. 2000, 247, 71–79. [Google Scholar]
- Hug, S.J.; Leupin, O. Iron-catalyzed oxidation of arsenic(III) by oxygen and by hydrogen peroxide: pH-dependent formation of oxidants in the Fenton reaction. Environ. Sci. Technol. 2003, 37, 2734–2742. [Google Scholar]
- Zhao, Z.; Jia, Y.; Xu, L.; Zhao, S. Adsorption and heterogeneous oxidation of As(III) on ferrihydrite. Water Res. 2011, 45, 6496–6504. [Google Scholar]
- Kim, S.O.; Lee, W.C.; Cho, H.G.; Lee, B.T.; Lee, P.K.; Choi, S.H. Equilibria, kinetics, and spectroscopic analyses on the uptake of aqueous arsenite by two-line ferrihydrite. Environ. Technol. 2014, 35, 251–261. [Google Scholar]
- Paikaray, S.; Essilfie-Dughan, J.; Göttlicher, J.; Pollok, K.; Perffer, S. Redox stability of As(III) on schwertmannite surfaces. J. Hazard. Mat. 2014, 265, 208–216. [Google Scholar]
- Johnston, R.B.; Singer, P.C. Redox reactions in the Fe-As-O2 system. Chemosphere 2007, 69, 517–525. [Google Scholar] [PubMed]
- Hug, S.J.; Canonica, L.; Wegelin, M.; Gechter, D.; von Gunten, U. Solar oxidation and removal of arsenic at circumneutral pH in iron containing waters. Environ. Sci. Technol. 2001, 35, 2114–2121. [Google Scholar] [PubMed]
- Roberts, L.C.; Hug, S.J.; Ruettimann, T.; Billah, M.; Khan, A.W.; Rahman, M.T. Arsenic removal with iron(II) and iron(III) waters with high silicate and phosphate concentrations. Environ. Sci. Technol. 2004, 38, 307–315. [Google Scholar]
- Wang, Y.; Morin, G.; Ona-Nguema, G.; Menguy, N.; Juillot, F.; Aubry, E.; Guyot, F.; Calas, G.; Brown, G.E., Jr. Arsenite sorption at the magnetite-water interface during aqueous precipitation of magnetite: EXAFS evidence for a new arsenite surface complex. Geochim. Cosmochim. Acta 2008, 72, 2573–2586. [Google Scholar]
- Su, C.; Puls, R.W. Arsenate and arsenite removal by zerovalent iron: Kinetics, redox transformation, and implications for in situ groundwater remediation. Environ. Sci. Technol. 2001, 35, 1487–1492. [Google Scholar]
- Wilkin, R.T.; Acree, S.D.; Ross, R.R.; Beak, D.G.; Lee, T.R. Performance of a zerovalent iron reactive barrier for the treatment of arsenic in groundwater: Part 1. Hydrogeochemical studies. J. Contam. Hydrol. 2009, 106, 1–14. [Google Scholar] [PubMed]
- Beak, D.G.; Wilkin, R.T. Performance of a zerovalent iron reactive barrier for the treatment of arsenic in groundwater: Part 2. Geochemical modeling and solid phase studies. J. Contam. Hydrol. 2009, 106, 15–28. [Google Scholar] [PubMed]
- Mercer, K.L.; Tobiason, J.E. Removal of arsenic from high ionic strength solutions: Effects of ionic strength, pH, and preformed versus in situ formed HFO. Environ. Sci. Technol. 2008, 42, 3797–3802. [Google Scholar] [PubMed]
- Mukiibi, M.; Ela, W.; Sáez, A. Effect of ferrous iron on arsenate sorption to amorphous ferric hydroxide. Ann. N. Y. Acad. Sci. 2008, 1140, 335–345. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| [As(V)]0/mM | pH | Eh/mV | [Fe2+]/mM | [Fe]/mM | Fe Removal/% | [As(V)]/mM | [As(III)]/mM | As Removal/% | As conc. in solids/mg kg−1 | Mineralogy | Solids Color | Magnetic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 | 6.59 | 30.8 | 21.9 | 27.8 | 89.55 | 0.053 | nd | 99.95 | 171,000 ± 6000 | amorphous HFO | gray | no |
| 50 | 6.23 | 53.7 | 69.6 | 62.3 | 76.58 | 0.018 | nd | 99.96 | 101,000 ± 500 | amorphous HFO | brown | no |
| 10 | 6.15 | 61.6 | 56 | 84 | 68.42 | nd | nd | 100 | 40,600 ± 6300 | amorphous HFO | brown | no |
| 5 | 5.78 | 86.3 | 69.8 | 91 | 65.79 | nd | nd | 100 | 17,400 ± 800 | lepido., goethite | brown | no |
| 1 | 5 | 187 | 96.2 | 95.5 | 64.1 | nd | nd | 100 | 3980 ± 230 | lepido., goethite | brown | no |
| 0.5 | 4.83 | 227 | 86.2 | 95.1 | 64.25 | nd | nd | 100 | 2070 ± 100 | goethite, lepido. | brown | no |
| 0 | 4.09 | 366 | 97.1 | 96.7 | 63.65 | nd | nd | nd | nd | goethite | brown | no |
| [As(III)]0/mM | pH | Eh/mV | [Fe2+]/mM | [Fe]/mM | Fe Removal/% | [As(V)]/mM | [As(III)]/mM | As Removal/% | As conc. in solids/mg kg−1 | Mineralogy | Solids Color | Magnetic |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 100 | 6.62 | 18.1 | 55.1 | 56.8 | 78.65 | 1.06 | 0.29 | 98.65 | 207,000 ± 7000 | amorphous HFO | gray | no |
| 50 | 6.34 | 56.8 | 47.9 | 75.6 | 71.58 | 0.22 | 0.32 | 98.92 | 136,300 ± 26,100 | SGR | green | no |
| 10 | 5.51 | 194 | 95.3 | 93.7 | 64.77 | 0.0039 | 0.071 | 99.25 | 31,600 ± 1150 | lepidocrocite | brown | no |
| 5 | 4.37 | 342 | 88.3 | 96.9 | 63.57 | 0.0024 | 0.047 | 99.01 | 14,900 ± 1700 | goethite, lepido. | brown | no |
| 1 | 3.73 | 417 | 92.2 | 98.1 | 63.12 | 0.00051 | 0.0070 | 99.25 | 3280±520 | goethite, lepido. | brown | no |
| 0.5 | 4.96 | 214 | 89.2 | 94 | 64.66 | nd | 0.0016 | 99.68 | 1850 ± 70 | goethite | brown | no |
| Coprecipitate Sample | Component | Fitted XANES Fraction |
|---|---|---|
| 100 mM As(III) | As(V) | 0.110 |
| As(III) | 0.890 | |
| 50 mM As(III) | As(V) | 0.131 |
| As(III) | 0.869 | |
| 10 mM As(III) | As(V) | 0.294 |
| As(III) | 0.706 | |
| 5.0 mM As(III) | As(V) | 0.509 |
| As(III) | 0.491 | |
| 1.0 mM As(III) | As(V) | 0.722 |
| As(III) | 0.278 | |
| 0.5 mM As(III) | As(V) | 0.722 |
| As(III) | 0.278 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, C.; Wilkin, R.T. Removal of Arsenate and Arsenite in Equimolar Ferrous and Ferric Sulfate Solutions through Mineral Coprecipitation: Formation of Sulfate Green Rust, Goethite, and Lepidocrocite. Soil Syst. 2020, 4, 68. https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems4040068
Su C, Wilkin RT. Removal of Arsenate and Arsenite in Equimolar Ferrous and Ferric Sulfate Solutions through Mineral Coprecipitation: Formation of Sulfate Green Rust, Goethite, and Lepidocrocite. Soil Systems. 2020; 4(4):68. https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems4040068
Chicago/Turabian StyleSu, Chunming, and Richard T. Wilkin. 2020. "Removal of Arsenate and Arsenite in Equimolar Ferrous and Ferric Sulfate Solutions through Mineral Coprecipitation: Formation of Sulfate Green Rust, Goethite, and Lepidocrocite" Soil Systems 4, no. 4: 68. https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems4040068