
Sorption Mechanisms of Chemicals in Soils

Department of Soil and Water Systems, University of Idaho, 875 Perimeter Drive MS 2340, Moscow, ID 83844-2340, USA
Soil Syst. 2021, 5(1), 13; https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems5010013
Submission received: 17 December 2020
/
Revised: 15 February 2021
/
Accepted: 17 February 2021
/
Published: 24 February 2021
(This article belongs to the Special Issue Sorption Processes in Soils and Sediments)
Abstract
:Sorption of chemicals onto soil particle surfaces is an important process controlling their availability for uptake by organisms and loss from soils to ground and surface waters. The mechanisms of chemical sorption are inner- and outer-sphere adsorption and precipitation onto mineral surfaces. Factors that determine the sorption behavior are properties of soil mineral and organic matter surfaces and properties of the sorbing chemicals (including valence, electron configuration, and hydrophobicity). Because soils are complex heterogeneous mixtures, measuring sorption mechanisms is challenging; however, advancements analytical methods have made direct determination of sorption mechanisms possible. In this review, historical and modern research that supports the mechanistic understanding of sorption mechanisms in soils is discussed. Sorption mechanisms covered include cation exchange, outer-sphere adsorption, inner-sphere adsorption, surface precipitation, and ternary adsorption complexes.
1. Historical Advancements in Adsorption Phenomenon in Soils
In 1789 Antoine Lavoisier published the book “Elementary Treatise of Chemistry,” which established the fundamentals of modern chemistry [1]. Armed with this new knowledge, early nineteenth-century scientists began researching the chemistry of soil-plant relationships. In 1821 Giuseppe Gazzeri conducted experiments to show that clay particles decolorized and deodorized liquid manure, retaining plant nutrients [2]. In the 1820s, Phillip Carl Sprengel advanced the theory that inorganic chemicals are key to plant nutrition [3]. In 1840, Justus von Leibig further advanced and supported Sprengel’s findings, which, in part, became known as the Law of Minimum, describing that plant growth is limited by the essential nutrient that has the lowest availability [3]. These early theories of plant nutrient requirements established the importance of soils for providing nutrients, but little was known of how chemicals reacted in soils, which came a few decades after Sprengel’s and Leibig’s theory.
It is now well known that the availability of chemicals in soils for uptake by an organism or transport out of soil is controlled by reactions on surfaces of soil minerals and soil organic matter (SOM). The chemicals interact with surfaces through sorption and desorption reactions, where the sorbed chemicals are distinct phases from the bulk solid and the bulk solution [4]. Sorption reactions include both adsorption and precipitation reactions on soil particle surfaces. Adsorbed chemicals have either attractive electrostatic forces holding them near the surface, or have direct chemical bonds with surface functional groups. Precipitation on soil particle surfaces creates new multi-nuclear solid phases.
In the nineteenth-century, there were many reports of the ability of soils to exchange ions between soil solids and solutions, including results from experiments on ion exchange by British farmer H.S. Thompson, who reported his findings to J. Thomas Way, a consulting Scientist to the Royal Agriculture Society of England. In 1850, Way published a foundational paper reporting the ion exchange phenomenon, which established this concept as an important process for plant nutrition in soils [5]. Way correctly predicted that the results would:
“…prove of great importance in modifying the theory and in confirming or improving the practice of many agricultural operations.”
Subsequent to Way’s report, foundational research on soil’s ability to adsorb and desorb chemicals firmly established cation exchange as an important process not only for agriculture, but also in natural ecosystems. Schuffelen [6], Thomas [7], Sposito [8], and Sparks [2] provide excellent reviews of the historical developments of cation exchange.
Following the discovery of cation exchange, investigators set out to understand and predict the details of the reaction processes and learn about the soil components that adsorb and release cations. Although J.T. Way and others proposed that soil clays were responsible for cation exchange, they lacked the tools to identify them. The discovery of X-ray diffraction (XRD) by Max von Laue and colleagues in 1912 provided a new tool for studying mineral structures. Nearly seventy years after the discovery of cation exchange in soils, Hendricks and Fry [9] and Kelley et al. [10] analyzed soil clay particles using XRD and chemical analysis and discovered that they are crystalline minerals belonging to the phyllosilicate group, and that they have isomorphic substitution of structural cations leading to overall charge deficit in the mineral. The charge deficit creates a permanent negative electrostatic charge on clay mineral surfaces that attracts cations and is the cause of cation exchange reactions.
By the 1930s, the discovery of soil chemical properties was in full swing, and the concepts of cation exchange had developed into a mature science that helped explain important soil properties, such as soil acidity, soil salinity, erosion of soil particles, and water infiltration. However, soils also adsorb anions and organic chemicals that are both ionic and non-ionic. Further, cation exchange is considered primarily an outer-sphere adsorption reaction promoted by electrostatic interactions between the cation and the charged surface, but many ions adsorb to soil particles through direct bonds that include covalent and ionic characteristics (Table 1). Chemical adsorption through direct bonding to mineral surfaces is called inner-sphere complexation, which occurs on soil clay minerals, oxide minerals, and SOM.
In this paper, mechanisms of sorption to soil particles are reviewed. This includes cation exchange, outer-sphere adsorption, inner-sphere adsorption (Figure 1), and precipitation on mineral surfaces. A thorough historical treatment of soil sorption phenomena is not given here; rather, the intent of this paper is to highlight research that shows direct evidence for the different sorption mechanisms that occur in soils. Two important tenets of sorption that underlie this review are:
- Chemical availability from soil solution for uptake by an organism or transport out of the soil is controlled by many distinct types of sorption processes that occur at the solid-solution interface, each with its own chemical energy that controls the distribution of the chemical between the solid and solution.
- Sorption amount can be evaluated indirectly by changes in the chemical composition of the solution, but accurate measurement of sorption mechanisms requires a multitude of investigation methods and is best supported using molecular-level tools that can directly measure sorbed chemical speciation [11,12].
2. Modern Concepts of Sorption
In soils, there are several different surfaces and types of chemicals that result in multiple sorption processes, making the measurement of sorption mechanisms difficult. However, because of its importance, a mechanistic understanding of sorption reactions is required for the effective management of chemicals in agricultural and natural ecosystems. Concepts of sorption are also used to manage and predict the physical processes of soils, such as aggregation and dispersion, which control erosion and soil water processes (e.g., infiltration and water holding capacity). For example, managing irrigation water salinity relies on using ratios of Na+, Mg2+, and Ca2+ in the irrigation water to predict which cations will adsorb when applied to soils because the different cations affect the separation distance between soil particles [29], which causes soils to aggregate and disperse.
Measurement and prediction of sorption reaction processes are challenging because the sorbed chemicals represent only a minor fraction of the mass of the soil; the solid mineral phase comprises the bulk of the soil mass. For example, the cation exchange capacity of the surface of a clay mineral-like montmorillonite is ~800 mmol (+) kg−1. If the exchange sites are saturated with Ca2+, the montmorillonite has 16,000 mg kg−1 of Ca2+ (i.e., 1.6% of the mineral mass is adsorbed Ca2+). The remaining mass of the montmorillonite is due to Si, O, Al, Mg, and Fe ions. Despite being a minor fraction of the mass, sorbed chemicals are often the most important species for controlling chemical availability to the soil solution.
Because soils contain numerous types of solids and chemicals, numerous different sorption mechanisms may be occurring simultaneously. Often, however, a certain solid phase and sorption reaction mechanism dominates the sorption of a particular chemical species because it is either the dominant type of solid in the soil or because the chemical has a high affinity for the mineral or organic matter sorption sites. For example, sorption of PO43− in an Oxisol is predominantly on iron and aluminum oxide minerals like hematite, goethite, and gibbsite through inner-sphere complexation [30].
Although understanding and predicting sorption processes in soils is often difficult, advancements in technology and data analysis methods have allowed for the development of analytical tools that can measure sorption mechanisms, providing new insights into the processes controlling chemical reactions at the solid-solution interface. In this review, an overview of classic and modern analysis of soil sorption phenomena is given.
3. Factors Controlling Sorption Mechanisms in Soils
A sorption reaction involves two reactants, the solid (sorbent) and the chemical species in solution (sorptive). The properties of the reactants control the specific physical and chemical interactions that occur between a sorbed chemical and the solid surface. The solid can have several types of reactive functional groups on its surfaces, can have varying specific surface areas, and can have permanent electrostatic charges associated with it. The chemicals in solution to be sorbed also have distinct characteristics that affect sorption mechanisms, including hydration sphere, ionic radius, valence, outer-shell electron configuration, covalent and ionic bonding characteristics, polarizability, hydrophobicity, molecular coordination, steric effects, and redox characteristics.
Table 2 summarizes the diverse types of adsorption surfaces and their respective characteristics. Even within the same solid, there are multiple types of adsorption sites. Montmorillonite, for example, has permanent layer charge adsorption sites on the interlayers created from isomorphic substitution and has unsatisfied bonds on its edges that adsorb chemicals through covalent bonding and electrostatic attraction. There are dozens of unique surface functional groups on the edges of montmorillonite, each with different adsorption affinities [31]. Another example is SOM, which has charged functional groups that can form both inner- and outer-sphere complexes and also has hydrophobic regions that can facilitate the adsorption of uncharged chemicals.
Soil mineral surfaces that have acidic or basic functional groups change their charge as soil solution pH changes, creating variable charged soils. SOM is also a contributor to variable surface charge in soils due to the weak acid and base characteristics of its functional groups. In contrast, soils in which clay minerals predominate have considerable amounts of permanently charged minerals—pH does not affect their surface charge. The permanent charge is created from isomorphic substitution and causes a negative layer charge on the clay surfaces.
On the edges of clay minerals, the oxygen functional groups are referred to as silanol and aluminol functional groups. Although these groups occur on all clays, on kaolinite, they are the main reactive surfaces for adsorption reactions (Figure 2). Iron, aluminum, and manganese oxides in soils also have reactive oxygen functional groups on their edges (Table 2), and because these minerals are often small, they have high specific surface areas and thus may predominate the solid-solution interface in soils. The reactivity of the surface oxygen functional groups is dependent on their coordination to the mineral surface (i.e., number of bonds between the oxygen and the mineral structural cations) and the type of cation to which they are coordinated. Reactivity of the surface functional groups can be quantitatively characterized using an acidity constant (pKa)—at least theoretically. Experimentally it is difficult to isolate a single type of functional group on mineral surfaces, so the measured surface pKa values are averages of all the different functional groups that exist on the surface [32,33].
The size of the physical space at the mineral surface is another important factor that affects adsorption. Minerals are often porous solids, and the physical constraints of small pores create forces on water and chemicals that affect molecular interactions of the sorbed chemicals [34].
Organic matter in soils has a very high specific surface area with numerous weak acid and base functional groups that can adsorb ions. The weak acid functional groups are carboxylic and phenolic acids that deprotonate, becoming negatively charged and can adsorb cations via electrostatic forces. The outer-shell electrons on the O functional groups are reactive and can share electrons with metal cations, and thus inner-sphere complexation is an important adsorption mechanism on SOM. A classic method for studying SOM adsorption properties is to separate the organic compounds from the soil by extraction, creating humic and fulvic acid separates [35,36]. Cation exchange capacities of humic and fulvic acids range from 4850 to 14,000 mmol (+)/kg [37]. Amine and thiol functional groups are also reactive sites for ion adsorption on SOM, including anions. For some metals, adsorption on amine and thiol functional groups is preferential; for example, Xia et al. [38] used X-ray absorption fine structure (XAFS) spectroscopy to show that Hg2+ preferentially adsorbs to thiol groups on humic acid.
Despite the usefulness of studying humic and fulvic acids because it isolates the SOM from the soil minerals, the chemical and physical properties of SOM are different when it is extracted [40]. Thus, the true adsorption behavior of SOM is best studied in-situ [37]. Helling et al. [41] studied CEC of 60 soils as a function of pH and used statistical analysis to determine that SOM has pH-dependent CEC: at pH < 3, CEC is ~600 mmol (+)/kg, and at pH 8, it is ~3500 mmol (+)/kg. XAFS spectroscopy has been used to investigate metal adsorption mechanisms on SOM (not extracted), e.g., [42,43,44,45]. Results show that metals preferentially adsorb by inner-sphere complexation onto carboxyl and phenol functional groups on the SOM and less onto the soil minerals. Thus, even in soils with small amounts of SOM, it can predominate cation exchange capacity and metal adsorption reactions [37].
Chemical characteristics also play a significant role in sorption mechanisms occurring in soils. For example, on the same mineral, alkali and alkaline earth metals like Na+, Mg2+, and Ca2+ may have different sorption mechanisms than many metals (e.g., Cu2+, Pb2+, and Zn2+). Sodium, Mg2+, and Ca2+ cations have strong hydration spheres and are mostly adsorbed as outer-sphere complexes on negatively charged surfaces because they do not share their electron orbitals with oxygen ligands during bonding. In contrast, many metal cations such as Cu2+, Pb2+, Zn2+, and Ni2+ adsorb by inner-sphere complexation with functional groups on SOM and edges of minerals.
Mechanistic surface complexation models differentiate adsorption complexes by relating the adsorbed chemical to the surface potential that it satisfies. The adsorbed chemicals are partitioned into charge-compensating regions next to the surface [4,46]. The net surface charge (σp) on a particle (mmol (±)/kg or C/m) is described by the charge-balance equation:
σp = σo + σH + σIS + σOS
The terms of this equation are described in Table 3 and illustrated in Figure 1. The charge originating from the mineral structural properties (σo and σH) is called an intrinsic charge because these charge components are inherent to the mineral, regardless of solution conditions [4,39]. The charge originating from inner-sphere or outer-sphere ion adsorption (σIS or σOS) is called Stern-layer charge. The net surface charge (σp) is balanced by counter ions adsorbed in a diffuse swarm adjacent to the mineral’s surface called a diffuse layer. The effective charge of the diffuse layer (σd) (and therefore its total electric potential) is of equal magnitude and opposite sign as the total particle surface charge (σp):
σp = σd
Thus, the net charge of the diffuse layer is directly proportional to the intrinsic and Stern-layer charges. The surface charge distribution model is useful for describing the amount of charge (and the associated surface electric potentials) associated with each type of adsorption because it allocates the surface charge into components that are based on adsorption mechanisms.
4. Outer-Sphere Adsorption
Cation exchange describes the adsorption and release of cations onto mineral surfaces based on the exchange of equivalent charges. Anion exchange is similar to cation exchange, and is an important reaction that determines the availability of anions in soils [47,48]; especially in soils where variable charged minerals dominate and soil pH is low [49]. Ion exchange is a surface interaction whereby the ions maintain their hydration sphere and are attracted to oppositely charged surfaces of soil particles (minerals or organic matter) via electrostatic forces. The adsorption mechanism is called outer-sphere complexation because the hydration sphere on the ion is maintained (Figure 1).
Many major soil cations react with soils through cation exchange reactions, including Na+, K+, Ca2+, or Mg2+. Soils in which cation exchange reactions predominate the adsorption processes are those that contain secondary clay minerals with permanent charge created by isomorphic substitution (e.g., moderately weathered soils). However, outer-sphere adsorption of ions on variable-charge mineral surfaces also occurs.
An example of a cation exchange reaction that occurs on soil clay minerals like montmorillonite is the exchange of Ca2+ ions adsorbed on a soil mineral surface by two Na+ cations in solution (Na-Ca exchange). The Na-Ca exchange reaction is written as
where CaX2 and NaX are the hydrated cations held onto the negatively charged surfaces (X) by electrostatic forces. The negatively charged surface is referred to as a double layer (or electric double layer) that includes the surface charge and a parallel (in theory) layer of adsorbed opposite charges. The double layer ion distribution and potential are modeled using the diffuse-double layer theory (DDL) [50], which includes electrostatic interactions and diffusion forces that create a distribution of ions in the solution on charged surfaces. A common mathematical solution to the DDL model is the Poisson–Boltzmann and Gouy–Chapman DDL model [50,51,52]. The DDL model provides a theoretical basis for predicting ion interactions with charged surfaces, as well as interactions of charged particles with each other. Applications of this model have been used to describe ion adsorption on soil particles [53,54] and to understand particle flocculation and dispersion in soils [55].
CaX2 + 2Na+ = 2NaX + Ca2+
A search for the keywords “diffuse double layer” and “soil” in Google Scholar reports 489 papers were published on this topic in 2019, showing the relevance and importance of this theory for understanding and predicting adsorption phenomena in soils, even after nearly a century of its first development. There are many modifications to the DDL model, and there is some debate on the applicability of DDL model equations for quantitative prediction in soil systems.
For modeling ion adsorption, the DDL model provides a basis for quantifying the electric potential at a charged surface [56]. However, many of the assumptions in the model are invalid in soil systems, and thus more descriptive surface adsorption models have been developed to better account for the physical distribution of adsorbed ions on particle surfaces, including models such as the Constant Capacitance model [57], the Triple Layer model [58], and the CD-MUSIC model [59,60]. These models include multiple modes of adsorption that are based on the mechanistic adsorption reactions illustrated in Figure 1. The models differentiate from each other in how they quantify the surface potential. Most of the surface charge adsorption models have been shown to be accurate in systems that are well defined, i.e., single mineral surfaces dispersed in homogeneous suspensions. However, applying theoretically-based adsorption models to adsorption reaction processes in soils and in field settings is limited by computational or physical limitations, and thus quantitative prediction is difficult [61]. Despite the limitations, surface complexation models are useful for studying how soil minerals in suspensions interact with ions, and results from surface complexation modeling studies allow for better predictions of adsorption behavior in nature. For example, Goldberg et al. [62,63] used soil pH and soil properties to predict constants for the Constant Capacitance model and accurately predicted the amount of borate adsorption in soils.
Quantitative prediction of cation exchange reactions has been researched since the early twentieth century. Models such as the Vanselow, Gaines–Thomas, and Gapon cation exchange model use classical equilibrium thermodynamic approaches to derive equilibrium exchange constants (exchange selectivity constants (Kex)) that predict cation partitioning between a solid and solution [8,64,65]. The models are used to predict soil processes; for example, the Gapon equation (KG) is used to derive the sodium adsorption ratio (SAR) equation and predicts how soil particle aggregation is affected by Na+, Mg2+, and Ca2+ concentrations in irrigation water [66].
While adsorption prediction models are accurate in some soils, the success of the model is not direct evidence of the ion adsorption mechanism. This requires corroboration using the measurement of either particle characteristics (such as electrophoretic mobility or proton adsorption and release) or, even better, spectroscopic and microscopic analyses that probes or images the adsorbed ions. However, molecular-level analysis of outer-sphere adsorbed ions is difficult because the ions maintain their hydration spheres when adsorbed onto the surface, and thus dehydrating the sample prior to analysis alters the adsorption mechanism. This prevents the use of a vacuum sample environment required for many spectroscopic methods, making detection of the adsorbed ion difficult unless a high-powered light source is used to increase signal to noise ratio from hydrated samples.
X-ray diffraction is one of the first tools used to measure the effects of different cations adsorbed in the interlayer on clay minerals [67,68]. The basal spacing (distance between basal planes, also called d-spacing) in the clay mineral changes when different ions adsorb in the interlayer. This is due to the effects of ion charge, size and hydration properties [69]. For example, adsorption of K+ cations in the interlayer of the clay mineral montmorillonite creates (001) d-spacings of 1.0 to 1.4 nm [70] (varying because of the temperature and dehydration treatment). In contrast, Na+ adsorption in the interlayer of montmorillonite causes d-spacing in the range of 1.2 to ~1.8 nm [71]. The cations K+, NH4+, and Cs+ have small or weak hydration spheres, and thus when they adsorb, they dehydrate, forming inner-sphere adsorption complexes with the ditrigonal oxygen atoms on the basal plane. This causes the interlayer of the clay minerals to collapse. Because the interlayer spacing varies as a function of the hydrated radius and charge of the adsorbed cations, XRD can be used to indirectly detect the hydration state of the adsorbed cations, which can be used to infer cation adsorption mechanisms (i.e., inner- our outer-sphere complexation).
Recent research used cryo-high resolution transmission electron microscopy (TEM) to monitor the hydration state of the montmorillonite interlayers as a function of interlayer ion composition [72,73]. By using high-resolution cryo-TEM together with small-angle X-ray spectroscopy, the interlayer distances of hydrated montmorillonite as a function of solution ion compositions was directly measured at a near-atomic-level resolution [72,73]. Imaging these clays with different adsorbed cations in a hydrated environment in such detail allows for direct observation of the effects of the adsorbed cations on the clay mineral structure and vice-versa. Specifically, cryo-TEM results advanced the current understanding of ion adsorption and clay hydration by showing that long-range electrostatic forces transfer across layers and are involved in the electrostatic adsorption of ions in the interlayers [73]. Whittaker et al. [72] observed that nano-scale regions in the interlayer have distinct hydration properties, depending on the type of cation adsorbed, and create an overall heterogeneous adsorption process on the clay minerals. These results improve our understanding of the swelling and structure of clays, and the number of water molecules in the interlayers as a function of both the adsorbed ions and the magnitude and location of the permanent charge on the mineral surfaces.
The electric potential and morphology of adsorbed chemicals on mineral surfaces have been measured using atomic force microscopy (AFM) [74,75,76]. AFM uses a sub-nanometer-sized tip that changes height as the electric potential between the tip and the mineral surface changes (Figure 3). The change in the height of the tip as it moves across the surface is related to the surface morphology. Zhai et al. [77] studied DNA adsorption on mica mineral surfaces by attaching a DNA fragment to an AFM tip. They concluded that the DNA adsorbs on the surface as an outer-sphere complex. Martin-Jimenez and Garcia [78] studied the position of adsorbed potassium ions on hydrated mica surfaces and made direct observations showing that the K+ cations adsorb in the Stern layer near the position of the maximum layer charge; confirming the concept of inner-sphere adsorbed K+ ions on tetrahedrally charged clay mineral surfaces. Siretanu et al. [75] measured Ca2+, Mg2+, Na+, and K+ adsorption on the basal plane of gibbsite nanoparticles using dynamic force spectroscopy to determine the distribution of the diffuse-layer charge and high-resolution AFM imaging to resolve the distribution of adsorbed divalent cations. Although theoretical models predict that there should be no surface charge or adsorption on the basal planes of gibbsite, AFM microscopy showed that the Ca2+ and Mg2+ cations adsorb as outer-sphere complexes on the surface, while Na+ and K+ do not adsorb on the gibbsite basal plane surface, indicating a strong preference of the surface for the divalent cations [75]. Araki et al. [79] used AFM to show that Cs+ cations, like K+, adsorb as inner-sphere complexes on montmorillonite surfaces, supporting the theory that the ion’s hydration sphere strength is an important characteristic in determining adsorption mechanisms.
Molecular simulations have been used to calculate mineral interfacial forces and potentials on hydrated mineral surfaces with adsorbed ions [81,82,83,84,85,86,87,88]. Approaches vary from using molecular dynamics to density function theory and Monte Carlo simulations. Ho et al. [89] modeled Na+, Ca2+, Ba2+, and Cl− adsorption on different surfaces of the aluminum oxide gibbsite and predicted that the adsorption mode is dependent on the mineral’s reactive surface. The model predicts that ions adsorb as outer-sphere complexes on some surfaces, while on others, they adsorb via inner-sphere complexes. For example, on the gibbsite (100) edge, Ca2+ ions adsorb as inner-sphere and outer-sphere complexes, whereas on the (001) surface, outer-sphere Ca2+ ions are the dominant species. The molecular simulations of Ca2+ adsorption on gibbsite agree with the adsorption mechanisms determined using AFM on the (001) plane [75]. Willemsen et al. [90] conducted molecular dynamic simulations of the hydrophobic organic molecule phthalate adsorption on smectites and predicted that the adsorption occurs both on exposed basal planes of the clay platelets, and in the interlayer. In the interlayer, the phthalate forms a bridge between adjacent clay surfaces and is attracted to the regions of the clay with the least amount of charge; the simulation results agreed with the d-spacing measured using XRD. Kobayashi et al. [91] used molecular dynamic simulations to show that alkali and alkaline earth metals adsorb on montmorillonite via inner-sphere complexes on two types of sites (one over a ditrigonal cavity and the other over isomorphically substituted Al), and also as outer-sphere complexes electrostatically attracted to the layer charge (Figure 4). The difference in the cation adsorption mode is attributed to the varying amount of electrostatic surface charge and the cation’s hydration energy.
The development of models that use first principles to understand adsorption behavior has been advancing both prediction capabilities and fundamental understanding of adsorption phenomena. The complexity of the models is also increasing; for example, Rahromostaqim and Sahimi [92] developed a molecular simulation model of a montmorillonite-chlorite mixed-layer mineral phase and predicted the effects of hydration, layer properties, and cation properties on adsorption. In another study, Zhang et al. [93] simulated clay mineral interactions with natural organic matter through adsorption reactions. This is an important reaction in soils where clays and SOM colloids are physically and chemically associated with each other. The molecular simulations of Zhang et al. [93] predicted that carboxylate functional groups of natural organic matter adsorb onto montmorillonite surfaces through direct bonding of carboxylate to clay mineral edges and through Ca2+ bridging on basal plane surfaces. The prediction of Ca2+ bridging agrees with the observations made using spectroscopic analysis of SOM-soil clay interactions by Sowers et al. [94]. As computing capability and molecular models advance, predicting soil adsorption behavior from first principles will be possible if the input model is sufficiently constrained.
XAFS spectroscopy and Fourier transform infrared (FTIR) spectroscopy are molecular investigation tools capable of probing the molecular structure of ions adsorbed on soils and minerals in a hydrated state. XAFS is advantageous because it uses X-rays tuned to specific energies that isolate the signals emitted from other elements in the sample, thus allowing for the probing of adsorbed phases on the soils, which are typically less than 1% of the sample mass. Papelis and Hayes [95] used XAFS spectroscopy to analyze Co2+ adsorption on the clay mineral montmorillonite. Strawn and Sparks [43] conducted similar experiments to investigate Pb2+ adsorption on montmorillonite. Most of the adsorption capacity of montmorillonite is on the interlayers and is due to the permanent charge created by isomorphic substitution in the octahedral sheet. However, on the mineral edges, partially coordinated oxygen ligands are available to form inner-sphere complexes with metals. Both Papelis and Hayes [95] and Strawn and Sparks [43] observed that under low background electrolyte concentrations of Na+ or Ca2+, the metal cations Co2+ and Pb2+ adsorb in the interlayers as hydrated ions (i.e., outer-sphere complexes), and adsorption is not pH-dependent because the interlayer adsorption sites have permanent charge. But, at high concentrations of background electrolytes, Co2+ and Pb2+, do not adsorb in the interlayers, but adsorb onto the edges of the clay minerals via inner-sphere adsorption complexes, and adsorption is pH-dependent. In another study, Vasconcelos et al. [96] used XAFS spectroscopic analysis to show that Cd2+ adsorption to kaolinite samples at pH 7 occurs by an outer-sphere complex with a single hydration sphere around the Cd2+ ion, and that it likely adsorbs to permanently charged basal plane sites created from the small amounts of isomorphic substitution in natural kaolinites [97]. At a higher pH (pH 9), Vasconcelos et al. [96] observed that the Cd2+ adsorbs to aluminol or silanol ligands on the kaolinite edges via inner-sphere complexation.
Recently, Wang et al. [98] combined XAFS spectroscopy, FTIR spectroscopy, and adsorption modeling to investigate SO42− adsorption mechanisms on hematite particles. Previous observations indicate that SO42− adsorption on hematite occurs as mostly inner-sphere complexes (see references in Wang et al. [98]), similar to PO43− adsorption [21]. However, using advanced spectroscopic analysis and surface complexation models, Wang et al. [98] observed that SO42- adsorbs on hematite as both inner-sphere and outer-sphere complexes (Figure 5). In fact, the outer-sphere complexes are more prevalent than the inner-sphere complexes under most conditions.
Xu et al. [99] used surface-sensitive spectroscopic methods (X-ray scattering) to measure AsO43- adsorption mechanisms on uncharged aluminum oxide (corundum) (001) surfaces. Arsenate adsorbs to most minerals through inner-sphere complexation on surface functional groups. However, on the corundum surface, Xu et al. [99] observed that AsO43- adsorbs as outer-sphere complexes. Electrostatic attraction, which is commonly considered to be the driving force for outer-sphere adsorption, is not a factor in the AsO43- adsorption on corundum (001) surfaces because they do not have surface charge. Instead, the molecular spectroscopic analysis showed that hydrogen-bonding between water molecules and AsO43- is the driving force for the outer-sphere adsorption on the corundum (001) surface. Xu et al. [99] determined that at high surface coverages, up to 40% of the AsO43- adsorption on aluminum oxides occurs as outer-sphere complexes. These molecular-scale observations provide mechanistic evidence that water-molecule dynamics are important factors in adsorption processes.
5. Inner-Sphere Adsorption
Early studies on adsorption in soils were focused mostly on cation adsorption on negatively charged clay minerals, and proposed that the cations are adsorbed as outer-sphere complexes attracted to the surfaces through electrostatic forces. Several early studies, however, recognized the ability of some soils to adsorb anions such as PO43− [100,101], and that the retention could be strong; but the adsorption mechanisms were not known (Sumner [102] provides a review of the early literature on anion exchange). It was later confirmed that anions and cations adsorb to varable charged fucnitonal groups on mineral edges. In 1952 Leeper [103] published a review of adsorption of metal cations in soils and used the term “X-factor” to describe metal adsorption to functional groups on soil minerals and SOM. Leeper [103] surmised that the metal cations complex to soils via chemical bonds in a similar way that metals bond with aqueous chelates, and that this was the reason that many metals have lower plant availability than other nutrient cations like Ca2+ and Mg2+ that adsorb as outer-sphere complexes. Additional support for the importance of surface functional groups came from research published in the 1960s by Sumner [104], who showed that in soils dominated by oxides and kaolinite clays, the mineral surfaces have variable charge, firmly establishing the concept that edges of variable charged minerals in soils are important for adsorption.
In 1967, Hingston et al. [105] monitored anion adsorption on goethite and the effects on solution pH and electrophoretic mobility. The anions included AsO43−, F−, and PO43−. They proposed that some anions adsorb by “specific adsorption” on mineral edges by entering the Helmholtz layer and forming direct bonds with the structural cations. The term specific adsorption refers to the formation of molecular bonds (inner-sphere complexes) between the adsorbed ion and the structural surface ions. In anion adsorption, the coordinating oxygen ligands surround structural Fe, Al, or Si cations at mineral edges. Hingston et al. [105] further noted that adsorption is pH-dependent.
It is now known that many ions adsorb on mineral surfaces through inner-sphere complexes that have covalent and ionic bond characteristics. The inner-sphere complexation is dependent on the ionic potential of the adsorbed chemical and the configuration of its outer-shell electrons (principally their electronegativity). There is no water between a chemical adsorbed as an inner-sphere complex and the surface functional groups, as is the case for outer-sphere adsorption. The surface functional groups occur on the edges of minerals or SOM (Table 2). Smaller particles have a greater specific surface area and more functional groups exposed to the soil solution because the edge to interior ratio increases as particles get smaller. Thus, the capacity of soils to adsorb ions as inner-sphere complexes on small colloids in soils is large.
Early reports of ions adsorbing as inner-sphere complexes by Hingston et al. [105], Stumm et al. [106], Huang and Stumm [107], and Schindler et al. [108] were based on the observations of changes in pH and particle surface charge measurements using electrophoretic mobility. Many anions and cations displace protons or hydroxides upon adsorption, which may change the particle surface charge. The early research findings provided evidence that oxide surfaces have reactive functional groups that behave as weak acids and bases, and that many ions adsorb to surface functional groups by inner-sphere complexation.
Since the 1970s, several researchers have studied pH-dependent adsorption of cations (e.g., Figure 6, [109,110] and references therein) and anions [111,112,113,114]. Surface complexation models were developed that include mechanistic adsorption reactions as either outer-sphere or inner-sphere complexation [58]. Modern surface complexation models include sorbate-specific adsorption mechanisms (e.g., bidentate or monodentate adsorption) that have been verified using spectroscopic analysis [115,116].
Spectroscopic techinques such as XAFS and FTIR are the most widely used methods for investigating ion adsorption mechanisms on soil and soil minerals. The spectroscopies probe the local molecular environment surrounding the adsorbed chemicals, thus resolving the molecular structure. In 1987, Hayes et al. [118] were amongst the first to use XAFS to study mechanisms of ion adsorption onto mineral surfaces. They studied selenate and selenite adsorption on goethite and observed that selenate adsorbs as an outer-sphere complex and selenite adsorbs as an inner-sphere complex [118]. It was later shown by Manceau and Charlet [119] and Peak and Sparks [120] using XAFS and FTIR spectroscopies that the adsorption mechanisms of selenate are dependent on the surface and solution conditions [119,120] and may be either inner- or outer-sphere complexes.
In recent decades, the development of brighter X-ray sources has allowed for more detailed analyses of ions adsorbed on surfaces. Schlegel et al. [121] and Furnare et al. [122] analyzed oriented clay minerals using polarized beams of the synchrotron X-ray to study the adsorption mechanisms of Co2+ on hectorite (a smectite) and Cu2+ on vermiculite, respectively. By rotating the crystallites through the polarized X-rays, they were able to measure the angular dependence of the bond orientation of Co2+ and Cu2+ adsorbed onto the clay minerals, allowing them to elucidate that the metals adsorb as inner-sphere complexes on the clay mineral edges, and as outer-sphere complexes on the interlayers.
Peacock and Sherman [123] used XAFS spectroscopy to probe the adsorption mechanisms of Cu2+ on goethite and made direct observations of inner-sphere complexes to Fe-O surface functional groups, and developed a surface complexation model that predicts the pH-dependent adsorption behavior. More recently, Tiberg et al. [124] investigated AsO43− adsorption in a mixed-mineral system consisting of poorly crystalline aluminum hydroxide and ferrihydrite. They also included competitive adsorption with PO43- in their study. XAFS spectroscopic analysis showed that the AsO43- adsorbs as inner-sphere complexes on the mineral surfaces (in agreement with Arai et al. [21] and Manceau [22]), and that the PO43− competes more effectively with AsO43- in a mixed-mineral system than in a single iron oxide system [124].
As discussed above, molecular simulation modeling has also successfully modeled inner-sphere complexation. Kubicki and Ohno [88] used density functional theory modeling to predict that salicylate in solution displaces PO43− adsorbed on goethite surfaces, causing PO43− release. These results provide an important understanding of the molecular processes by which plants increase PO43− availability by releasing low molecular weight organic acids into the rhizosphere.
Adsorption of most metals and oxyanions has been studied using spectroscopic analysis, and extensive knowledge exists on types of ions and surfaces in which adsorption occurs by inner-sphere complexation. As discussed above, the adsorption mode is dependent on the properties of the mineral surface and the solution conditions. Many of the transition metals adsorb as inner-sphere complexes on variable charged surfaces, including Ni2+, Cu2+, Zn2+, Co2+, and Hg2+. The oxyanions PO43−, SeO32−, and AsO43− and the environmentally prevalent metal Pb2+ also adsorb as inner-sphere complexes on variable charged surfaces. Because inner-sphere complexes involve covalent bonding, adsorption of these ions is strong and is thus an important factor for controlling chemical availability for plant uptake and leaching of chemicals out of the soil into ground or surface waters [125].
6. Multi-Nuclear Precipitation on Mineral Surfaces
Sorption of ions can often include precipitation reactions, whereby the solid phases precipitate on soil mineral surfaces [11,126] and are distinct from bulk precipitates because they have different crystallinities, compositions, morphologies, and solubilities. Surface precipitation reactions may be a homogenous solid on the mineral surface or include a solid mixture that consists of either a coprecipitate or a multi-nuclear precipitate incorporating ions from the solid surface on which the precipitate is forming. Soil minerals can catalyze the formation of solids on their surfaces, which is termed heterogeneous nucleation. When ions from the soil mineral surface are incorporated into the new solid forming on the surface, the product is a surface precipitate [24]. Precipitates on mineral surfaces are neoformed solid phases. Their formation is an important process that removes metals from the soil solution and thus limits metal mobility.
In 1985, Farley et al. [127] proposed a sorption model for metals on oxide mineral surfaces that include a solid-solution composition continuously varying between that of the original mineral solid and a pure precipitate of the sorbing cation. In 1987, Comans and Middelburg [128] used a similar model to predict solid-solution precipitates of metals (Zn2+, Co2+, Cd2+, and Mn2+) on calcite surfaces. The studies of Farley et al. [127] and Comans and Middelburg [128] relied on fitting mechanistic models to sorption data and do not make direct observations of the sorbate speciation. Sposito [11] summarizes the requisite approach for determining sorption mechanisms as follows:
“Molecular concepts can be studied only by molecular methods.”
One of the most useful techniques for measuring the speciation of ions sorbed on soil minerals by surface precipitation is spectroscopic analysis that uses wavelengths small enough to probe the molecular structure of chemicals sorbed on mineral surfaces. In 1985, Bleam and McBride [129] provided direct evidence for surface precipitation as a sorption reaction using electron spin resonance spectroscopy (ESR) to observe that Mn(II) adsorbs on aluminum and iron oxides as “surface clusters”. In 1990, Chisholmbrause et al. [130] used XAFS to investigate the mechanisms of Co2+ sorption on three different solids (aluminum oxide, titanium oxide, and kaolinite), and observed direct evidence of the presence of multi-nuclear sorption complexes at surface coverages below one monolayer of Co2+ atoms. Charlet and Manceau [131] used XAFS spectroscopy to probe the molecular structure of Cr(III) adsorbed on iron oxide surfaces (hydrous ferric oxide), and discovered that the Cr(III) sorbs as multi-nuclear complexes on the mineral’s surface. Using XAFS spectroscopy, Waychunas et al. [25] observed that AsO43− adsorbs on iron oxide surfaces as a coprecipitate. In 1994, Charlet and Manceau [132] measured Ni2+ and Co2+ sorbed on clay minerals using XAFS spectroscopy and observed that they adsorb as surface clusters with a clay-like local structure, suggesting precipitation of new hydrous silicate solids on the clay mineral surfaces.
Layered double hydroxides (LDHs) are a class of precipitates that have been extensively studied because they are important surface precipitates that occur in many environments. The structure of the LDH minerals is two metals incorporated into a hydroxide sheet, as in the Al hydroxide sheet of gibbsite, but one of the metals is divalent, and the overall layer becomes positively charged because of excess Al substitution. The positive charge is balanced by anions in the interlayer space between the hydroxide sheets. The LDH minerals form on mineral surfaces as surface precipitates (Figure 7). In 1995, Scheidegger and Sparks [133] were amongst the first to show that LDHs are the species of surface precipitate on clay mineral surfaces. It has been shown that LDH phases are common for metals such as Ni, Zn, and Co on surfaces of Al-rich minerals such as clay minerals (e.g., montmorillonite or kaolinite) [24,134]. Siebecker et al. [24] provides an excellent review of LDH phases and their formation and reaction properties.
Surface precipitation is an important mechanism controlling solubility of inorganic ions in soils. The solubility products of solids precipitated on minerals surfaces may be lower than those precipitated in bulk solution [24,135,136], and thus they can form under conditions when the bulk solutions are unsaturated with respect to homogenous solids.
Distinguishing between two-dimensional surface adsorption or three-dimensional precipitates on mineral surfaces is complex, particularly in soils that have multiple types of surfaces present and where sorption is heterogeneous throughout the soil. Using advanced spectroscopic tools is providing new insights into the different sorption mechanisms and can identify whether the sorption process is adsorption or surface precipitation.
7. Ternary Surface Complexes
A ternary surface complex consists of the adsorption of two interacting chemicals on a mineral or organic matter surface (Figure 8). Ternary (i.e., composed of three parts) refers to the two adsorbing chemicals and the soil particle adsorption site. Although more complex than binary adsorption systems often studied, adsorption via ternary complexation is important because in soils there are many different constituents that interact [137]. A ternary complex often involves the adsorption of anions and cations. The anions may be singular ions such as Cl−, oxyanions such as PO43− or SO42−, or dissolved organic compounds with negatively charged functional groups (including both natural and anthropogenic organic compounds). Ternary complexes are bonded to surfaces through either the cation or the anion, where the non-surface bound ion is held as a bridged ion (it is bridged to the surface through the adsorbed ion).
Mendez and Hiemstra [139] used mechanistic adsorption model fitting (CD-MUSIC) to predict that Mg2+, Ca2+, and PO43− ions in solution adsorb onto ferrihydrite surfaces via ternary complexes. Their model predicts more adsorption of ions as ternary complexes on ferrihydrite compared to systems with only the individual ions present. The type of complexes formed in the cation-PO43−-ferrihydrite systems are anion-bridged ternary complexes, e.g., ≡Fe-O-PO3-Ca (where the italics indicate the structural mineral ions). Tiberg and Gustafsson [140] used XAFS spectroscopy to show that Cd2+ and PO43− form ternary complexes on ferrihydrite surfaces and that the complex enhances Cd2+ adsorption.
Low molecular weight organic compounds are common anions that form ternary complexes with metals in the environment [138]. Flynn and Catalano [138] measured the molecular coordination of Ni2+ sorbed to iron oxides in the presence of oxalate (Figure 8). They used XAFS and FTIR spectroscopy to investigate the mechanisms of adsorption and observed Ni-oxalate ternary complexes on the iron oxide surfaces. However, in the presence of the oxalate less Ni2+ adsorbs. Strathmann and Myneni [141] studied Ni2+ adsorption on aluminum oxide mineral surfaces in the presence of fulvic acid using XAFS spectroscopy. They observed inner-sphere adsorption of the Ni2+ and also Ni2+-fulvic acid ternary complexation with the aluminol functional groups.
8. Conclusions
The fate of chemicals in soils is controlled by sorption reactions on surfaces that include adsorption as outer- and inner-sphere complexes or surface precipitation on mineral surfaces. For nearly two-hundred years, scientists have been pursuing knowledge of how chemicals interact with soil particles, spurred on by advancements of technology that allow for more detailed information on sorption processes to be gleaned from samples. Advancements in knowledge have been made from studying relatively pure, two-component systems consisting of only one type of solid, such as a clay mineral or oxide mineral, and the sorbing chemical of interests. Understanding the reaction mechanisms in these simplified systems allows for a detailed understanding of the reaction process and provides a theoretical basis for how the chemicals behave in the more complex soil environments. However, given the inherent complexity of soils, studying the mechanistic sorption processes in whole soils is difficult. But new advancements in spectroscopic, microscopic, and computing tools are allowing for new insights into the molecular processes in soils. With these tools, investigation of adsorption mechanisms in more complex systems can be undertaken to discover the adsorption mechanisms occurring in natural systems. Future research should also investigate the molecular interactions that occur in dynamic systems that include microbes and plant roots because the biological processes create varying gradients in chemical energy that cause changes in sorption mechanisms that need to be understood.
The technology and knowledge to achieve the goal of using soil physicochemical properties and chemical properties to a-priori quantitatively predict how a particular nutrient or contaminant will behave in the soil environment are not far off—in some cases, it is already possible. Armed with the knowledge of sorption mechanisms for a particular chemical, researchers are developing computer models that use approaches such as neural network analysis, deep learning, and artificial intelligence decision making to accurately predict chemical availability, fate, and transport, and how the ecosystem will respond to the chemical. Land and resource managers will be able to use this information to make management decisions to increase productivity and preserve ecosystem health. Thus, understanding detailed sorption mechanisms that occur in soils is key to developing effective resource management strategies.
Funding
This research is supported by the University of Idaho Agricultural Experiment Station USDA Hatch project (Mutli-state NC1187: Particulate Reactivity and Cycling in a Changing Environment: Implications for Agriculture and Human Health).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lavoisier, A. Elements of Chemistry in New Systematic Order, Containing All Modern Discoveries, Illustrated with 13 Copperplates, Translated from the French by Robert Kerr, 1st ed.; William Creech: Edinburgh, UK, 1790; Volume 1, p. 511. [Google Scholar]
- Sparks, D.L. The origins of agricultural chemistry: The forerunner of soil chemistry. In Footprints in Soil; Warkentin, B., Ed.; Elsevier Science: Amsterdam, The Netherlands, 2006; p. 572. [Google Scholar]
- Van der Ploeg, R.R.; Böhm, W.; Kirkham, M.B. On the origin of the theory of mineral nutrition of plants and the law of the minimum. Soil Sci. Soc. Am. J. 1999, 63, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Everett, D.H. Manual of symbols and terminology for physicochemical quantities and units, appendix II: Definitions, terminology and symbols in colloid and surface chemistry. Pure Appl. Chem. 1972, 31, 577–638. [Google Scholar] [CrossRef]
- Way, J.T. On the power of soils to absorb manure. J. R. Agric. Soc. Engl. 1850, 11, 313. [Google Scholar]
- Schuffelen, A.C. A few aspects of 50 years of soil chemistry. Geoderma 1974, 12, 281–297. [Google Scholar] [CrossRef]
- Thomas, G.W. Historical developments in soil chemistry—Ion-exchange. Soil Sci. Soc. Am. J. 1977, 41, 230–238. [Google Scholar] [CrossRef]
- Sposito, G. Cation exchange in soils: A historical and theoretical perspective. In Chemistry in the Soil Environment; American Society of Agronomy: Madison, WI, USA, 1981; pp. 13–30. [Google Scholar]
- Hendricks, S.B.; Fry, W.H. The results of X-ray and microscopical examinations of soil colloids. Soil Sci. 1930, 29, 457–479. [Google Scholar] [CrossRef]
- Kelley, W.P.; Dore, W.H.; Brown, S.M. The nature of the base-exchange material of bentonite, soils, and zeolites, as revealed by chemical investigation and X-ray analysis. Soil Sci. 1931, 31, 25–55. [Google Scholar] [CrossRef]
- Sposito, G. Distinguishing adsorption from surface precipitation. In Geochemical Processes at Mineral Surfaces; American Chemical Society: Washington, DC, USA, 1987; Volume 323, pp. 217–228. [Google Scholar]
- Veith, J.A.; Sposito, G. On the use of the Langmuir equation in the interpretation of “adsorption” phenomena. Soil Sci. Soc. Am. J. 1977, 41, 697–702. [Google Scholar] [CrossRef]
- Gebhardt, H.; Coleman, N.T. Anion adsorption by allophanic tropical soils: I. chloride adsorption. Soil Sci. Soc. Am. J. 1974, 38, 255–259. [Google Scholar] [CrossRef]
- Wang, P.G.; Ji, G.L.; Yu, T.R. Adsorption of chloride and nitrate by variable charge soils in relation to the electric charge of the soil. Z. Pflanz. Bodenkd. 1987, 150, 17–23. [Google Scholar] [CrossRef]
- Sokolova, T.A.; Alekseeva, S.A. Adsorption of sulfate ions by soils (A review). Eurasian Soil Sci. 2008, 41, 140–148. [Google Scholar] [CrossRef]
- Goldberg, S. Modeling selenite adsorption envelopes on oxides, clay minerals, and soils using the triple layer model. Soil Sci. Soc. Am. J. 2013, 77, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S. Competitive adsorption of arsenate and arsenite on oxides and clay minerals. Soil Sci. Soc. Am. J. 2002, 66, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Bradl, H.B. Adsorption of heavy metal ions on soils and soils constituents. J. Colloid Interface Sci. 2004, 277. [Google Scholar] [CrossRef]
- Peng, L.; Liu, P.; Feng, X.; Wang, Z.; Cheng, T.; Liang, Y.; Lin, Z.; Shi, Z. Kinetics of heavy metal adsorption and desorption in soil: Developing a unified model based on chemical speciation. Geochim. Cosmochim. Acta 2018, 224, 282–300. [Google Scholar] [CrossRef]
- Arai, Y.; Sparks, D.L. Phosphate reaction dynamics in soils and soil components: A multiscale approach. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 2007; Volume 94, pp. 135–179. [Google Scholar]
- Arai, Y.; Sparks, D.L.; Davis, J.A. Arsenate adsorption mechanisms at the allophane-water interface. Environ. Sci. Technol. 2005, 39, 2537–2544. [Google Scholar] [CrossRef]
- Manceau, A. The mechanism of anion adsorption on iron oxides: Evidence for the bonding of arsenate tetrahedra on free Fe(O, OH)6 edges. Geochim. Cosmochim. Acta 1995, 59, 3647–3653. [Google Scholar] [CrossRef]
- Sawhney, B.L. Selective sorption and fixation of cations by clay minerals: A review. Clays Clay Miner. 1972, 20, 93–100. [Google Scholar] [CrossRef]
- Siebecker, M.G.; Li, W.; Sparks, D.L. The important role of layered double hydroxides in soil chemical processes and remediation: What we have learned over the past 20 years. In Advances in Agronomy; Elsevier: Amsterdam, The Netherlands; Academic Press Inc: San Diego, CA, USA, 2018; Volume 147, pp. 1–59. [Google Scholar]
- Waychunas, G.A.; Rea, B.A.; Fuller, C.C.; Davis, J.A. Surface-chemistry of ferrihydrite.1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim. Cosmochim. Acta 1993, 57, 2251–2269. [Google Scholar] [CrossRef]
- Voice, T.C.; Weber, W.J. Sorption of hydrophobic compounds by sediments, soils and suspended solids—I. Theory and background. Water Res. 1983, 17, 1433–1441. [Google Scholar] [CrossRef] [Green Version]
- Kravchenko, A.N.; Guber, A.K. Soil pores and their contributions to soil carbon processes. Geoderma 2017, 287, 31–39. [Google Scholar] [CrossRef]
- Strawn, D.; Bohn, H.L.; O’Connor, G.A. Soil Chemistry, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2020. [Google Scholar]
- Allison, L.E.; Al, E. Diagnosis and Improvement of Saline and Alkali Soils; USDA, United States Salinity Laboratory: Riverside, CA, USA, 1954; Volume 60.
- Parfitt, R.L. Phosphate adsorption on an oxisol. Soil Sci. Soc. Am. J. 1977, 41, 1064–1067. [Google Scholar] [CrossRef]
- Tournassat, C.; Davis, J.A.; Chiaberge, C.; Grangeon, S.; Bourg, I.C. Modeling the acid-base properties of montmorillonite edge surfaces. Environ. Sci. Technol. 2016, 50, 13436–13445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickmore, B.R.; Tadanier, C.J.; Rosso, K.M.; Monn, W.D.; Eggett, D.L. Bond-valence methods for pKa prediction: Critical reanalysis and a new approach. Geochim. Cosmochim. Acta 2004, 68, 2025–2042. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H.; Bolt, G.H. Multisite proton adsorption modeling at the solid/solution interface of (hydr)oxides: A new approach: I. model description and evaluation of intrinsic reaction constants. J. Colloid Interface Sci. 1989, 133, 91–104. [Google Scholar] [CrossRef]
- Zachara, J.; Brantley, S.; Chorover, J.; Ewing, R.; Kerisit, S.; Liu, C.; Perfect, E.; Rother, G.; Stack, A.G. Internal domains of natural porous media revealed: Critical locations for transport, storage, and chemical reaction. Environ. Sci. Technol. 2016, 50, 2811–2829. [Google Scholar] [CrossRef] [PubMed]
- Posner, A.M. Humic acids extracted by various reagents from a soil yield inorganic components and titration curves. J. Soil Sci. 1966, 17, 65–78. [Google Scholar] [CrossRef]
- Lees, H. A note on the copper-retaining power of a humic acid from peat soil. Biochem. J. 1950, 46, 450–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, F.J. Humus Chemistry: Genesis, Composition, Reactions; Wiley: New York, NY, USA, 1982; p. 443. [Google Scholar]
- Xia, K.; Skyllberg, U.L.; Bleam, W.F.; Bloom, P.R.; Nater, E.A.; Helmke, P.A. X-ray absorption spectroscopic evidence for the complexation of Hg (II) by reduced sulfur in soil humic substances. Environ. Sci. Technol. 1999, 33, 257–261. [Google Scholar] [CrossRef]
- Sposito, G. Characterization of particle surface charge. In Environmental Particles; Buffle, J., van Leeuwen, H.P., Eds.; Lewis Publishers: Boca Raton, FL, USA, 1992; Volume 1, p. 291. [Google Scholar]
- Kleber, M.; Lehmann, J. Humic substances extracted by alkali are invalid proxies for the dynamics and functions of organic matter in terrestrial and aquatic ecosystems. J. Environ. Qual. 2019, 48, 207–216. [Google Scholar] [CrossRef]
- Helling, C.S.; Chesters, G.; Corey, R.B. Contribution of organic matter and clay to soil cation-exchange capacity as affected by the pH of the saturating solution. Soil Sci. Soc. Am. J. 1964, 28, 517–520. [Google Scholar] [CrossRef]
- Strawn, D.G.; Baker, L.L. Speciation of Cu in a contaminated agricultural soil measured by XAFS, μ-XAFS, and μ-XRF. Environ. Sci. Technol. 2008, 42, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Strawn, D.G.; Sparks, D.L. The use of XAFS to distinguish between inner- and outer-sphere lead adsorption complexes on montmorillonite. J. Colloid Interface Sci. 1999, 216, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manceau, A.; Boisset, M.-C.; Sarret, G.; Hazemann, J.-L.; Mench, M.; Cambier, P.; Prost, R. Direct determination of lead speciation in contaminated soils by EXAFS spectroscopy. Environ. Sci. Technol. 1996, 30, 1540–1552. [Google Scholar] [CrossRef]
- Shi, Z.; Peltier, E.; Sparks, D.L. Kinetics of Ni sorption in soils: Roles of soil organic matter and Ni precipitation. Environ. Sci. Technol. 2012, 46, 2212–2219. [Google Scholar] [CrossRef]
- Anderson, S.J.; Sposito, G. Proton surface-charge density in soils with structural and pH-dependent charge. Soil Sci. Soc. Am. J. 1992, 56, 1437–1443. [Google Scholar] [CrossRef]
- Sparks, D.L. Environmental Soil Chemistry, 2nd ed.; Academic Press: Amsterdam, The Netherlands; Boston, MA, USA, 2003; p. 352. [Google Scholar]
- Zelazny, L.W.; He, L.; Vanwormhoudt, A. Charge analysis of soils and anion exchange. In Methods of Soil Analysis; Soil Science Society of America: Madison, WI, USA, 1996; pp. 1231–1253. [Google Scholar]
- Eick, M.J.; Brady, W.D.; Lynch, C.K. Charge properties and nitrate adsorption of some acid southeastern soils. J. Environ. Qual. 1999, 28, 138–144. [Google Scholar] [CrossRef]
- Singh, U.; Uehara, G. Electrochemistry of the double layer: Principles and applications to soils. In Soil Physical Chemistry, 2nd ed.; Sparks, D.L., Ed.; CRC Press: New York, NY, USA, 1988. [Google Scholar]
- Quirk, J.P. Interparticle forces: A basis for the interpretation of soil physical behavior. In Advances in Agronomy; Sparks, D.L., Ed.; Academic Press: Cambridge, MA, USA, 1994; Volume 53, pp. 121–183. [Google Scholar]
- Bolt, G.H. Chapter 1: The ionic distribution in the diffuse double layer. In Developments in Soil Science; Bolt, G.H., Ed.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 5, pp. 1–26. [Google Scholar]
- Bolt, G.H.; van Riemsdijk, W.H. Chapter 13: Ion adsorption on inorganic variable charge constituents. In Developments in Soil Science; Bolt, G.H., Ed.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 5, pp. 459–504. [Google Scholar]
- Barak, P. Double layer theory prediction of Al-Ca exchange on clay and soil. J. Colloid Interface Sci. 1989, 133, 479–490. [Google Scholar] [CrossRef]
- Quirk, J.P. Particle interaction and soil swelling. Isr. J. Chem. 1968, 6, 213–234. [Google Scholar] [CrossRef]
- Westall, J. Chemical equilibrium including adsorption on charged surfaces. In Particulates in Water; American Chemical Society: Washington, DC, USA, 1980; Volume 189, pp. 33–44. [Google Scholar]
- Goldberg, S. Constant Capacitance Model. In Emerging Technologies in Hazardous Waste Management III; American Chemical Society: Washington, DC, USA, 1993; Volume 518, pp. 278–307. [Google Scholar]
- Goldberg, S. Use of surface complexation models in soil chemical systems. In Advances in Agronomy; Sparks, D.L., Ed.; Soil Science Society of America: Madison, WI, USA, 1992; Volume 47, pp. 233–330. [Google Scholar]
- Goldberg, S.; Criscenti, L. Modeling adsorption of metals and metalloids by soil components. In Biophysico-Chemical Processes of Heavy Metals and Metalloids in Soil Environments; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2007; pp. 215–264. [Google Scholar]
- Hiemstra, T.; van Riemsdijk, W.H. A surface structural approach to ion adsorption: The charge distribution (CD) model. J. Colloid Interface Sci. 1996, 179, 488–508. [Google Scholar] [CrossRef]
- Gustafsson, J.P. Modelling competitive anion adsorption on oxide minerals and an allophane-containing soil. Eur. J. Soil Sci. 2001, 52, 639–653. [Google Scholar] [CrossRef]
- Goldberg, S.; Corwin, D.L.; Shouse, P.J.; Suarez, D.L. Prediction of boron adsorption by field samples of diverse textures. Soil Sci. Soc. Am. J. 2005, 69, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, S.; Lesch, S.M.; Suarez, D.L. Predicting boron adsorption by soils using soil chemical parameters in the constant capacitance model. Soil Sci. Soc. Am. J. 2000, 64, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Kelley, W.P. Cation Exchange in Soils; Reinhold Publishing Corporation: New York, NY, USA; Chapman & Hall: London, UK, 1948; p. 144. [Google Scholar]
- Kelley, W.P. Review of investigations on cation exchange and semiarid soils. Soil Sci. 1964, 97, 80–88. [Google Scholar] [CrossRef]
- Richards, L.A. Diagnosis and Improvement of Saline and Alkali Soils; US Department of Agriculture: Washington, DC, USA, 1954; p. 166.
- Norrish, K. The swelling of montmorillonite. Discuss. Faraday Soc. 1954, 18, 120–134. [Google Scholar] [CrossRef]
- Norrish, K.; Quirk, J.P. Crystalline swelling of montmorillonite—Use of electrolytes to control swelling. Nature 1954, 173, 255–256. [Google Scholar] [CrossRef]
- Moore, D.M.; Reynolds, R.C. X-ray Diffraction and the Identification and Analysis of Clay Minerals; Oxford University Press: Oxford, UK; New York, NY, USA, 1989; pp. 227–260. [Google Scholar]
- Harris, W.; Norman White, G. X-ray diffraction techniques for soil mineral identification. In Methods of Soil Analysis Part 5—Mineralogical Methods; Soil Science Society of America: Madison, WI, USA, 2008; pp. 81–115. [Google Scholar]
- Hensen, E.J.M.; Smit, B. Why clays swell. J. Phys. Chem. B 2002, 106, 12664–12667. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, M.L.; Lammers, L.N.; Carrero, S.; Gilbert, B.; Banfield, J.F. Ion exchange selectivity in clay is controlled by nanoscale chemical-mechanical coupling. Proc. Natl. Acad. Sci. USA 2019, 116, 22052–22057. [Google Scholar] [CrossRef]
- Tester, C.C.; Aloni, S.; Gilbert, B.; Banfield, J.F. Short- and long-range attractive forces that influence the structure of montmorillonite osmotic hydrates. Langmuir 2016, 32, 12039–12046. [Google Scholar] [CrossRef] [Green Version]
- Fukuma, T. Water distribution at solid/liquid interfaces visualized by frequency modulation atomic force microscopy. Sci. Technol. Adv. Mater. 2010, 11, 033003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siretanu, I.; Ebeling, D.; Andersson, M.P.; Stipp, S.L.S.; Philipse, A.; Stuart, M.C.; van den Ende, D.; Mugele, F. Direct observation of ionic structure at solid-liquid interfaces: A deep look into the Stern Layer. Sci. Rep. 2014, 4, 4956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Andersson, M.P.; van den Ende, D.; Mugele, F.; Siretanu, I. Probing the surface charge on the basal planes of kaolinite particles with high-resolution atomic force microscopy. Langmuir 2017, 33, 14226–14237. [Google Scholar] [CrossRef]
- Zhai, H.; Wang, L.; Putnis, C.V. Molecular-scale investigations reveal noncovalent bonding underlying the adsorption of environmental DNA on mica. Environ. Sci. Technol. 2019, 53, 11251–11259. [Google Scholar] [CrossRef] [PubMed]
- Martin-Jimenez, D.; Garcia, R. Identification of single adsorbed cations on mica-liquid interfaces by 3D force microscopy. J. Phys. Chem. Lett. 2017, 8, 5707–5711. [Google Scholar] [CrossRef] [Green Version]
- Araki, Y.; Satoh, H.; Okumura, M.; Onishi, H. Localization of cesium on montmorillonite surface investigated by frequency modulation atomic force microscopy. Surf. Sci. 2017, 665, 32–36. [Google Scholar] [CrossRef] [Green Version]
- Xian, Z.; Hao, Y.; Zhao, Y.; Song, S. Quantitative determination of isomorphous substitutions on clay mineral surfaces through AFM imaging: A case of mica. Colloids Surf. A Physicochem. Eng. Asp. 2017, 533, 55–60. [Google Scholar] [CrossRef]
- Rotenberg, B.; Marry, V.; Malikova, N.; Turq, P. Molecular simulation of aqueous solutions at clay surfaces. J. Phys. Condens. Matter 2010, 22, 284114. [Google Scholar] [CrossRef] [PubMed]
- Le Crom, S.; Tournassat, C.; Robinet, J.-C.; Marry, V. Influence of polarizability on the prediction of the electrical double layer structure in a clay mesopore: A molecular dynamics study. J. Phys. Chem. C 2020, 124, 6221–6232. [Google Scholar] [CrossRef]
- Greathouse, J.A.; Refson, K.; Sposito, G. Molecular dynamics simulation of water mobility in magnesium-smectite hydrates. J. Am. Chem. Soc. 2000, 122, 11459–11464. [Google Scholar] [CrossRef]
- Teppen, B.J.; Rasmussen, K.; Bertsch, P.M.; Miller, D.M.; Schäfer, L. Molecular dynamics modeling of clay minerals. 1. Gibbsite, kaolinite, pyrophyllite, and beidellite. J. Phys. Chem. B 1997, 101, 1579–1587. [Google Scholar] [CrossRef]
- Chatterjee, A.; Iwasaki, T.; Ebina, T.; Miyamoto, A. A DFT study on clay-cation-water interaction in montmorillonite and beidellite. Comput. Mater. Sci. 1999, 14, 119–124. [Google Scholar] [CrossRef]
- Sposito, G.; Skipper, N.T.; Sutton, R.; Park, S.-h.; Soper, A.K.; Greathouse, J.A. Surface geochemistry of the clay minerals. Proc. Natl. Acad. Sci. USA 1999, 96, 3358–3364. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Oyabu, N.; Kimura, K.; Ido, S.; Suzuki, K.; Imai, T.; Tagami, K.; Tsukada, M.; Yamada, H. Visualization of hydration layers on muscovite mica in aqueous solution by frequency-modulation atomic force microscopy. J. Chem. Phys. 2013, 138, 184704. [Google Scholar] [CrossRef] [Green Version]
- Kubicki, J.D.; Ohno, T. Integrating density functional theory modeling with experimental data to understand and predict sorption reactions: Exchange of salicylate for phosphate on goethite. Soil Syst. 2020, 4, 27. [Google Scholar] [CrossRef]
- Ho, T.A.; Greathouse, J.A.; Lee, A.S.; Criscenti, L.J. Enhanced ion adsorption on mineral nanoparticles. Langmuir 2018, 34, 5926–5934. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, J.A.R.; Myneni, S.C.B.; Bourg, I.C. Molecular dynamics simulations of the adsorption of phthalate esters on smectite clay surfaces. J. Phys. Chem. C 2019, 123, 13624–13636. [Google Scholar] [CrossRef]
- Kobayashi, K.; Liang, Y.; Murata, S.; Matsuoka, T.; Takahashi, S.; Nishi, N.; Sakka, T. Ion distribution and hydration structure in the stern layer on muscovite surface. Langmuir 2017, 33, 3892–3899. [Google Scholar] [CrossRef]
- Rahromostaqim, M.; Sahimi, M. Molecular dynamics study of the effect of layer charge and interlayer cations on swelling of mixed-layer chlorite-montmorillonite clays. J. Phys. Chem. C 2020, 124, 2553–2561. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Zhang, C.; Lu, X. A combined first principles and classical molecular dynamics study of clay-soil organic matters (SOMs) interactions. Geochim. Cosmochim. Acta 2020, 291, 110–125. [Google Scholar] [CrossRef]
- Sowers, T.D.; Wani, R.P.; Coward, E.K.; Fischel, M.H.H.; Betts, A.R.; Douglas, T.A.; Duckworth, O.W.; Sparks, D.L. Spatially resolved organomineral interactions across a permafrost chronosequence. Environ. Sci. Technol. 2020, 54, 2951–2960. [Google Scholar] [CrossRef] [PubMed]
- Papelis, C.; Hayes, K.F. Distinguishing between interlayer and external sorption sites of clay minerals using X-ray absorption spectroscopy. Colloids Surf. A Physicochem. Eng. Asp. 1996, 107, 89–96. [Google Scholar] [CrossRef]
- Vasconcelos, I.F.; Haack, E.A.; Maurice, P.A.; Bunker, B.A. EXAFS analysis of cadmium (II) adsorption to kaolinite. Chem. Geol. 2008, 249, 237–249. [Google Scholar] [CrossRef]
- Vasconcelos, I.F.; Bunker, B.A.; Cygan, R.T. Molecular dynamics modeling of ion adsorption to the basal surfaces of kaolinite. J. Phys. Chem. C 2007, 111, 6753–6762. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Z.; Peak, D.; Tang, Y.; Feng, X.; Zhu, M. Quantification of coexisting inner- and outer-sphere complexation of sulfate on hematite surfaces. ACS Earth Space Chem. 2018, 2, 387–398. [Google Scholar] [CrossRef]
- Xu, T.; Stubbs, J.E.; Eng, P.J.; Catalano, J.G. Response of interfacial water to arsenate adsorption on corundum (0 0 1) surfaces: Effects of pH and adsorbate surface coverage. Geochim. Cosmochim. Acta 2018, 239, 198–212. [Google Scholar] [CrossRef]
- Roszmann, C.A. Retention of phosphorus by soil colloids. Soil Sci. 1927, 24, 465–474. [Google Scholar] [CrossRef]
- Ravikovitch, S. Anion exchange: I. adsorption of the phosphoric acid ions by soils. Soil Sci. 1934, 38, 219–240. [Google Scholar] [CrossRef]
- Sumner, M.E. Soil chemistry: Past, present and future. In Future Prospects for Soil Chemistry: Proceedings of a Symposium Sponsored by Division S-2, Soil Chemistry of the Soil Science Society of America in St. Louis, MO, USA, 30–31 October 1995; Huang, P.M., Sparks, D.L., Boyd, S.A., Eds.; Soil Science Society of America: Madison, WI, USA, 1998; p. 233. [Google Scholar]
- Leeper, G.W. Factors affecting availability of inorganic nutrients in soils with special reference to micronutrient metals. Annu. Rev. Plant Phys. 1952, 3. [Google Scholar] [CrossRef]
- Sumner, M.E. Effect of iron oxides on positive and negative charges in clays and soils. Clay Miner. 1963, 5, 218–226. [Google Scholar] [CrossRef]
- Hingston, F.J.; Atkinson, R.J.; Posner, A.M.; Quirk, J.P. Specific adsorption of anions. Nature 1967, 215, 1459–1461. [Google Scholar] [CrossRef]
- Stumm, W.; Huang, C.; Jenkins, S. Specific chemical interaction affecting the stability of dispersed systems. Croat. Chem. Acta 1970, 42, 223–245. [Google Scholar]
- Huang, C.-P.; Stumm, W. Specific adsorption of cations on hydrous γ-Al2O3. J. Colloid Interface Sci. 1973, 43, 409–420. [Google Scholar] [CrossRef]
- Schindler, P.W.; Fürst, B.; Dick, R.; Wolf, P.U. Ligand properties of surface silanol groups. I. Surface complex formation with Fe3+, Cu2+, Cd2+, and Pb2+. J. Colloid Interface Sci. 1976, 55, 469–475. [Google Scholar] [CrossRef]
- Forbes, E.A.; Posner, A.M.; Quirk, J.P. The specific adsorption of divalent Cd, Co, Cu, Pb, and Zn on goethite. J. Soil Sci. 1976, 27, 154–166. [Google Scholar] [CrossRef]
- Sauve, S.; Hendershot, W.; Allen, H.E. Solid-solution partitioning of metals in contaminated soils: Dependence on pH, total metal burden, and organic matter. Environ. Sci. Technol. 2000, 34, 1125–1131. [Google Scholar] [CrossRef]
- Manning, B.A.; Goldberg, S. Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil Sci. Soc. Am. J. 1996, 60, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Cabrera, F.; Madrid, L.; de Arambarri, P. Adsorption of phosphate by various oxides: Theoretical treatment of the adsorption envelope. J. Soil Sci. 1977, 28, 306–313. [Google Scholar] [CrossRef] [Green Version]
- Hingston, F.J.; Posner, A.M.; Quirk, J.P. Competitive adsorption of negatively charged ligands on oxide surfaces. Discuss. Faraday Soc. 1971, 334–342. [Google Scholar] [CrossRef]
- Parfitt, R.L.; Russell, J.D. Adsorption on hydrous oxides. IV. Mechanisms of adsorption of various ions on goethite. J. Soil Sci. 1977, 28, 297–305. [Google Scholar] [CrossRef]
- Hiemstra, T.; van Riemsdijk, W.H. Surface structural ion adsorption modeling of competitive binding of oxyanions by metal (hydr)oxides. J. Colloid Interface Sci. 1999, 210, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Fukushi, K.; Sverjensky, D.A. A surface complexation model for sulfate and selenate on iron oxides consistent with spectroscopic and theoretical molecular evidence. Geochim. Cosmochim. Acta 2007, 71. [Google Scholar] [CrossRef]
- McKenzie, R. The adsorption of lead and other heavy metals on oxides of manganese and iron. Soil Res. 1980, 18, 61–73. [Google Scholar] [CrossRef]
- Hayes, K.F.; Roe, A.L.; Brown, G.E., Jr.; Hodgson, K.O.; Leckie, J.O.; Parks, G.A. In situ X-ray absorption study of surface complexes: Selenium oxyanions on α-FeOOH. Science 1987, 238, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Manceau, A.; Charlet, L. The mechanism of selenate adsorption on goethite and hydrous ferric oxide. J. Colloid Interface Sci. 1994, 168, 87–93. [Google Scholar] [CrossRef]
- Peak, D.; Sparks, D.L. Mechanisms of selenate adsorption on iron oxdes and hyroxides. Environ. Sci. Technol. 2002, 36, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, M.L.; Manceau, A.; Chateigner, D.; Charlet, L. Sorption of metal Ions on clay minerals: I. Polarized EXAFS evidence for the adsorption of Co on the edges of hectorite particles. J. Colloid Interface Sci. 1999, 215, 140–158. [Google Scholar] [CrossRef]
- Furnare, L.J.; Vailionis, A.; Strawn, D.G. Molecular-level investigation into copper complexes on vermiculite: Effect of reduction of structural iron on copper complexation. J. Colloid Interface Sci. 2005, 289. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.L.; Sherman, D.M. Copper (II) sorption onto goethite, hematite and lepidocrocite: A surface complexation model based on ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta 2004, 68, 2623–2637. [Google Scholar] [CrossRef]
- Tiberg, C.; Sjöstedt, C.; Eriksson, A.K.; Klysubun, W.; Gustafsson, J.P. Phosphate competition with arsenate on poorly crystalline iron and aluminum (hydr)oxide mixtures. Chemosphere 2020, 255, 126937. [Google Scholar] [CrossRef] [PubMed]
- Barrow, N. The description of desorption of phosphate from soil. J. Soil Sci. 2006, 30, 259–270. [Google Scholar] [CrossRef]
- Corey, R.B. Sorption vs precipitaiton. In Adsorption of Inorgnaics at Solid-Liquid Interfaces; Anderson, M.A., Rubin, A.J., Eds.; Ann Arbor Science: Ann Arbor, MI, USA, 1980; pp. 161–182. [Google Scholar]
- Farley, K.J.; Dzombak, D.A.; Morel, F.M.M. A surface precipitation model for the sorption of cations on metal oxides. J. Colloid Interface Sci. 1985, 106, 226–242. [Google Scholar] [CrossRef]
- Comans, R.N.J.; Middelburg, J.J. Sorption of trace metals on calcite: Applicability of the surface precipitation model. Geochim. Cosmochim. Acta 1987, 51, 2587–2591. [Google Scholar] [CrossRef] [Green Version]
- Bleam, W.F.; McBride, M.B. Cluster formation versus isolated-site adsorption—A study of Mn (II) and Mg (II) adsorption on boehmite and goethite. J. Colloid Interface Sci. 1985, 103, 124–132. [Google Scholar] [CrossRef]
- Chisholmbrause, C.J.; Oday, P.A.; Brown, G.E.; Parks, G.A. Evidence for multinuclear metal-ion complexes at solid water interfaces from X-ray absorption-spectroscopy. Nature 1990, 348, 528–531. [Google Scholar] [CrossRef]
- Charlet, L.; Manceau, A. X-ray absorption spectroscopic study of the sorption of Cr (III) at the oxide water interface. 2. adsorption, coprecipitation, and surface precipitation on hydrous ferric-oxide. J. Colloid Interface Sci. 1992, 148, 443–458. [Google Scholar] [CrossRef]
- Charlet, L.; Manceau, A. Evidence for the neoformation of clays upon sorption of Co (II) and Ni (II) on silicates. Geochim. Cosmochim. Acta 1994, 58, 2577–2582. [Google Scholar] [CrossRef]
- Scheidegger, A.M.; Sparks, D.L. Kinetics of the formation and the dissolution of nickel surface precipitates on pyrophyllite. Chem. Geol. 1996, 132, 157–164. [Google Scholar] [CrossRef]
- Ford, R. Potential formation of secondary hydrotalcite-like precipitates during Zn and Cu sorption to pyrophyllite. Mineral. Mag. 1998, 462–463. [Google Scholar] [CrossRef]
- Zhu, C. Estimation of surface precipitation constants for sorption of divalent metals onto hydrous ferric oxide and calcite. Chem. Geol. 2002, 188, 23–32. [Google Scholar] [CrossRef]
- Peltier, E.; Allada, R.; Navrotsky, A.; Sparks, D.L. Nickel solubility and precipitation in soils: A thermodynamic study. Clays Clay Miner. 2006, 54, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Fu, Q.; Qiu, G.; Liu, Y.; Hu, H.; Huang, Q.; Violante, A. Influence of low molecular weight anionic ligands on the sorption of heavy metals by soil constituents: A review. Environ. Chem. Lett. 2019, 17, 1271–1280. [Google Scholar] [CrossRef]
- Flynn, E.D.; Catalano, J.G. Competitive and cooperative effects during nickel adsorption to iron oxides in the presence of oxalate. Environ. Sci. Technol. 2017, 51, 9792–9799. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.C.; Hiemstra, T. Ternary complex formation of phosphate with Ca and Mg Ions binding to ferrihydrite: Experiments and mechanisms. ACS Earth Space Chem. 2020, 4, 545–557. [Google Scholar] [CrossRef]
- Tiberg, C.; Gustafsson, J.P. Phosphate effects on cadmium (II) sorption to ferrihydrite. J. Colloid Interface Sci. 2016, 471, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strathmann, T.J.; Myneni, S.C.B. Effect of soil fulvic acid on nickel (II) sorption and bonding at the aqueous-boehmite (γ-AlOOH) interface. Environ. Sci. Technol. 2005, 39, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Illustration showing examples of ion adsorption as inner- and outer-sphere complexes on a mineral surface. Ions in the diffuse-double layer are hydrated (water molecules not shown). Surface charge components (σ) are shown (see Table 3). Adapted from Strawn et al. [28] with permission.
Figure 1.
Illustration showing examples of ion adsorption as inner- and outer-sphere complexes on a mineral surface. Ions in the diffuse-double layer are hydrated (water molecules not shown). Surface charge components (σ) are shown (see Table 3). Adapted from Strawn et al. [28] with permission.

Figure 2.
Edge of kaolinite mineral showing the silanol and aluminol functional groups and their protonation and deprotonation states. From Strawn et al. [28] with permission.
Figure 2.
Edge of kaolinite mineral showing the silanol and aluminol functional groups and their protonation and deprotonation states. From Strawn et al. [28] with permission.

Figure 3.
Illustration of atomic force microscopic imaging of mica surface showing cations adsorbed on charged surface sites. Adapted from Xian et al. [80] with permission.
Figure 3.
Illustration of atomic force microscopic imaging of mica surface showing cations adsorbed on charged surface sites. Adapted from Xian et al. [80] with permission.

Figure 4.
Illustration of the molecular dynamic model of cation distribution on a charged clay mineral surface (mica). See text for a description of adsorption mechanisms. Image adapted from Kobayashi et al. [91] with permission.
Figure 4.
Illustration of the molecular dynamic model of cation distribution on a charged clay mineral surface (mica). See text for a description of adsorption mechanisms. Image adapted from Kobayashi et al. [91] with permission.

Figure 5.
Illustration of mechanisms of SO42− adsorption on hematite (left). Amounts of SO42− adsorption on hematite (µmmol/g) at different pH (right). Changing ionic strength affects the amount of outer-sphere adsorbed SO42− at the different suspension pH values. At the highest ionic strength, the NO3− anions outcompete SO42− for the outer-sphere adsorption sites, but have minimal effect on the SO42− inner-sphere adsorption because NO3- does not adsorb via inner-sphere complexation on hematite. Image adapted from Wang et al. [98] with permission.
Figure 5.
Illustration of mechanisms of SO42− adsorption on hematite (left). Amounts of SO42− adsorption on hematite (µmmol/g) at different pH (right). Changing ionic strength affects the amount of outer-sphere adsorbed SO42− at the different suspension pH values. At the highest ionic strength, the NO3− anions outcompete SO42− for the outer-sphere adsorption sites, but have minimal effect on the SO42− inner-sphere adsorption because NO3- does not adsorb via inner-sphere complexation on hematite. Image adapted from Wang et al. [98] with permission.

Figure 6.
pH-dependent adsorption of metals on goethite. Source: modified from McKenzie [117] with permission.
Figure 6.
pH-dependent adsorption of metals on goethite. Source: modified from McKenzie [117] with permission.

Figure 7.
Two views (top of (001) plane in panel (A) and side view in panel (B)) of layered double hydroxide mineral surface precipitates (green, blue, and red) on a clay mineral (blue and red). Image adapted from [24] with permission.
Figure 7.
Two views (top of (001) plane in panel (A) and side view in panel (B)) of layered double hydroxide mineral surface precipitates (green, blue, and red) on a clay mineral (blue and red). Image adapted from [24] with permission.

Figure 8.
Ternary adsorption. Image adopted from Flynn and Catalano [138] with permission. Aqueous Ni-oxalate complexation is also shown.
Figure 8.
Ternary adsorption. Image adopted from Flynn and Catalano [138] with permission. Aqueous Ni-oxalate complexation is also shown.


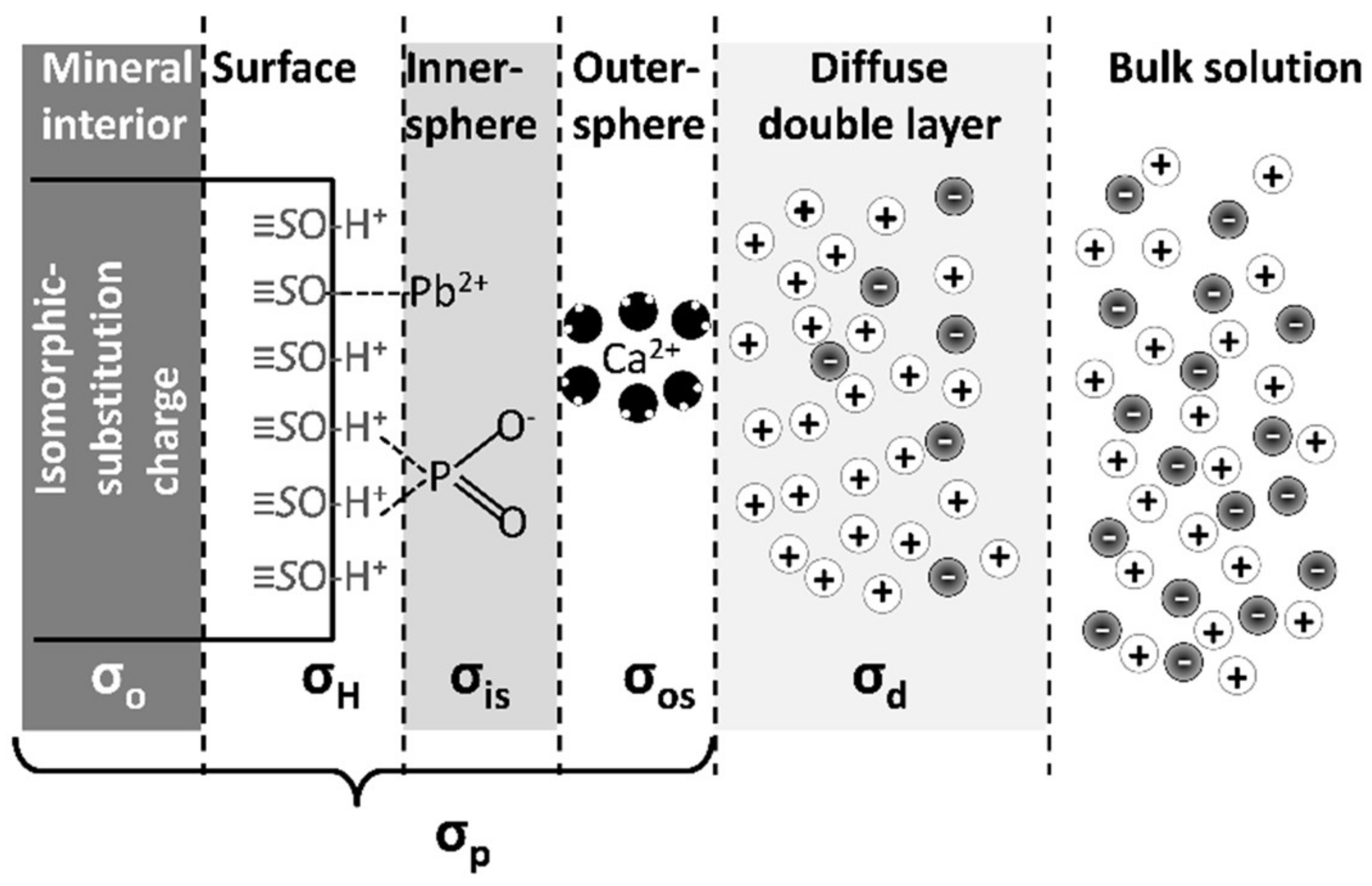{kind=link}
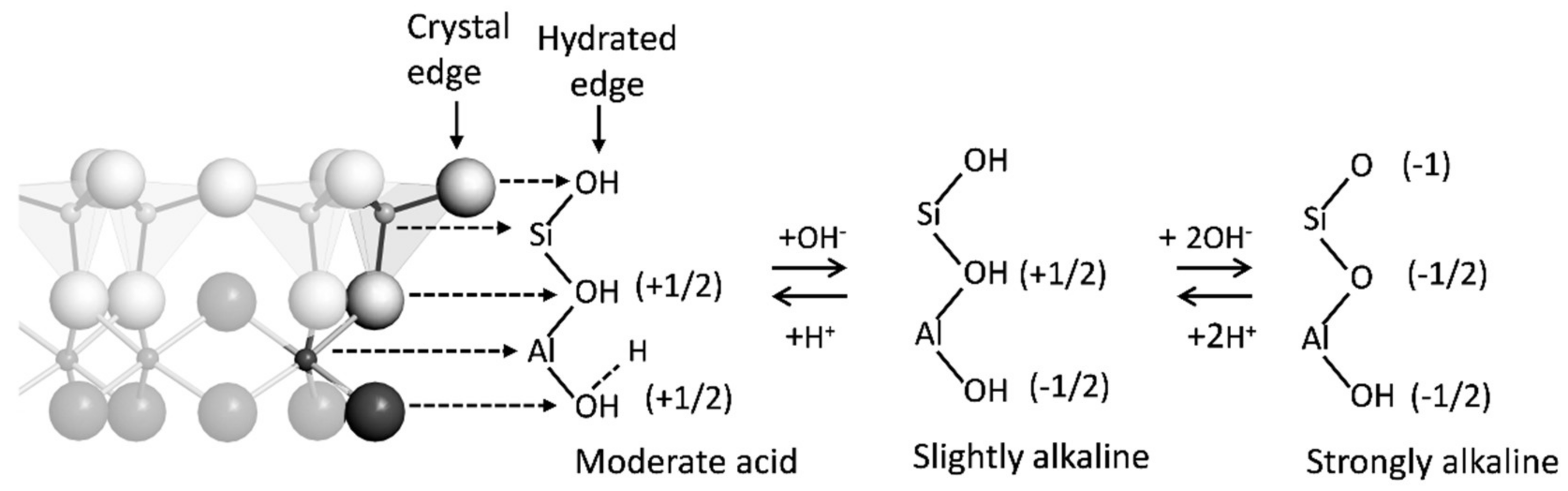{kind=link}
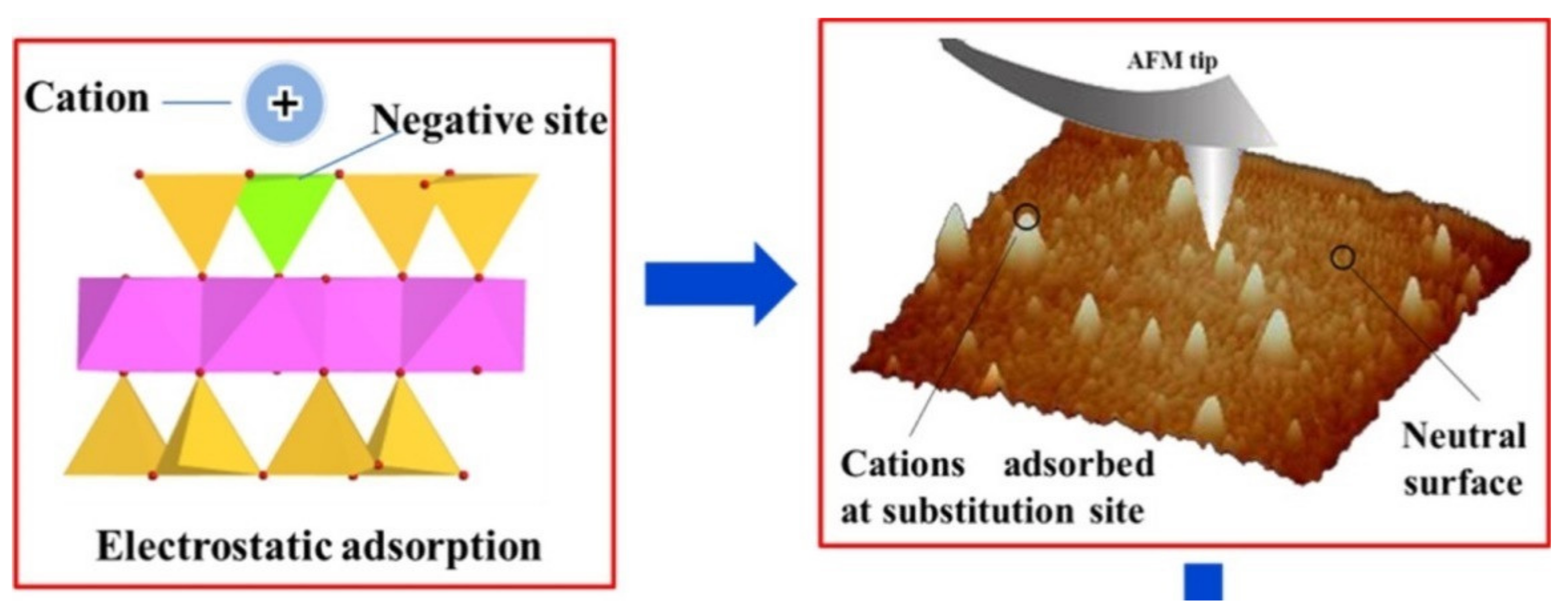{kind=link}
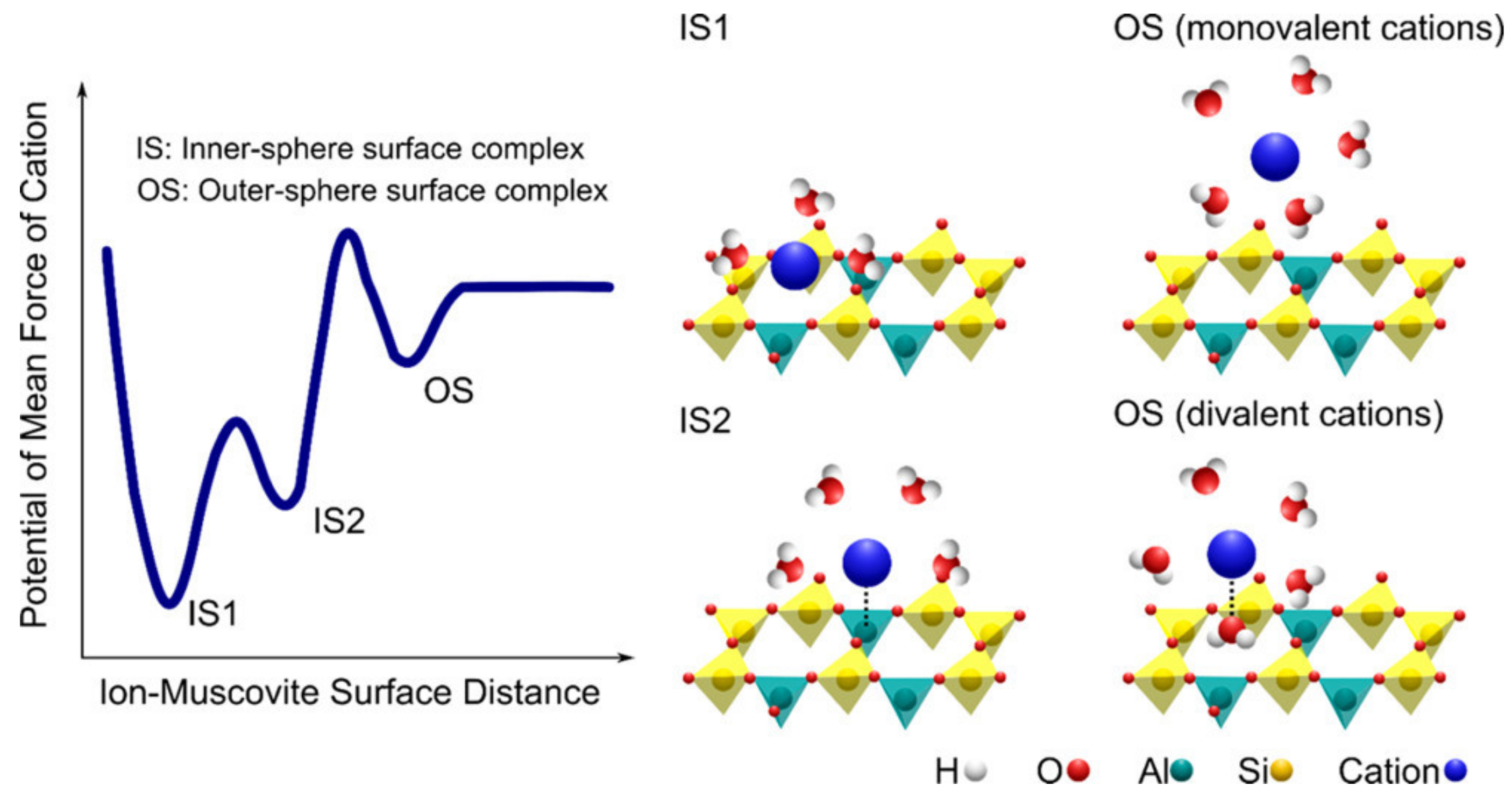{kind=link}
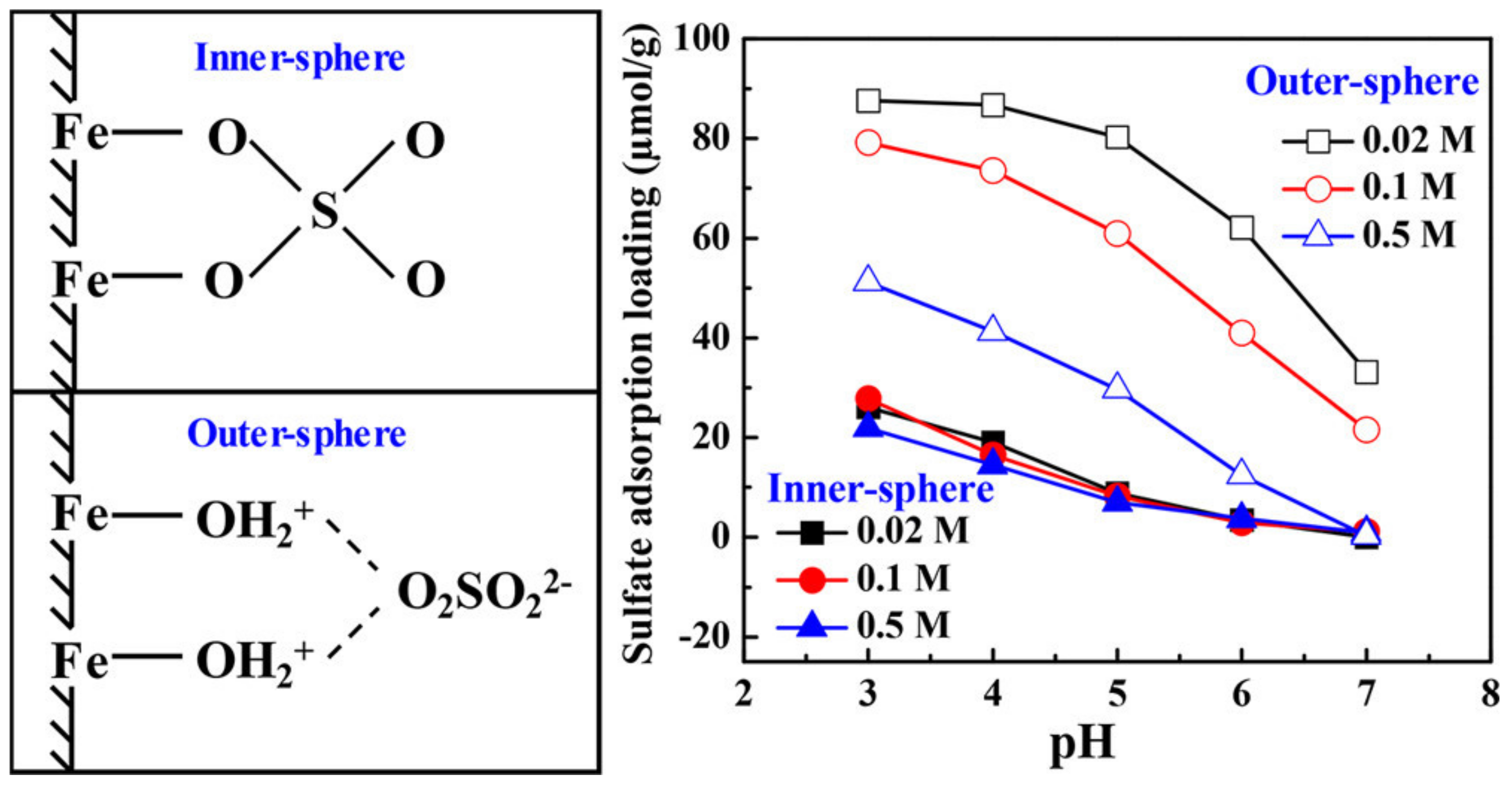{kind=link}
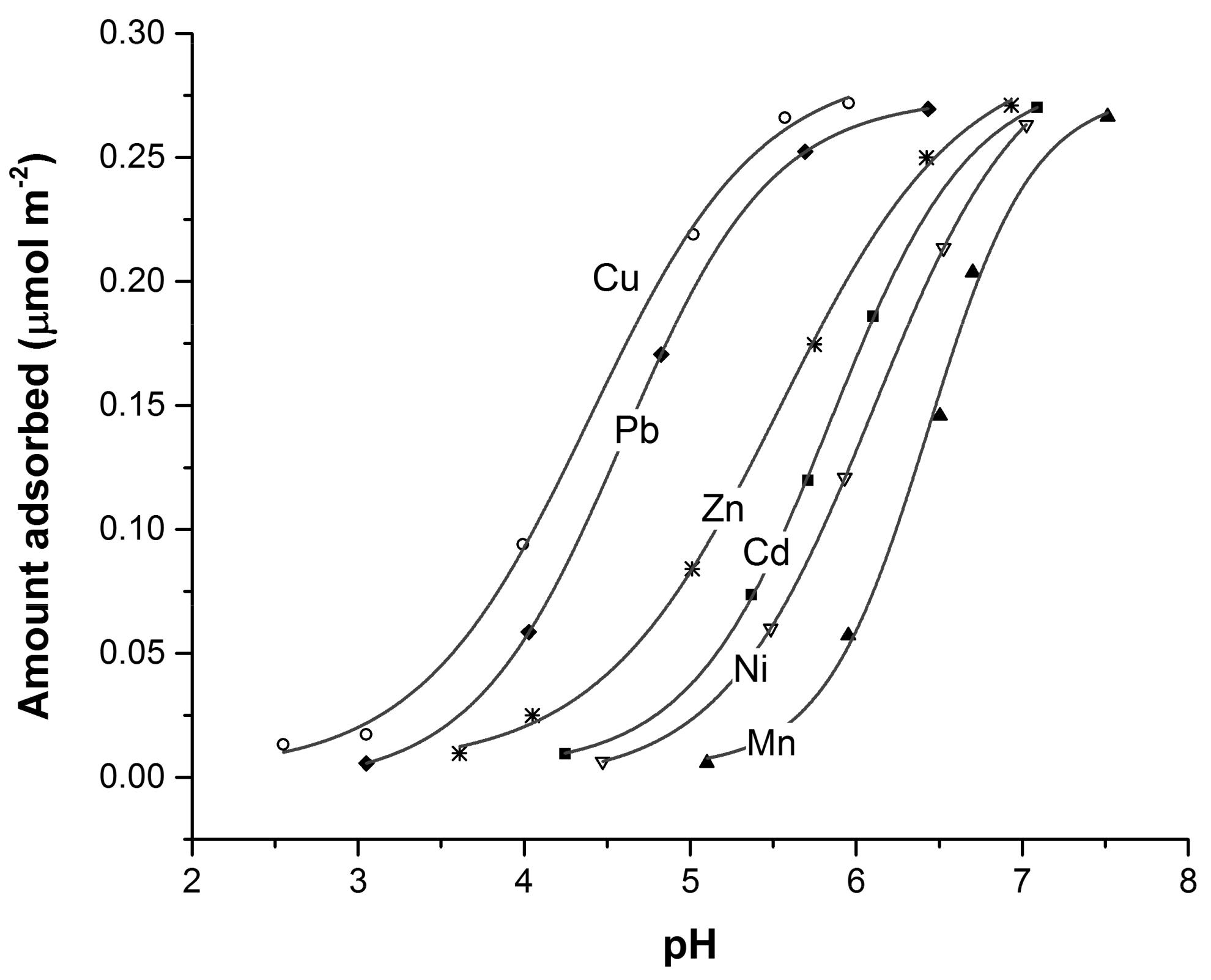{kind=link}
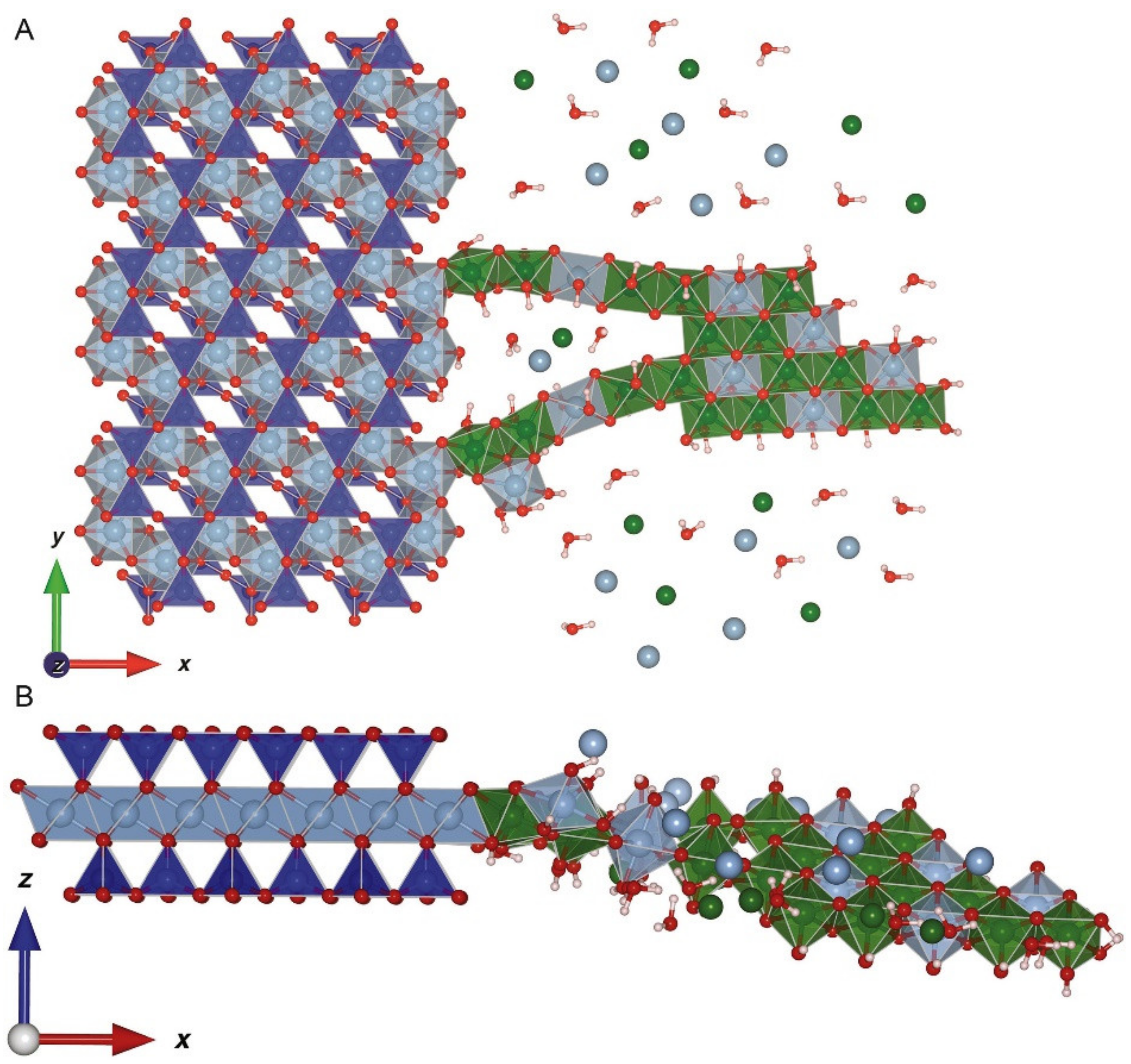{kind=link}
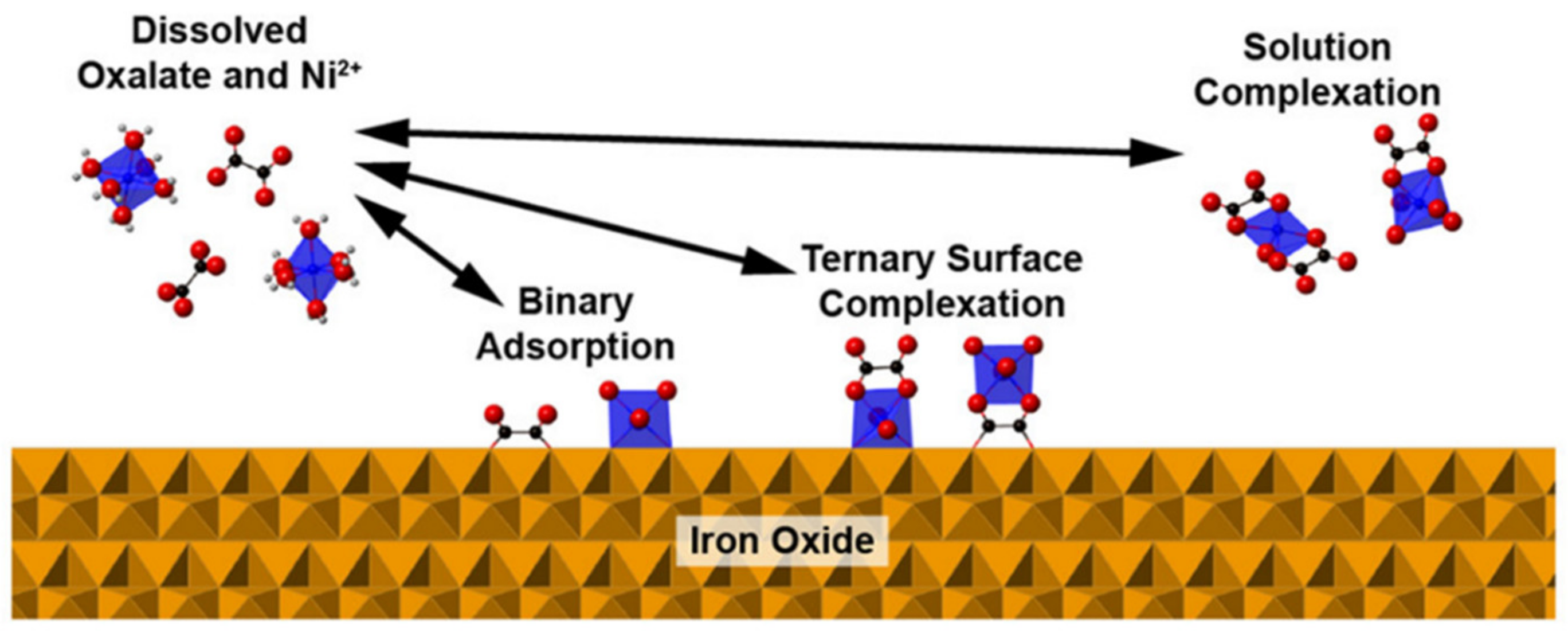{kind=link}
Table 1.
Sorption mechanisms that occur on soil particles.
| Sorption Mode | Molecular Mechanism | Soil Particles Involved | Examples 1 |
|---|---|---|---|
| Outer-sphere adsorption | Electrostatic attraction of opposite charges At least one water molecule exists between the surface and the adsorbed chemical | Clay minerals with permanent charge, edges of all soil minerals (esp. oxides) that become charged, charged functional groups on SOM | Cations in clay mineral interlayers, alkali and alkaline earth metals on mineral edges and SOM functional groups, anions Cl− [13], NO3− [14], SO42− [15], SeO42− [16], and AsO33− [17] |
| Inner-sphere adsorption—edges and SOM | Sharing of electrons in covalent-type bonds | Soil mineral edges with reactive O ligands and carboxylic acid and phenolic acid functional groups sites on SOM | Metals capable of forming covalent-type bonds [18,19], e.g., Pb2+, Cu2+, Ni2+, and Zn2+, and anions PO43− [20] and AsO43− [21,22] |
| Inner-sphere adsorption—basal planes | Ionic-type bond between dehydrated cations and permanently charged oxygen atoms on basal planes of clay minerals | Clay minerals with tetrahedral isomorphic substitution: vermiculite and illite | Alkali metals with weak hydration spheres [23] (K+, Cs+) and ammonium (NH4+) |
| Precipitation on mineral surfaces | Small multi-nuclear complexes formed on the surfaces of minerals | All soil minerals | Metals, especially transition metals [24], oxyanions AsO43− [25] |
| Hydrophobic partitioning | van der Waal forces between hydrophobic chemicals reduces interactions to polar water molecules reducing entropy | Uncharged regions of SOM and some uncharged mineral surfaces, e.g., basal plane of kaolinite | Organic compounds, e.g., pesticides, industrial compounds (DNAPL), hydrophobic molecules released from biota [26] |
| Absorption | Uptake of solutions into pore space, mainly hydrogen bonding and capillary forces, some Weak van der Waal forces | Porous solids (aggregates, crystallites, or SOM) in which solutes become entrapped | Aqueous and non-aqueous liquids that become physically isolated from the bulk solution [27] |
1 Sorption mechanisms are examples of common sorption modes for chemicals. Chemical sorption mechanisms vary depending on the solution and solid properties. SOM: soil organic matter.
Table 2.
Types of adsorption sites on minerals and the surface charge components.
| Adsorption Surface Types | Details | Surface Charge Components 1 | Examples |
|---|---|---|---|
| Permanent negative charge in the interlayer of clay minerals created from isomorphic substitution | Delocalized electrostatic charge that attracts cations. In some cases, cations with weak hydration spheres dehydrate | σo + σOS | Common soil clay minerals, e.g., montmorillonitevermiculite, illite (frayed edges) |
| Hydroxyl ligands on edges of minerals | Unsatisfied bonds on mineral edges are Bronsted acid and bases that gain or lose protons causing pH-dependent charge and adsorption | σH + σIS + σOS |
|
| Weak acid and base functional groups on SOM | Carboxyl, phenol, amine, and thiol functional groups | σH + σIS + σOS | SOM functional groups developed during biomolecule degradation |
| Hydrophobic regions on SOM | Uncharged regions of SOM such as alkanes and aromatic rings. | none |
|
Table 3.
Surface charge components of soil minerals.
| Surface Charge Component | Description |
|---|---|
| σp | Total charge on the surface of a particle, not including the ions in the diffuse double layer |
| σo | Permanent surface charge of the particle due to isomorphic substitution |
| σH | Charge on the surface of a particle due to protonation and deprotonation of oxygen functional groups on the edge of the mineral |
| σIS | Charge on the surface caused by adsorption of ions via inner-sphere adsorption |
| σOS | Charge on the surface caused by adsorption of ions via outer-sphere adsorption; not including diffuse layer ions |
| σd | Total charge of ions in the diffuse double layer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Strawn, D.G. Sorption Mechanisms of Chemicals in Soils. Soil Syst. 2021, 5, 13. https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems5010013
AMA Style
Strawn DG. Sorption Mechanisms of Chemicals in Soils. Soil Systems. 2021; 5(1):13. https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems5010013
Chicago/Turabian StyleStrawn, Daniel G. 2021. "Sorption Mechanisms of Chemicals in Soils" Soil Systems 5, no. 1: 13. https://0-doi-org.brum.beds.ac.uk/10.3390/soilsystems5010013