Monoterpenoids: The Next Frontier in the Treatment of Chronic Pain?



1
Department of Pharmacology and Pharmaceutical Sciences, School of Pharmacy, University of Southern California, Los Angeles, CA 90089, USA
2
Dipartimento di Scienze della Salute, Università “Magna Græcia” di Catanzaro, 88100 Catanzaro, Italy
*
Author to whom correspondence should be addressed.
J 2020, 3(2), 195-214; https://0-doi-org.brum.beds.ac.uk/10.3390/j3020016
Submission received: 27 April 2020
/
Revised: 22 May 2020
/
Accepted: 26 May 2020
/
Published: 28 May 2020
(This article belongs to the Special Issue Pain and Chronic Pain)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Ointments and lotions from natural extracts have a long tradition of being used in folk medicines against pain conditions. Monoterpenoids are among the major constituents of several natural topical remedies. The field of chronic pain is one of the most investigated for new active molecular entities. This review will discuss several molecular mechanisms against which monoterpenoids have been proven to be good candidates for the topical treatment of chronic pain.
1. Introduction
Monoterpenoids are plant-derived or synthetic 10-carbon-containing compounds. Monoterpenoids are unknowingly used in perfumes and other preparations by millions of people every day. Many representatives from this class have been characterized from the extracts of several plants traditionally used to relieve pain and treat minor diseases [1]. Their relatively low molecular weights as well as their intrinsic lipophilicity make these molecules suitable for skin permeation and suggest a possible role in topical treatments. The efficacy of the compounds in causing pain to subside mostly relies on their ability to inhibit transient receptor potential cation channels in the skin (TRP) [1]. TRP channels are the most abundant pain receptors in the body and are most plentiful in the skin [2]. Topical application of medicines has been proven useful in the treatment of several conditions, ranging from minor injuries to chronic pain [3,4,5].
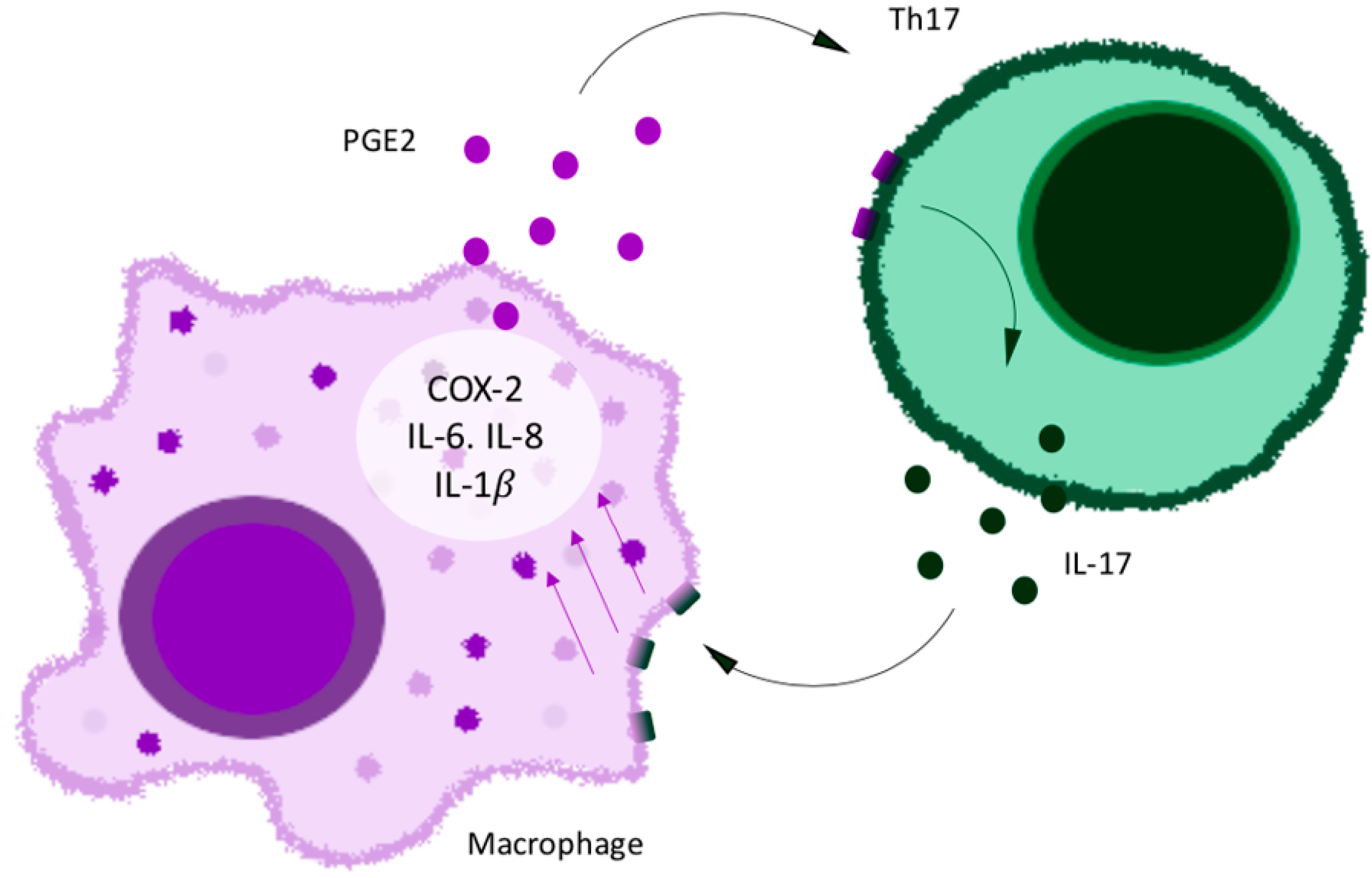
The mechanisms of chronic pain make this condition particularly hard to treat. Brain neurons may become sensitized to pain in chronic pain conditions. Several interconnected processes in the skin cause a self-perpetuating pain chemokine cycle that results in chronic pain (Figure 1). These interconnected processes involve TRP channels, cyclooxygenase 2, prostaglandins, macrophages, chemokines, skin resident T cells, neutrophils, and interleukins [5]. Damage to a sensory neuron or other cell in the skin releases chemokines that attract and activate macrophages and neutrophils. Cyclooxygenase 2 becomes induced in macrophages by this process and releases prostaglandins into the skin. Macrophages also release their own chemokines that transactivate TRP channels in sensory neurons. Neutrophils release leukotrienes that activate TRP channels to cause pain [6]. This may be the basis of increased pain sensitivity in some patients. Chemokines induce the production of IL-17 by skin resident T cells (Figure 2). IL-17 induces chemokine production by sensory neurons and macrophages. Prostaglandins cause pain by binding to prostaglandin receptors and enhance the activation of TRP channels.
Monoterpenoids are also known to deactivate TRP channels [1]. This review will also address the ability of topically applied monoterpenoids to modulate skin cyclooxygenase 2, prostaglandin production, chemokine release, and IL-17 actions. The current work will evaluate the evidence that topical monoterpenoids can interfere with the chemokine cycle in the skin and provide relief in chronic pain conditions.
2. The Topical Management of Chronic Pain
Topical preparations based on natural products have a long tradition of being applied in the treatment of several conditions, especially pain, in many cultures [7,8,9]. It is very interesting how many of these traditional remedies against pain seem to contain an abundance of monoterpenoids in their extracts. The Chumash Native Americans of California have traditional medicines containing monoterpenoids that are used to cure chronic pain [7,8,9]. There are several mechanisms involved in the actions of monoterpenoids in chronic pain.
Chronic pain involves a complex mechanism that includes several receptors and endogenous compounds. Usually, pain is referred to an area on the body surface, even though it may originate in a tissue fairly remote from that area. This is because branches of afferent nociceptors form synapses in the spinal cord and receive pain signals from the skin.
Skin, in fact, can be considered as an immuno-endocrine organ that can influence a broad range of physiological and pathophysiological functions, such as inflammation. The central nervous system (CNS) is directly or indirectly connected to the skin thanks to primary afferent and autonomic nerves, which release neuromodulators and activate specific receptors on many target cells in the skin. These mediators are barely present in normal conditions, but a dramatic increase is registered upon the actions of different stimuli, like trauma or inflammation [10].
In addition, an interactive network is likely to exist between sensory nerves and immune cells. Mast cells, derivatives of mononuclear precursors, are usually concentrated in the dermal–epidermal junction around blood vessels and nerves. In some conditions, they can migrate to the epidermis as well, since chemokines attract them. The IgE-mediated immunological stimulation of mast cells induces the liberation of arachidonic acid, which can trigger both the cyclooxygenase enzymes (COX) or the 5-LOX pathways, with the production of prostaglandin D2 and leukotriene C4 [11]. All of the accumulation of these mediators induces the activation of nociceptors, especially TRP channels. The noxious input, then, reaches the spinal cord and, through the dorsal root ganglia, is transmitted to specific areas in the CNS, resulting in the perception of pain, burning, or itching.
In chronic inflammation and pain, prolonged skin nociceptor activation may lead to their sensitization, with a reduction in the threshold for action potentials, an increase in response to a stimulus, and an apparent spontaneous activity of nociceptors, especially TRP channels. The two main manifestations are hyperalgesia (exaggerated response to a noxious stimulus) and allodynia (perception of pain without a noxious stimulus). This is the result of a pathological neural plasticity in response to the injury [12].
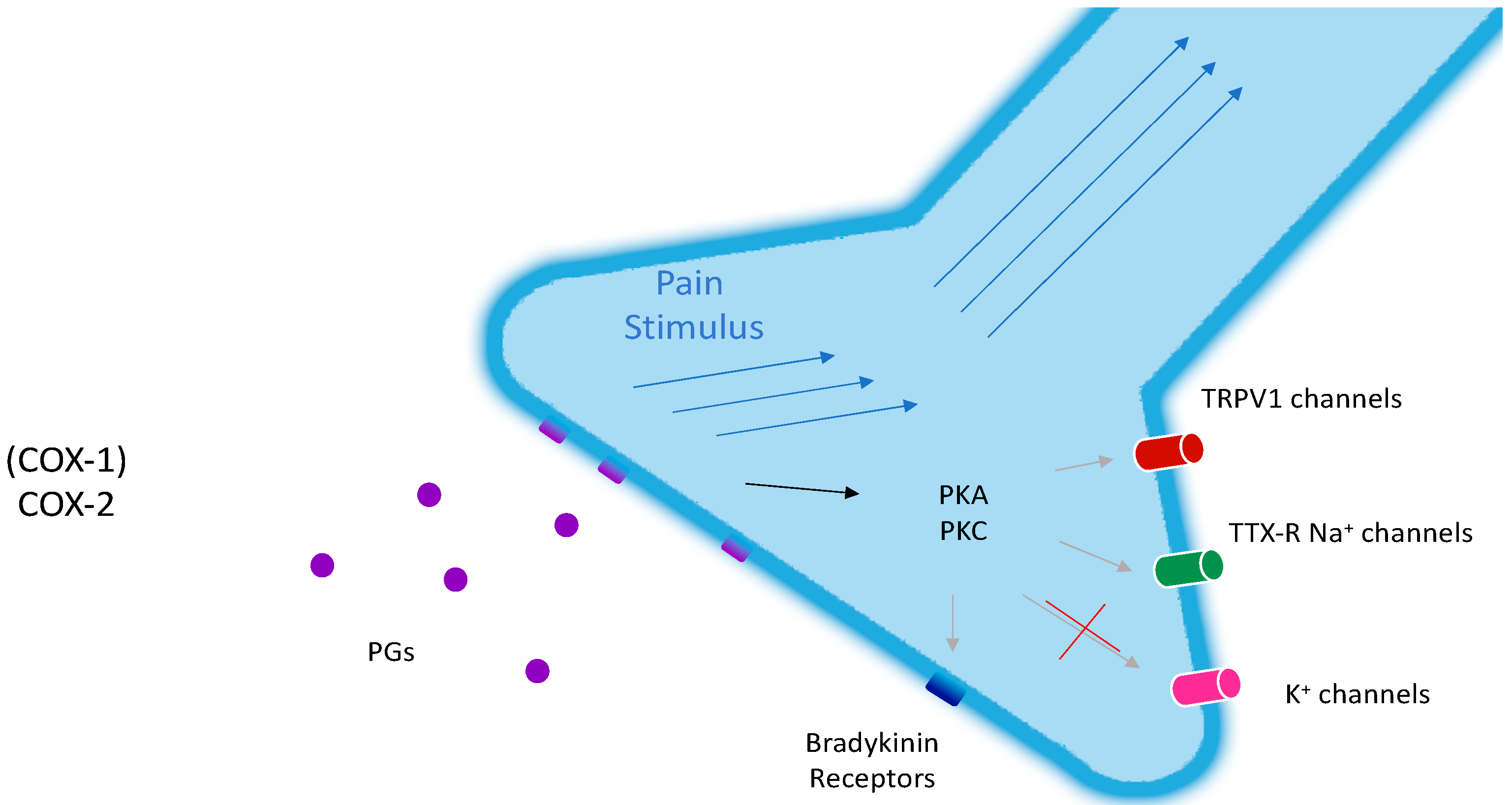
Prostanoids are strong sensitizing agents, both peripherally and centrally. The peripheral mechanisms include an activation of TTX-R (tetrodotoxin-resistant) Na+ channels and an inhibition of voltage-gated potassium currents. The indirect effects are the enhancement of the sensitivity of sensory neurons to bradykinin, which acts on B2-receptors, and to capsaicin, which acts on TRPV1-receptors (vanilloid receptors, Figure 1).
COX-2 is the main source of prostanoids in injured tissues, even though the role of COX-1 should not be underestimated (Figure 3). In the CNS, both isoenzymes are present. However, during inflammation, there is a great up-regulation of COX-2 in the skin and spinal cord, meaning that COX-2 is responsible for the increase in prostaglandin E-2 (PGE-2) levels in the skin, which produces central sensitivity [12]. During chronic pain, the induction of COX-2 can last for years, thus making the skin a pain-producing organ.
Although the pathophysiology of pain is nowadays clear, chronic pain is often difficult to treat. Primarily, the etiology of chronic pain is only partially recognized for nociceptive conditions (such as osteoarthritis) or neuropathic conditions (such as post-herpetic neuralgia). Chronic pain conditions are usually treated with oral non-steroidal anti-inflammatory drugs (NSAIDs) or opioids, with obvious consequences in terms of adverse effects, especially for elderly patients (>65 years), who are the most affected by these diseases.
Data suggest that topical therapies may offer a safer, well-tolerated, and effective alternative to systemic therapies in the treatment of patients with chronic, localized musculoskeletal and neuropathic pain. A topical administration allows a lower daily dose and a site-specific drug delivery, avoiding first-pass metabolism, drug interactions, and systemic side effects [13]. The majority of topical monoterpenoids evaporate from the skin.
Topical administration is particularly suitable for patients with musculoskeletal pain because the drug can penetrate muscle, synovium, and joint tissue below the site of application. Concentrations achieved here are at least equivalent to those obtained with oral administration, although very variable [14]. In fact, penetration depth changes on account of several factors; for example, variability in subcutaneous vasculature.
The primary barrier to drug delivery through the skin is provided by the stratum corneum, located on the outer surface. The epidermis below is a viable tissue devoid of blood vessels. Just below the dermal–epidermal junction, the dermis contains capillary loops that can take up administered drugs for systemic distribution [15].
So, subcutaneous concentrations with topical administration exceed the ones achieved by oral administration, but synovial concentrations are equivalent [14]. This is because trans-synovial transport is a slow process that depends on the pharmacological characteristics of the drug and on the properties of the joint and joint space [12].
Several NSAIDs are available as topical formulations. In patients with osteoarthritis, topical diclofenac, ibuprofen, or ketoprofen have shown efficacy similar to that of oral preparations. In addition, in neuropathic pain, including post-herpetic neuralgia (PHN), complex regional pain syndrome (CRPS), and diabetic peripheral neuropathic pain (DPNP), topical capsaicin or lidocaine are effective treatments [16]. A micro-emulsion for the topical delivery of celecoxib has also been described [17]. In addition, many topical monoterpenoid preparations are available to treat pain, such as Tiger Balm and Salon Pas.
The use of topical analgesics could be advantageous in palliative care, where the main goal is relief from chronic pain in patients with cancer. In this condition, the non-oral route of administration avoids side effects and interactions with other drugs. Although some adverse effects—mainly cutaneous (hopefully minimal, like rashes, pruritus, etc.)—can occur with topical formulations, they are undoubtedly superior in terms of systemic side effects and offer a safer, more effective way to treat chronic pain.
3. Terpenes Are Suitable Candidates for Skin-Permeation Enhancement
For a successful transdermal drug delivery, compounds need to reversibly interact with the stratum corneum, altering its highly ordered structure [18]. Terpenes have been classified as a safe and potent class of penetration enhancers to help transdermal drug delivery for both lipophilic and hydrophilic compounds [19]. Their mechanism of action exploits the interaction with the lipids in the stratum corneum, leading to their extraction and/or fluidization or interaction with keratin.
From this perspective, oxygenated terpenes are more suitable because of their functional groups that may help with H-bond interactions in the lipid bilayer [19]. Another mechanism may be the improvement of the solubility of a drug in the stratum corneum [20]. The capability of a terpene to enhance skin permeation depends on its chemical structure (a chain terpene will move more easily into the bilayer than a ring-shaped one [20]) and on other physicochemical properties [21].
Lipophilicity: Expressed as the partition coefficient (octanol/water), LogP, this is the most important feature, given that the stratum corneum offers a more polar environment than octanol [18]. Therefore, non-polar terpenes like limonene are more suitable for the enhancement of the permeation of lipophilic drugs than the related oxygenated compounds and vice versa. Usually, amphiphilic terpenes are the perfect compromise to enter the highly packed bilayer [19].
Size and chirality: It has been observed that smaller and (-) isomers perform better.
Boiling point and energy of vaporization: They are inversely related to the enhancement capability; the lower the values of these parameters, the higher the permeation rate.
Degree of unsaturation: This factor depends on the individual terpenes.
Nevertheless, very often, permeation studies may be misleading when just the stratum corneum is considered. When the latter is removed, in fact, permeation through epidermal and dermal tissue is the real rate-limiting step. The Enhancement Index (EI) takes this peculiarity into consideration, and it may hence be used to get a better idea of the maximum achievable drug permeation [18].
Furthermore, according to the apparent permeability coefficient (p (app)) values, selected substances are ranked in the following order: Monoterpene hydrocarbons < monoterpene alcohols < monoterpene ketones < phenylpropanoids [22].
As far as concentrations are concerned, it has been shown that the accepted range should be 1%–5% in transdermal drug delivery systems [19]. Of course, other compounds (e.g., ethanol [23]) may synergistically act to improve the permeation, allowing a decrease in single terpene concentrations in the future.
Another important feature to consider is the type of vehicle in which monoterpenes are provided: Cutaneous accumulation of terpenes is higher when they are applied in pure essential oils than in topical vehicles, leaving no doubt about the cooperation of the terpenes in skin permeation [22,24].
It should be mentioned that human skin models are considered superior to animal models, such as hairless pigs or rats [25]. Availability of human skin is limited. Animal models are used for preliminary studies and are followed up by human models as necessary [25].
Overall, terpenes are non-toxic, safe, and non-irritant, and are very effective permeation enhancers; they hence provide an extremely powerful tool in the harsh field of transdermal and transmucosal drug delivery.
4. Monoterpenoids Have Great Potential in the Treatment of Chronic Pain
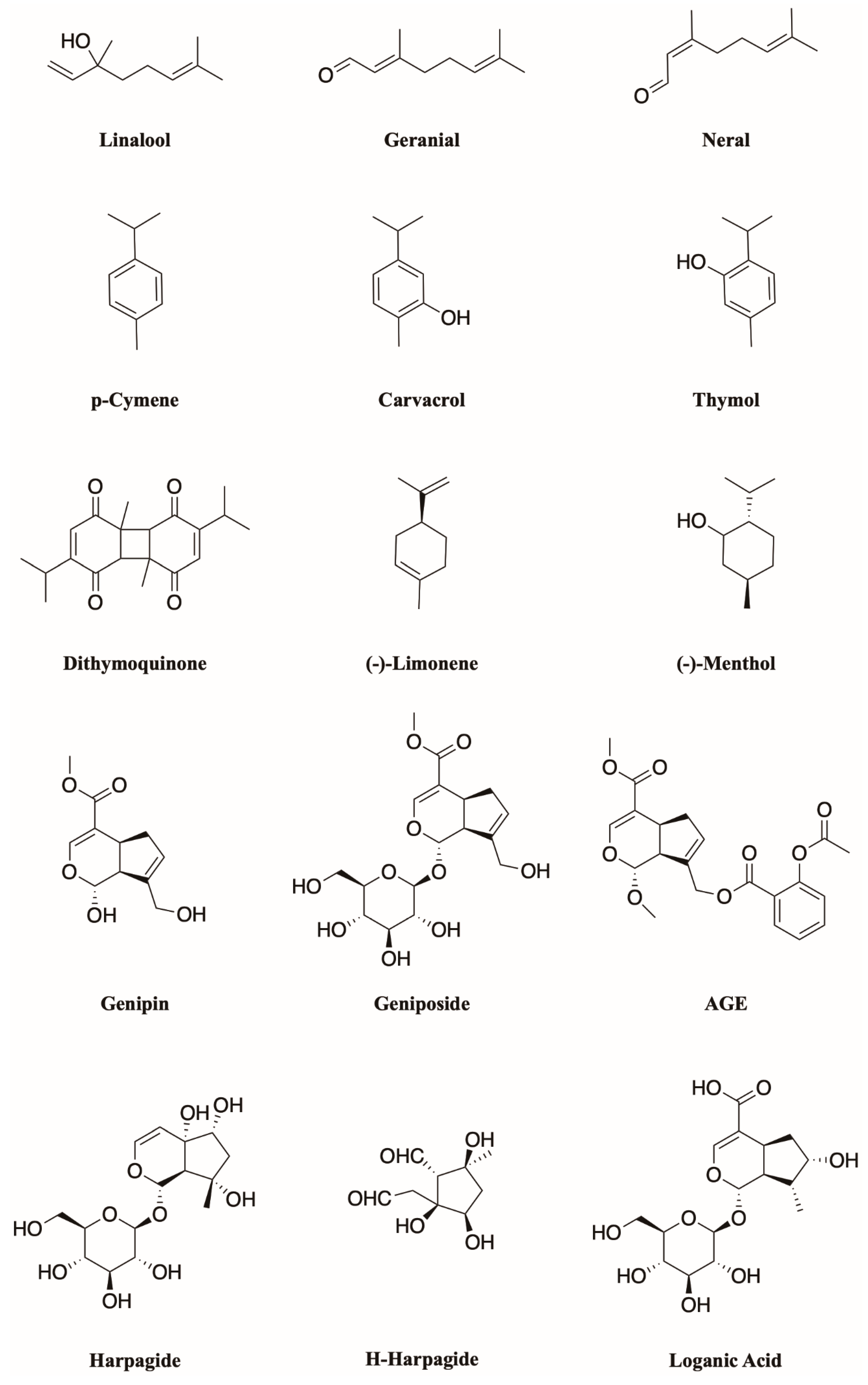
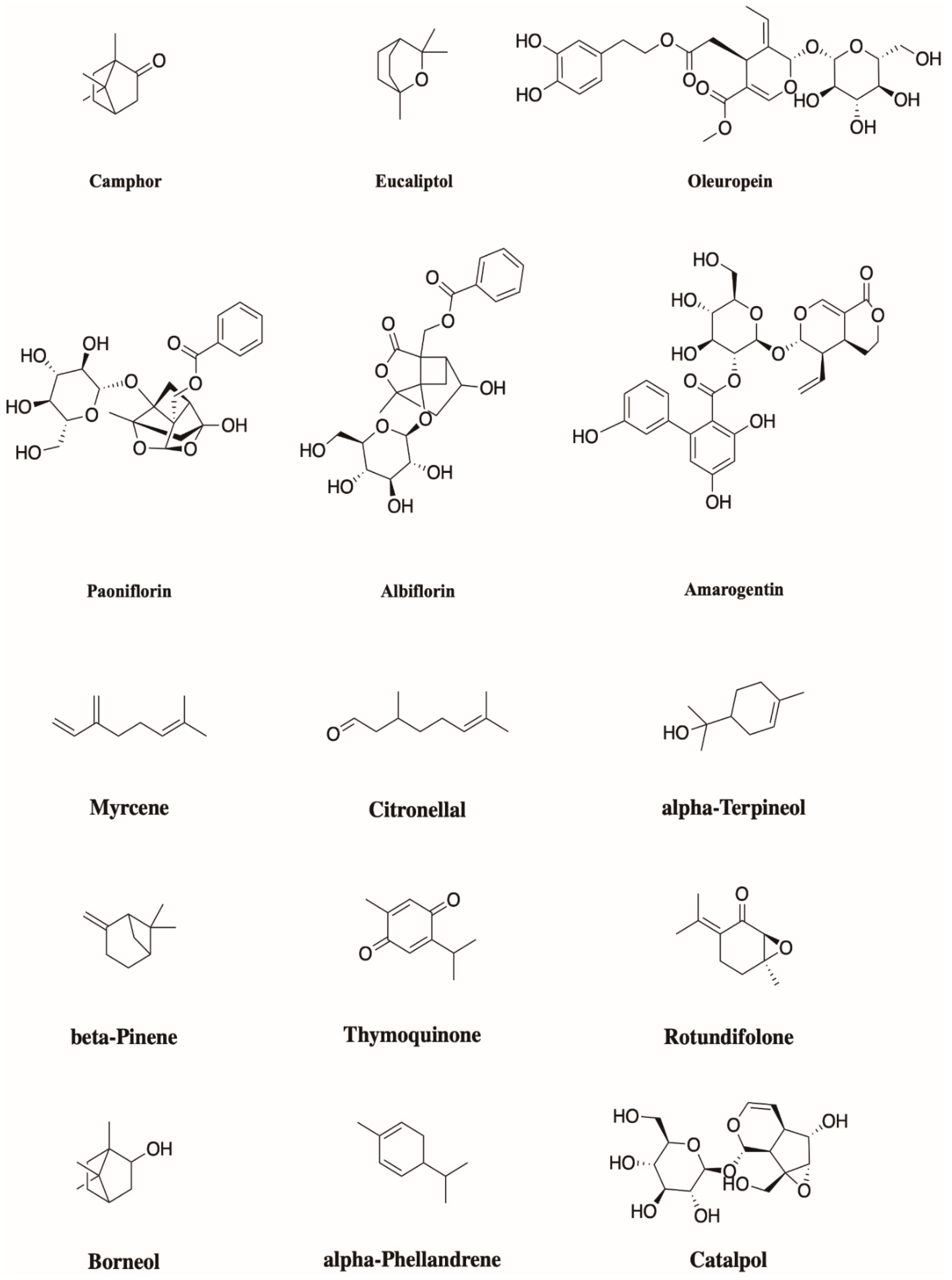
Monoterpenoids (Figure 4) are widely occurring natural compounds found in higher plants, algae, and fungi, and they generally show typical odor and taste. Structurally, they are the result of the head-to-tail condensation of two isoprene units; then, starting from this 10-carbon backbone, several enzymatic reactions, such as hydroxylations, dehydrogenations, double bonds and/or carbonyl reductions, isomerizations, and conjugations lead to extensive biodiversity [26], with about 20,000 different terpene metabolites known. To sum up, they can be divided into three main subgroups: Acyclic, monocyclic, and bicyclic, depending on their arrangement open chains, closed chains, or two joined rings, respectively.
They are widely known for their numerous biological activities, which include antimicrobial, hypotensive, antipruritic, antigerminative, antiplasmodial, antiesophageal cancer, anticandidal, and, especially interesting concerning this review, anti-inflammatory activities [27].
As for their anti-inflammatory activities, more than one mechanism of action has been suggested, and up to ten different systems seem to be modulated by linalool, the most studied monoterpene. Extensive reviews have already explored the available literature for the analgesic and anti-inflammatory properties of these compounds [28,29,30].
Here, we propose an innovative approach: The most important pain-related pathways in which monoterpenes have showed efficacy will be briefly discussed. Afterward, the IL-17 pathway and the chemokine axis will be described as poorly explored but very promising future targets for more research involving the monoterpene activity spectrum. Similarly, the inhibitory potential of monoterpenes against COX-2- and TRP-mediated stimuli will be discussed.
The opioid system is an important inhibitory system in nociception, which acts through two main pathways: Central and peripheral. At a central level, opioid agonists activate pain-control descending pathways as well as potassium channels and inhibit voltage-dependent calcium channels [31]. These are generally modulated by acyclic monoterpenes, including myrcene, linalool, citronellal, and citronellol. Exceptions are represented by α-terpineol, paeoniflorin, β-pinene, thymoquinone, p-cymene, and rotundifolone. Opioids also induce chemokine formation, which can make chronic pain worse and can cause opioid-induced hyperalgesia [5].
The inhibition of the signaling cascade starting with the NF-kB transcription factor is a common feature of the majority of the monoterpenes that exert both anti-inflammatory and anticancer activity [32]. Consequences include, obviously, the reduction in inducible Nitric Oxide Synthase (iNOS) and COX-2 expression.
α-Pinene and linalool showed efficacy in inhibiting COX-2 expression [33]. Citral (geranial) and carvacrol act by inhibiting COX-2 expression and activating the peroxisome proliferator-activated receptors (PPAR) α and γ. The latter are ligand-dependent transcription factors that are also known to modulate COX-2 expression, and vice versa [25]. Other important molecules involved in this pathway are (+)-pulegone, α-terpineol, tymoquinone, and 1,8-cineole (eucalyptol) [27].
Nitric oxide (NO) is considered a pro-inflammatory mediator. Rather than on the release of NO itself, many monoterpenes act on the reduction of the expression levels of the inducible Nitric Oxide Synthase (iNOS) mostly through the NF-κB pathway. Examples are citral, limonene, geraniol, and linalool [30].
Activated macrophages release inflammatory cytokines that bind to the receptors maintaining the inflammatory response. Numerous monoterpenes show a significant capability to inhibit, for example, the expression of TNF-α, among which are linaool, catalpol, carvacrol, citronellol, geniposide, paeoniflorin, 1,8-cineole (eucalyptol), borneol, α-terpineol, etc. [28,29,30]. The same compounds very often show a great ability to decrease other cytokines like Il-1β, IL-6, IL-4, and IL-8.
Glutamate is an excitatory amino acid, and glutamate-mediated events may also contribute to the peripheral nociception and inflammation [34]. Diverse monoterpenes act on this neurotransmission, such as linalool (N-methyl-d-aspartate (NMDA) receptors), myrcene, carvacrol, α-phellandrene, paeoniflorin, and citronellal [28]; moreover, gentiopicroside shows a peculiar capability to down-regulate GluN2B receptors [30].
Many other neurotransmission systems that are involved and interwoven often appear as the targets of monoterpenoid action. These include the serotoninergic system (citral acts on 5HT2A and 5HT3 receptors [30]), the adrenergic system (myrcene and paeoniflorin, which act on α2 receptors [30]), the cholinergic system, since painful stimuli are known to increase acetylcholine in the spinal cord (γ-terpinene [35], α-phellandrene, linaool [28], and geraniol have the capability to reduce acetylcholinesterase activity [30]), the GABA-ergic system (borneol, carvacrol, menthol, linalool, and α-terpineol act on GABA-A receptors [28,30]), and the adenosinergic and dopaminergic systems as well [28,30]. Most of these receptors occur in the skin and are involved in pain sensation in the skin [3].
5. Pro-Inflammatory Cytokine IL-17 and Chemokines as New Targets against the Mechanisms of Pain and Chronic Pain
Interleukin-17 (IL-17) is a Th1 cytokine secreted from activated CD4 cells; therefore, it is one of the molecules that may serve as a mediator of the T cell response to pathogens [36]. This subset of T-Helper cells was identified because of its capability to produce IL-17A, IL-17F, and IL-22 [37]. The IL-17 polypeptide consists of a 19 amino acid signal sequence followed by a 136 amino acid mature segment. It contains at least one N-glycosylation site and six cysteine residues that form intermolecular bonds during dimerization [38]. Apart from IL-17(A), five additional IL-17 members have been described (termed IL-17B to IL-17F), which all have conservations in their C-terminal region.
They are a family of pro-inflammatory cytokines, and are so called because of their capability to promote the production of CXC chemokines, Matrix Metalloprotineases (MMPs), Granulocyte-colony stimulating factor (G-CSF), and increased intracellular adhesion molecule 1 (ICAM-1) from tissues, regulating neutrophil homeostasis as well [39]. They promote a great variety of inflammation processes, like the induction of nitric oxide (NO) production, with an increase in the inducible nitric oxide synthase (iNOS) [39,40,41]. They also induce COX-2, and one of the novel signaling pathways suggests the involvement of both DNA-binding and RNA-binding proteins [40,42]. Moreover, the autocrine IL-17A production by Th17 cells is critical to sustain the proinflammatory loop, and key elements involved in Th17 cell differentiation and function are prostaglandins themselves [43,44,45].
Among the numerous target genes of Il-17-dependent stimulation, there are the genes of IL-1β, IL-6, IL-8, and, especially, TNF-α. In fact, IL-17 is able to induce NF-κB transcription factors in fibroblasts, macrophages, chondrocytes, intestinal epithelial cells, and colonic and pancreatic myofibroblasts [46]. All of the above-mentioned biological activities and signaling pathways have been widely shown to be augmented by the synergistic action of TNF-α [36,47,48].
The receptor for IL-17A (IL-17R) is a single-pass transmembrane protein of approximately 130 kDa. Five members of this receptor family have been identified so far: IL-17R, IL-17RH1, IL-17RL (receptor like), IL-17RD, and IL-17RE, and they show a widespread distribution in disparate tissues, such as in articular cartilage, bone, meniscus, brain, hematopoietic tissue, kidneys, lungs, skin, and intestines [49]. Recent studies analyzing the IL-17RA cytoplasmic tail have uncovered motifs that share features with a signaling moiety found in Toll-Like Receptors and IL-1R [39], but signal transduction pathways have not been clearly established yet.
Resident cells in the skin that contribute to innate responses include keratinocytes, epidermal Langerhans cells, and dermal dendritic cells (DDCs). The latter promote the development of specialized effector T cells, like CD4+ T helper type 1 (Th1) and CD4+ T helper type 2 (Th2) cells, as well as the novel aforementioned subset called Th17 cells [50]. In addition, in mucosal and epithelial surfaces like the skin, another significant source of IL-17 is the gamma-delta T cell population, which may be important in the early neutrophil response [39]. Th17 cells should be able to enter the skin by their preferential expression of chemokine receptors CCR4 and CCR6 [51], regulating chemotaxis by the chemokines CCL17 (cutaneous venules) and CCL20 (keratinocytes, dermal fibroblasts, and endothelial cells) [50].
There is no evidence of IL-17 expression in normal skin, but there is in skin lesions, allergic contact dermatitis, and psoriasis [46]. Both Th1 and Th17 cytokines have been found in psoriatic skin with unregulated expression of β-defensin-2 (BD-2), psoriasin, cathelicidin, and calprotectin [52]. In keratinocytes, IL-17 induces IL-6, IL-8, GROα, GM-CSF, and ICAM-1 by acting either directly or amplifying the actions of IFN-γ, IL-4, and TNFα [46].
High levels of IL-17 were found in the rheumatoid synovia of patients with rheumatoid arthritis (RA), but not with osteoarthritis, causing inflammation and bone degradation due to the stimulated expression of several chemokines, such as CXCL8 (IL-8), CXCL1 (KC/GRO-α), CXCL2 (MIP2α/GRO-β), CCL20 (MIP-3α), CCL2 (MCP1), and CCL7 (MCP3) [52,53]. Moreover, in synoviocytes, IL-17 synergizes with IL-1β- and TNFα-induced production of cytokines, including IL-6, IL-8, growth-related oncogene α (GROα), leukemia inhibitory factor (LIF), and MIP-3α [46]. Increased NO level, as well as the release of cartilage proteoglycans, glycosaminoglycans (GAGs) and collagen fragments, contribute to matrix degradation [49]. In conclusion, T cells with inflammatory effects on epithelial, endothelial, and fibroblast cells express IL-17.
IL-17 has what it takes to be a potential therapeutic target [54]; hence, the effect of monoterpenoids on the regulation of proinflammatory cytokines could be an interesting topic for further research. To date, only poor data is available that revolve around the mechanisms of monoterpenoid activity on cytokine expression pathways or against their receptors. Likely, some of the most effective compounds might be able to interact at different stages of the inflammatory pathways.
p-Cymene, carvacrol, and thymol are all monoterpenoids isolated from the leaves of Lippia sidoides Cham., a plant endowed with anti-microbial activity [55]. Their application in the treatment of emphysema in mice showed positive outcomes and resulted in the reduction of expressed IL-17 along with other cytokines [56].
Oleuropein can be retrieved from the leaves of olive trees [57]. It is a phenolic seco-iridoid recently patented for the treatment of type 2 diabetes [58]. The anti-oxidant and anti-inflammatory effects of oleuropein have been demonstrated [59]. The compound was proven to suppress IL-17 expression in bowel cells from ulcerative colitis patients [60].
Amarogentin is a seco-iridoid glycoside found in the root of Gentiana lutea L. [61]. The compound is an inhibitor of Topoisomerase I and has anti-leishmanial properties in animal models [62]. Recent studies demonstrated the ability of amarogentin to inhibit histamine and TNFα synergetic stimulation of CXCL8 expression in keratinocytes [63]. However, these results highlight how the inhibition of the expression and activity of the mediators of inflammation consists of an intricate network of inter-dependent pathways that cannot be considered separately.
Geniposide, an iridoid glycoside, and its aglycone, genipin, derived from the fruits of Gardenia jasminoides J.Ellis, both seem to be good candidates as inhibitors of inflammation [64]. As a matter of fact, genipin is able to significantly reduce the expression of CXCL1 and inhibits autophagy-dependent inflammasome activation [65], whilst geniposide affects CXCL8 production by blocking p38 MAPK and the ERK 1/2 pathway [66]. Geniposide was also able to inhibit IL-17 expression in rats with adjuvant arthritis [67].
Paeoniflorin was reported as an inhibitor of the secretion of CXCL1 and CCL2 chemokines from microglia [68] as well as for its ability to regulate CXCR3 expression in macrophages and to interfere with CXCL11 and CXCR3 interactions [69]. The inhibition of CXCL8 release was also documented [70]. One of the suggested mechanisms for the suppression of chemokine release after an inflammatory stimulus is the blockade of TNFα-induced NF-κB and ERK 1/2 activation by phosphorylation [71]. The anti-inflammatory effect might be also mediated by the inhibitory potential of the compound against abnormal IL-17 production, as reported by Dai et al. in 2015 [72]. In addition, albiflorin, an isomer of paeoniflorin, showed efficacy in inhibiting CXCL1 expression [73]. Paeoniflorin and albiflorin are monoterpene glycosides extracted from the roots of Paeonia lactiflora. The plant is known in traditional Chinese medicine as bai shao yao, and has been widely used for the treatment of several forms of pain and menstrual disorders [74].
Linalool (Figure 4) is a mixture of two isomers of a monoterpene alcohol mostly found in the plant families of Lamiaceae, Lauraceae, and Rutaceae. The raceme mixture is commonly used as a scent in the perfume industry. A recent study led by Ma et al. demonstrated that linalool inhibits CXCL8 and CCL2 release in tissues by affecting the NF-B pathway [75]. Interestingly, a computational approach attempted in 2015, aimed at the discovery of new targets and lead compounds for the treatment of oral cancer, actually resulted in the selection of linalool, along with its isomer β-linalool, as potential lead compounds for the inhibition of CXCR4 [76].
6. COX-2: Will a Target Ever Be Obsolete
The synthesis of prostaglandins G and H is the second step of a biosynthetic path known as the prostanoid synthetic pathway, which starts with the synthesis of prostaglandin G from arachidonic acid, dihomo-γ-linolenic acid, or EPA, performed by the cyclooxygenase enzymes (COX) [77]. The production of prostanoids must not be labeled as an exclusive pathologic condition even if it is involved in some mechanisms of inflammation. Cells produce these compounds as a physiological activity, such as platelet aggregation, regulation of gastric and intestinal secretion, broncho-dilation, and constriction and maintenance of vascular tone.
The role of PGE2 is particularly relevant, for it has a key role in the genesis of pain [78]. It is widely accepted that COX-1 production of prostanoids is aimed at “housekeeping functions”, while COX-2 expression and enzymatic activity would be induced, in several tissues, under stress conditions to synthesize prostanoids as intermediates in inflammation and cancer processes [79].
Traditional NSAIDs inhibit both COX-1 and -2, even if they may show some differences concerning binding affinity [77]. Due to the inhibition of constitutive COX-1, which is expressed in the epithelium of the stomach, the synthesis of prostanoids decreases, thus triggering a fall in the production of gastric mucus secretion as well as an increased acid and pepsin secretion [77,79].
Selective COX-2 inhibitors have been thoroughly researched through the years. Many compounds are available on the market, and the class of drugs known as Coxibs holds a certain importance [80,81]. Heart attacks and other cardiac problems have resulted from these agents.
The significant side effects associated with traditional NSAIDs and some COX-2-selective inhibitor consumption is still prompting researchers to investigate new classes of molecules that may be good candidates as COX-2-selective inhibitors [82]. As a matter of fact, COX-2 remains one of the most investigated molecular targets.
The tertiary structure of COX-2 features three different domains [83]. The N-terminal region consists of the Epidermal Growth Factor (EGF) domain, the central section was identified as the Membrane Binding Domain (MBD), whilst the catalytic domain, which contains the two active sites (peroxidase (POX) and cyclooxygenase (COX)), folds into the globular shape comprising the C-terminal region [83,84,85].
The COX-1 active site is an L-shaped, 25 Ǻ long, hydrophobic channel. The COX-2 channel is 20% larger than that of COX-1, since Ile523 in the COX-1 active site is replaced by the smaller Val523 in COX-2. The lack of a methyl group on the amino acid 523 opens a sort of pocket on the side of the L-shaped channel, therefore making it a Y-shaped channel. This is why bulkier ligands can fit into the active site of COX-2 better than into that of COX-1. In both isoforms’ active sites, amino acids Tyr385 and Ser530 have a central role in binding the ligand [83,84,85].
Carvacrol is a phenolic monoterpene that can be retrieved from the extracts of several plants, such as Nigella sativa L. [86], Origanum vulgare L. [87], and Thymus vulgaris L. [88]. Several remedies in traditional medicines feature one of these plants for their activity in the treatment of inflammatory states and pain. Carvacrol was active in the inhibition of both COX-1 and COX-2 activity. Its efficacy in vitro was comparable to that of indomethacin. Nonetheless, the compound would be still ranked among the traditional NSAIDs. The small phenolic structure is obviously not a good choice for selective inhibition [89].
Similar studies about thymol and thymoquinones from Nigella sativa L. were reported by Marsik et al. According to the obtained data, thymol showed a better affinity for COX-1 despite being an excellent inhibitor of COX-2. The thymoquinones, on the other hand, inhibited COX-2 with a certain selectivity; dithymoquinone in particular showed very poor efficacy against COX-1. The scaffold of dithymoquinone is quite a bit bulkier than the other compounds, and this explains why it would hardly fit into the COX-1 channel [90]. Nigella sativa L. is quite known for its uses as an anti-inflammatory traditional remedy [86].
The aforementioned amarogentin was also the focus of an in silico evaluation as a possible ligand/inhibitor of both of the cyclooxygenase isoforms. In the study, the binding affinity of amarogentin against the enzymes was predicted along with the stability of the ligand–protein complex and the binding free energy. In all three experiments, amarogentin was a good candidate for the selective inhibition of COX-2; in fact, the COX-2–amarogentin complex demonstrated higher stability over time, and the gap in the comparison of the binding free energies (−52.35 Kcal/mol for COX-2 versus −8.57 Kcal/mol for COX-1) suggested an important potential in selectivity. After all, amarogentin is a secoiridoid glycoside with a large scaffold composed of an iridoid subunit joined to a trihydroxy-biphenyl carboxylic acid moiety by a glucose molecule, which allows amarogentin to bind in the cavity of COX-2 in a similar way to that of selective inhibitors [91].
Another computational approach was attempted on the outcome of a test performed on a subset of 16 iridoids isolated from Penstemon barbatus (Cav.) Roth, Castilleja tenuiflora Benth, Cresentia alata Kunth, and Vitex mollis Kunth. The four plants occur in the ancient tradition of Mexican folk medicine. The compounds’ subset was previously subjected to an in vitro screening against the enzymes. Among the tested compounds, loganic acid was the one to show an important efficacy in inhibiting COX-2 and only a mild effect against COX-1. Subsequently, the molecule was subjected to an in silico evaluation of its theoretical affinity that basically confirmed the biological assay outcomes. The same results provided an interesting prospect on how some factors seem to be relevant in the binding of monoterpenoids to COX-2, such as the presence and orientation of a methyl and a hydroxyl group and the presence of a carboxylic acid moiety [92].
A similar protocol was applied to evaluate the activity of H-harpagide, an iridoid monoterpene that is actually a metabolite obtained by the hydrolysis of Harpagide and Harpagoside in plants such as Scrophularia ningpoensis Hemsl. [93], Scrophularia buergeriana Miq. [94], and Harpagophytum procumbens (Burch.) DC. ex Meisn. [95] and catalyzed by β-glucosidase. The parent compounds did not show any anti-inflammatory activity. H-harpagide, on the other hand, was able to selectively inhibit COX-2 over COX-1, suggesting that the anti-inflammatory activity associated with the mentioned plants may be due to the presence of the active metabolite [96].
An innovative approach was attempted by Sun et al. in 2016. The team designed several compounds by merging NSAID scaffolds with methylated genipin. The obtained data showed how the conjugation of aspirin with genipin (to obtain a compound labeled as AGE) was likely the most promising approach to obtain selectivity. The inhibitory activity of AGE was hence tested against the enzymes, and the results confirmed the computational prediction (the IC50 against COX-2 was 0.36 M, whilst that against COX-1 was 9 M) [97].
7. Monoterpenoids Show a Wide Activity Spectrum in Modulating Several Receptor Subtypes of the TRPCC Family
There is always more evidence about the involvement of transient receptor potential cation channels (TRPCC) in the modulation of immediate and delayed inflammation and immune responses [98].
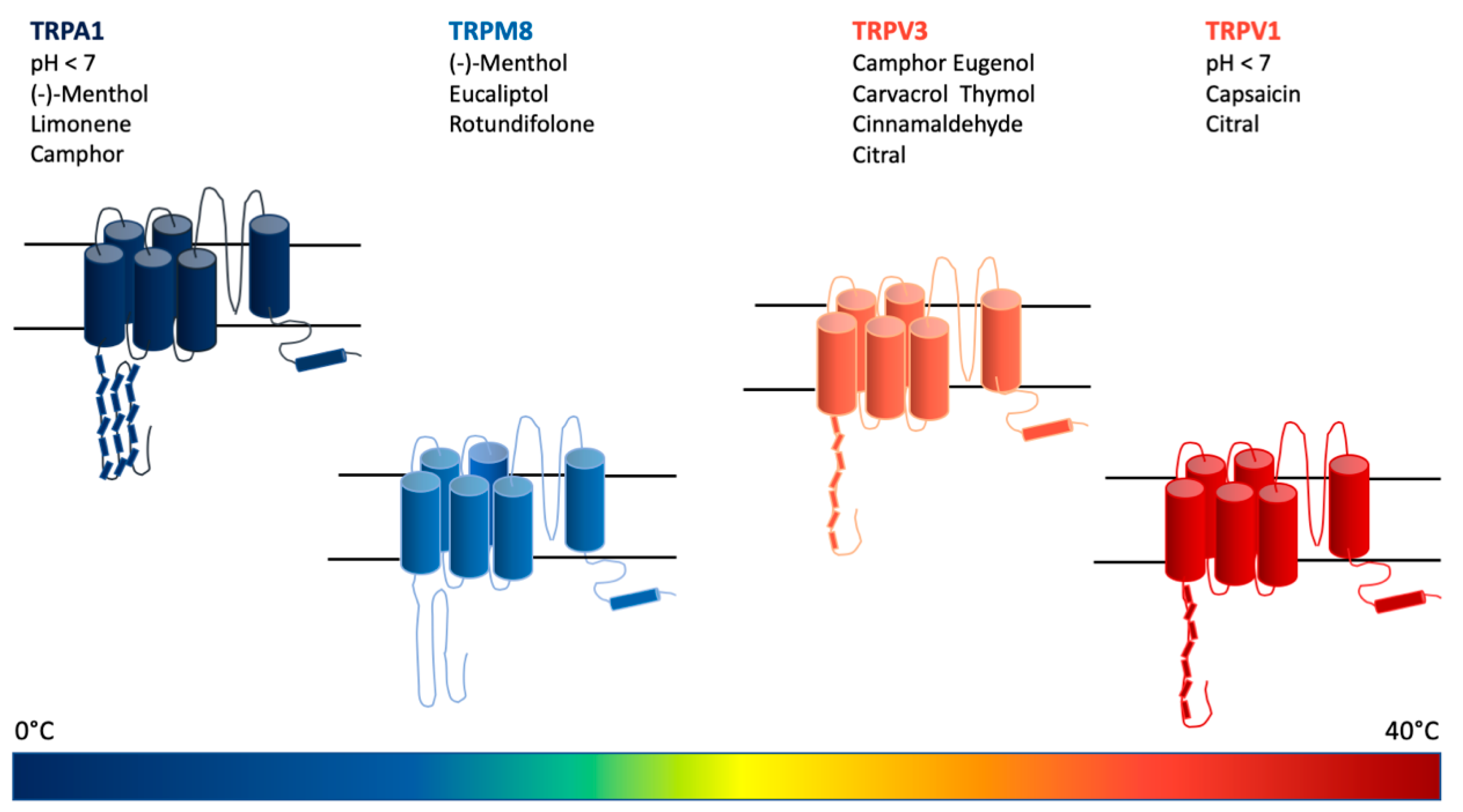
TRP channels are cation channels composed of four subunits gathered together around a central pore (Figure 5). Each subunit consists of six transmembrane domains [99]. Six sub-families of TRP channels have been discovered in mammals: Canonical TRP (TRPC), vanilloid TRP (TRPV), melastatin TRP (TRPM), ankyrin TRPA1, polycystic TRP (TRPP), and mucolipin TRP (TRPML) [100].
Generally, TRP channels are ubiquitous, as they are involved in the perception of either external or internal stimuli (thermosensation, mechanosensation, and perception of osmotic pressure, as well as nociception and other kinds of modifications of the surrounding environment), and they also respond to the stimulation induced by endogenous chemical mediators (G-proteins, diacylglycerol (DAG), etc.). Recently, several receptors of the TRP family have been identified as potential targets for the treatment of pain.
TRPV1 belongs to a family of non-selective cation channels. They are thermosensitive channels involved in the perception of “hot” stimuli and acidic pH. These channels are very responsive to activation by capsaicin, and they are distributed in both the central and peripherical nervous systems, as well as in keratinocytes [101]. TRPV1 seems to be involved in several pain states and is strictly associated with skin inflammatory disease [102,103]. A study performed in 2014 suggested that citral, which is a mixture of the monoterpenes geranial and neral, acts as a partial agonist on TRPV1 channels, and it is hence able to reduce noxious stimuli caused by other agonists [104].
TRPV3, on the other hand, is sensitive to warm temperatures and to a certain amount of chemicals, such as camphor and eugenol. The presence of these receptors in keratinocytes in addition to neuronal tissues was reported. This subtype of channel is subjected to a peculiar phenomenon known as sensitization. After a protracted stimulus, the receptor becomes more sensitive and the related responses are amplified. Camphor is a monoterpenoid found in the wood of Cinnamomum camphora (L.) J.Presl [105]. Its effects as a topical analgesic have been known for centuries. Camphor is an agonist of TRPV3 channels. The prolonged exposition of TRPV3 to camphor results in the desensitization of the receptor and in a diminished calcium influx. A study demonstrated that the TRPV1 receptors hold more than one site where agonists can bind. In the case of camphor, it seems that the molecule’s property of inducing desensitization is actually related to its site of interaction with the receptor [106].
A study led in 2007 revealed several monoterpenoids as more potent agonists of TRPV3 than camphor. The obtained results allowed the authors to draw a structure–activity relationship pattern concerning the binding of monoterpenoids to TRPV3 channels. Accordingly, the presence of ketone moieties on the test molecules diminished the activation profile when compared to hydroxyl-bearing compounds. Similarly, the lack of hydroxyl functions dramatically decreased the activation potential. The position of the hydroxyl moieties was also crucial, especially when it came to non-aromatic compounds [107].
TRPM8 channels are sodium- and calcium-permeable, and are activated by an ample range of cold temperatures. They are mainly expressed in the peripheral nervous system. Cool-inducing chemicals, such as menthol, are known agonists of these receptors. Their involvement in the inhibition of inflammatory pain is likely due to the cold sensation that they trigger that would hence simply soothe the pain as cold temperatures do. Nevertheless, some pain conditions such as cold hypersensitivity are likely associated with the induced hyperexpression or hyperstimulation of these receptors.
Eucalyptol is a monoterpenoid found in many species of the genus Eucalyptus. The compound is active in triggering ion currents mediated by TRPM8 channels, hence inducing the “cold sensation” typically mediated by this kind of receptor. As a result, eucalyptol seems to be able to reduce painful states induced by inflammatory conditions [108].
(-)-Menthol is a cool-inducing monoterpenoid mainly found in the leaves of Mentha arvensis L. [109]. The compound is probably the most acknowledged agonist of TRPM8. The analgesic potential related to menthol is strictly limited to “heat” pain, such as pain-inducing inflammatory states or burning and itching sensations. In the case of neuropathic pain or other kinds of cold-related painful states, the application of menthol did not show any improving effect, but rather worsened the conditions [110].
Rotundifolone is a monoterpenoid component from the extract of Mentha x villosa Huds. [111] whose activity on TRPM8 receptors was comparable to that of (-)-menthol. Interestingly, rotundifolone also showed a strict selectivity for this subtype of receptor [112].
In a study led in 2010, several synthetic derivatives of menthol were tested for their activation of TRPM8 channels. As a result, bulkier derivatives, particularly featuring large moieties built on the hydroxyl function, showed higher potency and selectivity in the activation of TRPM8 channels [113].
TRPA1 channels are mostly expressed in the nervous system and in keratinocytes [99]. They are voltage-dependent Ca2+ channels. Human channels are responsive to acidic pH and to obnoxious cold temperatures. The involvement of TRPA1 in several inflammatory pain conditions, such as neuropathic pain, headache, and osteoarthritis, has been reported [114]. Moreover, recent findings asserted that lipopolysaccharides (LPS) stimulates “irritation” via the activation of TRPA1 channels [115].
(-)-Menthol, (-)-limonene, and camphor have a peculiar effect on TRPA1 channels. (-)-Limonene is the S enantiomer of limonene, which is less common and occurs in plants from the Mentha genus [116]. At lower dosages, they activate the channel, hence inducing or enhancing a painful sensation. Interestingly, at higher concentrations, or after systemic administration in the case of limonene, they show anti-nociceptive activity, which is also mediated by the inhibition of TRPA1 channels [117,118,119]. Several mechanisms have been proposed, but the most accepted explanation is that, after an initial activation, the channels would shift into an “inactive” state that would be reversed into “active” after the wash-out of the agonist compounds.
Most of the aforementioned monoterpenoids have low selectivity for the several subtypes of the TRP family. Their antinociceptive and anti-inflammatory effects are likely triggered by a synergistic activity mediated by the activation/desensitization of several channels or by the induction/inhibition of multiple pathways [120].
8. Conclusions
The importance of chemokines in chronic pain is beginning to be recognized. Chemokines are produced in the skin during an initial painful event. Chemokines are involved in the enhancement of pain and inflammation in the skin. They increase prostaglandin production and IL-17 production and transactivate TRP channels. Prostaglandins and IL-17 increase chemokine production in the skin. This is the basis of the pain chemokine cycle that produces chronic pain.
Skin sensory neurons send signals to the brain that induce chemokine production in the brain [121]. Brain chemokines modulate the activities of descending neurons that release chemokines in the skin. Therefore, the brain is involved in perpetuating the pain chemokine cycle.
Monoterpenoids can inhibit the pain chemokine cycle at several points, as discussed above, and are clearly good candidates for the development of new drugs that are able to target TRP channels. At least four subtypes of TRP cation channels are involved in nociception. Skin TRP channels are critical to pain sensation. However, inhibition of spinal TRP channels by monoterpenoids such as thymol, carvacrol, and cineole may be important as well [122].
Inflammation can be involved in some chronic pain conditions, such as fibromyalgia [123]. Skin mast cells can be stimulated by cytokines released from macrophages in the skin. Mast cells then release inflammatory factors. It is not known if monoterpenoids can interrupt this macrophage mast cell inflammation in the skin.
As a general perspective, monoterpenoids have the ability to treat pain and produce long-term benefits with respect to chronic pain. Future research may lead to the development of more effective therapies, likely formulated as mixtures of different compounds, for the treatment of pain and chronic pain.
Author Contributions
Conceptualization, F.P., A.C. and J.D.A.; writing—original draft preparation, F.P., A.C. and J.D.A.; writing—review and editing, F.P., A.C. and J.D.A.; visualization, F.P., A.C. and J.D.A.; supervision, J.D.A. All authors have read and agree to the published version of the manuscript.
Funding
The present work was co-funded by the European Committee, FESR FSE 2014-2020, and by Regione Calabria. The author (A.C.) is the only one responsible for this publication, and the European Committee and Regione Calabria decline any responsibility concerning the use of the information disclosed in the paper.
Acknowledgments
The authors thank the Chumash Native Americans of California for teaching them the importance of topical pain relief with monoterpenoid mixtures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Premkumar, L.S. Transient receptor potential channels as targets for phytochemicals. ACS Chem. Neurosci. 2014, 5, 1117–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran, M.M.; McAlexander, M.A.; Bíró, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Wang, X. Control of pain with topical plant medicines. Asian Pacific J. Tropical Biomed. 2015, 5, 93–95. [Google Scholar] [CrossRef] [Green Version]
- Adams, J. The effects of yin, yang and qi in the skin on pain. Medicines 2016, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J. Chronic pain can it be cured? J. Pharmaceut. Drug Devel. 2017, 4, 105–109. [Google Scholar]
- Fernandes, E.; Vong, C.; Quek, S.; Cheong, J.; Awal, S.; Gentry, C.; Aubdool, A.; Liang, L.; Bodkin, J.; Bevan, S.; et al. Superoxide generation and leukocyte accumulation: Key elements in the mediation of leukotriene B ₄ -induced itch by transient receptor potential ankyrin 1 and transient receptor potential vanilloid 1. FASEB J. 2013, 27, 1664–1673. [Google Scholar] [CrossRef]
- Adams, J.D.; Guhr, S.; Villaseñor, E. Salvia mellifera—How Does It Alleviate Chronic Pain? Medicines (Basel) 2019, 6, 33. [Google Scholar]
- Adams, J. Chronic pain two cures. OBM Integ. Comp. Med. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, P.; Wong, V.; Williams, T.; Garcia, C.; Adams, J. Chemical Composition and Antinociceptive Activity of California Sagebrush (Artemisia californica). J. Pharmacog. Phytother. 2013, 5, 1–11. [Google Scholar]
- Roosterman, D.; Goerge, T.; Schneider, S.W.; Bunnett, N.W.; Steinhoff, M. Neuronal control of skin function: The skin as a neuroimmunoendocrine organ. Physiol. Rev. 2006, 86, 1309–1379. [Google Scholar] [CrossRef]
- Church, M.K.; Clough, G.F. Human skin mast cells: In vitro and in vivo studies. Ann. Allergy Asthma Immunol. 1999, 83, 471–475. [Google Scholar] [CrossRef]
- Burian, M.; Geisslinger, G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol. Ther. 2005, 107, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Moody, M.L. Topical medications in the treatment of pain. Pain Med. News 2010, 15–31. Available online: http://www.nationalsalesforce.org/wp-content/uploads/2014/08/Topical-Medications-in-the-Treatment-of-Pain.pdf (accessed on 27 May 2020).
- Heyneman, C.A.; Lawless-Liday, C.; Wall, G.C. Oral versus topical NSAIDs in rheumatic diseases: A comparison. Drugs 2000, 60, 555–574. [Google Scholar] [CrossRef]
- Prausnitz, M.R.; Mitragotri, S.; Langer, R. Current status and future potential of transdermal drug delivery. Nat. Rev. Drug Discov. 2004, 3, 115–124. [Google Scholar] [CrossRef]
- Stanos, S.P.; Galluzzi, K.E. Topical therapies in the management of chronic pain. Postgrad. Med. 2013, 125, 25–33. [Google Scholar] [CrossRef]
- Subramanian, N.; Ghosal, S.K.; Moulik, S.P. Topical delivery of celecoxib using microemulsion. Acta Pol. Pharm. 2004, 61, 335–341. [Google Scholar]
- Williams, A.C.; Barry, B.W. The enhancement index concept applied to terpene penetration enhancers for human skin and model lipophilic (oestradiol) and hydrophilic (Sfluorouracil) drugs. Int. J. Pharmaceut. 1991, 74, 157–168. [Google Scholar] [CrossRef]
- Chen, J.; Jiang, Q.D.; Chai, Y.P.; Zhang, H.; Peng, P.; Yang, X.X. Natural Terpenes as Penetration Enhancers for Transdermal Drug Delivery. Molecules 2016, 21, 1709. [Google Scholar] [CrossRef] [Green Version]
- Sapra, B.; Jain, S.; Tiwary, A.K. Percutaneous permeation enhancement by terpenes: Mechanistic view. AAPS J. 2008, 10, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Aqil, M.; Ahad, A.; Sultana, Y.; Ali, A. Status of terpenes as skin penetration enhancers. Drug Discov. Today 2007, 12, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Schaefer, U.; Sporer, F.; Reichling, J. Comparative study on the in vitro human skin permeation of monoterpenes and phenylpropanoids applied in rose oil and in form of neat single compounds. Pharmazie 2010, 65, 102–105. [Google Scholar] [PubMed]
- Magnussona, B.M.; Runn, P.; Karlsson, K.; Koskinen, L.O.D. Terpenes and ethanol enhance the transdermal permeation of the tripeptide thyrotropin releasing hormone in human epidermis. Int. J. Pharmaceutics. 1997, 157, 113–121. [Google Scholar] [CrossRef]
- Cal, K. Skin penetration of terpenes from essential oils and topical vehicles. Planta Med. 2006, 72, 311–316. [Google Scholar] [CrossRef]
- Shereen, A.; Yousef, E.A.; Pastore, M.N.; Telaprolu, K.; Mohammed, Y.H.; Namjoshi, S.; Grice, J.E.; Roberts, M.S. Skin models for the testing of transdermal drugs. Clin. Pharmacol. 2016, 8, 163–167. [Google Scholar]
- Davis, E.M. Advances in the Enzymology of Monoterpene Cyclization Reactions. Compr. Nat. Prod. II 2010, 1, 585–608. [Google Scholar]
- Tchimene, M.K.; Okunji, C.O.; Iwu, M.M.; Kuete, V. Monoterpenes and Related Compounds from the Medicinal Plants of Africa. In Medicinal Plant Research in Africa; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1–32. [Google Scholar]
- Guimarães, A.G.; Quintans, J.S.; Quintans, L.J. Monoterpenes with analgesic activity—A systematic review. Phytother. Res. 2013, 27, 1–15. [Google Scholar] [CrossRef]
- De Cássia da Silveira e Sá, R.; Andrade, L.N.; de Sousa, D.P. A review on anti-inflammatory activity of monoterpenes. Molecules 2013, 18, 1227–1254. [Google Scholar] [CrossRef]
- Gouveia, D.N.; Pina, L.T.S.; Rabelo, T.K.; da Rocha Santos, W.B.; Quintans, J.S.S.; Guimaraes, A.G. Monoterpenes as Perspective to Chronic Pain Management: A Systematic Review. Curr. Drug Targets 2018, 19, 960–972. [Google Scholar] [CrossRef]
- Passos, F.F.; Lopes, E.M.; de Araújo, J.M.; de Sousa, D.P.; Veras, L.M.; Leite, J.R.; Almeida, F.R. Involvement of Cholinergic and Opioid System in γ-Terpinene-Mediated Antinociception. Evid. Based Complement. Alternat. Med. 2015, 2015, 829414. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Lehtonen, M.; Suuronen, T.; Kaarniranta, K.; Huuskonen, J. Terpenoids: Natural inhibitors of NF-kappaB signaling with anti-inflammatory and anticancer potential. Cell Mol. Life Sci. 2008, 65, 2979–2999. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Yang, Y.J.; Li, Y.S.; Zhang, W.K.; Tang, H.B. α-Pinene, linalool, and 1-octanol contribute to the topical anti-inflammatory and analgesic activities of frankincense by inhibiting COX-2. J. Ethnopharmacol. 2016, 179, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.H.; Chang, Y.C.; Jean, Y.H. Excitatory amino acid glutamate: Role in peripheral nociceptive transduction and inflammation in experimental and clinical osteoarthritis. Osteoarthr. Cartil. 2015, 23, 2009–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, N.S.; Houghton, P.J.; Theobald, A.; Jenner, P.; Perry, E.K. In-vitro inhibition of human erythrocyte acetylcholinesterase by salvia lavandulaefolia essential oil and constituent terpenes. J. Pharm. Pharmacol. 2000, 52, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef]
- Zhang, X.; Angkasekwinai, P.; Dong, C.; Tang, H. Structure and function of interleukin-17 family cytokines. Protein. Cell 2011, 2, 26–40. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.J.; Gaffen, S.L. Interleukin-17: A novel inflammatory cytokine that bridges innate and adaptive immunity. Front. Biosci. 2008, 13, 170–177. [Google Scholar] [CrossRef]
- Shalom-Barak, T.; Quach, J.; Lotz, M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J. Biol. Chem. 1998, 273, 27467–27473. [Google Scholar] [CrossRef] [Green Version]
- Lotz, M.; Blanco, F.J.; von Kempis, J.; Dudler, J.; Maier, R.; Villiger, P.M.; Geng, Y. Cytokine regulation of chondrocyte functions. J. Rheumatol. Suppl. 1995, 43, 104–108. [Google Scholar]
- Faour, W.H.; Mancini, A.; He, Q.W.; Di Battista, J.A. T-cell-derived interleukin-17 regulates the level and stability of cyclooxygenase-2 (COX-2) mRNA through restricted activation of the p38 mitogen-activated protein kinase cascade: Role of distal sequences in the 3’-untranslated region of COX-2 mRNA. J. Biol. Chem. 2003, 278, 26897–26907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulissen, S.M.; van Hamburg, J.P.; Davelaar, N.; Asmawidjaja, P.S.; Hazes, J.M.; Lubberts, E. Synovial fibroblasts directly induce Th17 pathogenicity via the cyclooxygenase/prostaglandin E2 pathway, independent of IL-23. J. Immunol. 2013, 191, 1364–1372. [Google Scholar] [CrossRef] [Green Version]
- Boniface, K.; Bak-Jensen, K.S.; Li, Y.; Blumenschein, W.M.; McGeachy, M.J.; McClanahan, T.K.; McKenzie, B.S.; Kastelein, R.A.; Cua, D.J.; de Waal Malefyt, R. Prostaglandin E2 regulates Th17 cell diff erentiation and function through cyclicAMP and EP2/EP4 receptor signaling. J. Exp. Med. 2008, 206, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, C.; Sakata, D.; Esaki, Y.; Li, Y.; Matsuoka, T.; Kuroiwa, K.; Sugimoto, Y.; Narumiya, S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 2009, 15, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witowski, J.; Ksiazek, K.; Jorres, A. Interleukin-17: A mediator of inflammatory responses. Cell. Molec. Life Sci. 2004, 61, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Nadiv, O.; Beer, Y. Interleukin-17 Enhances Tumor Necrosis Factor a–Induced Synthesis of Interleukins 1, 6, and 8 in Skin and Synovial Fibroblasts. Arthritis Rheum. 2001, 44, 2176–2184. [Google Scholar] [CrossRef]
- Stamp, L.K.; Cleland, L.G.; James, M.J. Upregulation of Synoviocyte COX-2 Through Interactions with T Lymphocytes: Role of Interleukin 17 and Tumor Necrosis Factor-α. J. Rheumatol. 2004, 31, 146–1254. [Google Scholar]
- Moseley, T.T.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Van Beelen, A.J.; Teunissen, M.B.; Kapsenberg, M.L.; de Jong, E.C. Interleukin-17 in inflammatory skin disorders. Curr. Opin. Allergy Clin. Immunol. 2007, 7, 374–381. [Google Scholar] [CrossRef]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef]
- Shabgah, A.G.; Fattahi, E.; Shahneh, F.Z. Interleukin-17 in human inflammatory diseases. Postepy Dermatol. Alergol. 2014, 31, 256–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakura, Y.; Nakae, S.; Saijo, S.; Ishigame, H. The roles of IL-17A in inflammatory immune responses and host defense against pathogens. Immunol. Rev. 2008, 226, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Dumont, F.J. IL-17 cytokine/receptor families: Emerging targets for the modulation of inflammatory responses. Expert Opin. Ther. Pat. 2003, 13, 287–303. [Google Scholar] [CrossRef]
- Botelho, M.A.; Nogueira, N.A.; Bastos, G.M.; Fonseca, S.G.; Lemos, T.L.; Matos, F.J.; Montenegro, D.; Heukelbach, J.; Rao, V.S.; Brito, G.A. Antimicrobial activity of the essential oil from Lippia sidoides, carvacrol and thymol against oral pathogens. Braz. J. Med. Biol. Res. 2007, 40, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Games, E.; Guerreiro, M.; Santana, F.R.; Pinheiro, N.M.; de Oliveira, E.A.; Lopes, F.D.; Olivo, C.R.; Tibério, I.F.; Martins, M.A.; Lago, J.H.; et al. Structurally Related Monoterpenes p-Cymene, Carvacrol and Thymol Isolated from Essential Oil from Leaves of Lippia sidoides Cham. (Verbenaceae) Protect Mice against Elastase-Induced Emphysema. Molecules 2016, 21, 1390. [Google Scholar] [CrossRef] [Green Version]
- Omar, S.H. Oleuropein in olive and its pharmacological effects. Sci. Pharm. 2010, 78, 133–154. [Google Scholar] [CrossRef] [Green Version]
- De Bock, M.; Hodgkinson, S.; Cutfield, W.; Schlothauer, R. Methods and Uses of an Extract from Olive Leaf in Management of Type 2 Diabetes. U.S. Patent WO2014038962A1, 13 August 2015. [Google Scholar]
- Qabaha, K.; Al-Rimawi, F.; Qasem, A.; Naser, S.A. Oleuropein Is Responsible for the Major Anti-Inflammatory Effects of Olive Leaf Extract. J. Med. Food 2018, 21, 302–305. [Google Scholar] [CrossRef]
- Larussa, T.; Oliverio, M.; Suraci, E.; Greco, M.; Placida, R.; Gervasi, S.; Marasco, R.; Imeneo, M.; Paolino, D.; Tucci, L.; et al. Oleuropein Decreases Cyclooxygenase-2 and Interleukin-17 Expression and Attenuates Inflammatory Damage in Colonic Samples from Ulcerative Colitis Patients. Nutrients 2017, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Citová, I.; Ganzera, M.; Stuppner, H.; Solich, P. Determination of gentisin, isogentisin, and amarogentin in Gentiana lutea L. by capillary electrophoresis. J. Sep. Sci. 2008, 31, 195–200. [Google Scholar] [CrossRef]
- Ray, S.; Majumder, H.K.; Chakravarty, A.K.; Mukhopadhyay, S.; Gil, R.R.; Cordell, G.A. Amarogentin, a naturally occurring secoiridoid glycoside and a newly recognized inhibitor of topoisomerase I from Leishmania donovani. J. Nat. Prod. 1996, 59, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Wölfle, U.; Haarhaus, B.; Schempp, C.M. Amarogentin Displays Immunomodulatory Effects in Human Mast Cells and Keratinocytes. Mediat. Inflamm. 2015, 2015, 630128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, T.; Taguchi, H. The Constituents of Gardenia jasminoides Geniposide and Genipin-gentiobioside. Chem. Pharm. Bull. 1973, 21, 2684–2688. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.X.; Du, C.T.; Chen, W.; Lei, Q.Q.; Li, N.; Qi, S.; Zhang, X.J.; Hu, G.Q.; Deng, X.M.; Han, W.Y.; et al. Genipin inhibits NLRP3 and NLRC4 inflammasome activation via autophagy suppression. Sci. Rep. 2015, 5, 17935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.T.; He, J.L.; Li, W.M.; Yang, Z.; Wang, Y.X.; Yin, J.; Du, Y.G.; Yu, C. Geniposide inhibits interleukin-6 and interleukin-8 production in lipopolysaccharide-induced human umbilical vein endothelial cells by blocking p38 and ERK1/2 signaling pathways. Inflamm. Res. 2010, 59, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.M.; Wu, H.; Li, H.; Chen, J.; Chen, J.Y.; Hu, S.L.; Shen, C. Effects and mechanisms of Geniposide on rats with adjuvant arthritis. Int. Immunopharmacol. 2014, 20, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, J.; Wang, P.; Xue, Y. Paeoniflorin attenuates Aβ1-42-induced inflammation and chemotaxis of microglia in vitro and inhibits NF-κB- and VEGF/Flt-1 signaling pathways. Brain Res. 2015, 1618, 149–158. [Google Scholar] [CrossRef]
- Liu, C.; Cheng, Z.; Wang, Y.; Dai, X.; Zhang, J.; Xue, D. Paeoniflorin exerts a nephroprotective effect on concanavalin A-induced damage through inhibition of macrophage infiltration. Diagn. Pathol. 2015, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.G.; Lin, J.L.; Niu, Q.X.; Wang, H.M.; Zhou, Y.C.; Chen, S.Y.; Liang, G.W. Paeoniflorin diminishes ConA-induced IL-8 production in primary human hepatic sinusoidal endothelial cells in the involvement of ERK1/2 and Akt phosphorylation. Int. J. Biochem. Cell Biol. 2015, 62, 93–100. [Google Scholar] [CrossRef]
- Chen, T.; Guo, Z.; Jiao, X.; Jia, B.; Zhang, Y.; Li, J.; Huang, X.; Liu, H. Peoniflorin suppresses tumor necrosis factor-α induced chemokine production in human dermal microvascular endothelial cells by blocking nuclear factor-κB and ERK pathway. Arch. Dermatol. Res. 2011, 303, 351–360. [Google Scholar] [CrossRef]
- Dai, X.; Wang, L.; Jia, X.; Chang, Y.; Wu, H.; Wang, C.; Wei, W. Paeoniflorin regulates the function of human peripheral blood mononuclear cells stimulated by rhIL-1β by up-regulating Treg expression. Immunopharmacol. Immunotox. 2015, 37, 252–257. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, L.; Wang, J.; Wang, C.; Yang, Z.; Zhu, Y.; Zhang, J. Paeoniflorin and Albiflorin Attenuate Neuropathic Pain via MAPK Pathway in Chronic Constriction Injury Rats. Evid. Based Complement. Alternat. Med. 2016, 2016, 8082753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Khoo, C.S.; Hennell, J.R.; Pearson, J.L.; Jarouche, M.; Halstead, C.W.; Bensoussan, A. LC determination of albiflorin and paeoniflorin in Bai Shao (Paeonia lactiflora) as a raw herb and dried aqueous extract. J. AOAC Int. 2009, 92, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Xu, H.; Wu, J.; Qu, C.; Sun, F.; Xu, S. Linalool inhibits cigarette smoke-induced lung inflammation by inhibiting NF-κB activation. Int. Immunopharmacol. 2015, 29, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Randhawa, V.; Kumar Singh, A.; Acharya, V. A systematic approach to prioritize drug targets using machine learning, a molecular descriptor-based classification model, and high-throughput screening of plant derived molecules: A case study in oral cancer. Mol. Biosyst. 2015, 11, 3362–3377. [Google Scholar] [CrossRef]
- Brunton, L.; Chubner, B.; Knollmann, B. Goodman & Gilman’s The Pharmacological Basis Of Therapeutics, 12th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Funk, C.D. Prostaglandins and leukotrienes: Advances in eicosanoid biology. Science 2001, 294, 1871–1875. [Google Scholar] [CrossRef] [Green Version]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenses 1 and 2. Ann. Rev. Pharmacol. Tox. 1998, 38, 97–120. [Google Scholar] [CrossRef]
- Bollettino d’Informazione Sui Farmaci. 2000, Lug-Ago. Available online: http://www.quadernidellasalute.it/imgs/C_17_pubblicazioni_217_allegato.pdf (accessed on 27 May 2020).
- Chen, L.C.; Ashcroft, D.M. Risk of myocardial infarction associated with selective COX-2 inhibitors: Meta-analysis of randomised controlled trials. Pharmacoepidemiol. Drug Saf. 2007, 16, 762–772. [Google Scholar] [CrossRef]
- Coricello, A.; El-Magboub, A.; Luna, M.; Ferrario, A.; Haworth, I.S.; Gomer, C.J.; Aiello, F.; Adams, J.D. Rational drug design and synthesis of new α-Santonin derivatives as potential COX-2 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 993–996. [Google Scholar] [CrossRef]
- Garavito, R.; Mulichak, A. The structure of mammalian cyclooxygneases. Ann. Rev. Biophys. Biomolecular Structures. 2003, 32, 183–206. [Google Scholar] [CrossRef]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazdair, M.R. The Protective Effects of Nigella sativa and Its Constituents on Induced Neurotoxicity. J. Toxicol. 2015, 2015, 841823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirino, I.C.; Menezes-Silva, S.M.; Silva, H.T.; de Souza, E.L.; Siqueira-Júnior, J.P. The Essential Oil from Origanum vulgare L. and Its Individual Constituents Carvacrol and Thymol Enhance the Effect of Tetracycline against Staphylococcus aureus. Chemotherapy 2014, 60, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Fachini-Queiroz, F.C.; Kummer, R.; Estevão-Silva, C.F.; Carvalho, M.D.; Cunha, J.M.; Grespan, R.; Bersani-Amado, C.A.; Cuman, R.K. Effects of Thymol and Carvacrol, Constituents of Thymus vulgaris L. Essential Oil, on the Inflammatory Response. Evid. Based Complement. Alternat. Med. 2012, 2012, 657026. [Google Scholar] [CrossRef] [Green Version]
- Landa, P.; Kokoska, L.; Pribylova, M.; Vanek, T.; Marsik, P. In vitro Anti-inflammatory Activity of Carvacrol: Inhibitory Effect on COX-2 Catalyzed Prostaglandin E2 Biosynthesis. Arch. Pharmaceut. Res. 2009, 32, 75–78. [Google Scholar] [CrossRef]
- Marsik, P.; Kokoska, L.; Landa, P.; Nepovim, A.; Soudek, P.; Vanek, T. In vitro inhibitory effects of thymol and quinones of Nigella sativa seeds on cyclooxygenase-1- and -2-catalyzed prostaglandin E2 biosyntheses. Planta. Med. 2005, 71, 739–742. [Google Scholar] [CrossRef]
- Shukla, S.; Bafna, K.; Sundar, D.; Thorat, S.S. The bitter barricading of prostaglandin biosynthesis pathway: Understanding the molecular mechanism of selective cyclooxygenase-2 inhibition by amarogentin, a secoiridoid glycoside from Swertia chirayita. PLoS ONE 2014, 9, e90637. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Cisneros, M.; Rios, M.Y.; Aguilar-Guadarrama, A.B.; Rao, P.P.; Aburto-Amar, R.; Rodríguez-López, V. In vitro COX-1 and COX-2 enzyme inhibitory activities of iridoids from Penstemon barbatus, Castilleja tenuiflora, Cresentia alata and Vitex mollis. Bioorg. Med. Chem. Lett. 2015, 25, 4505–4508. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, S.; Gao, W.; Zhu, D. Iridoid glycosides from Scrophularia ningpoensis. Phytochem 1999, 50, 101–104. [Google Scholar] [CrossRef]
- Lin, S.; Jiang, S.; Li, Y.; Zeng, J.; Zhu, D. Two novel iridoids from Scrophularia buergeriana. Tet. Lett. 2000, 41, 1069–1071. [Google Scholar] [CrossRef]
- Qi, J.; Chen, J.J.; Cheng, Z.H.; Zhou, J.H.; Yu, B.Y.; Qiu, S.X. Iridoid glycosides from Harpagophytum procumbens D.C. (devil’s claw). Phytochemistry 2006, 67, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Feng, L.; Jia, Q.; Xu, J.; Wang, R.; Wang, Z.; Wu, Y.; Li, Y. Effects of b-glucosidase hydrolyzed products of harpagide and harpagoside on cyclooxygenase-2 (COX-2) in vitro. Bioorg. Med. Chem 2011, 19, 4882–4886. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Wu, H.; Zhang, Y.Z.; Wang, R.; Wang, W.Y.; Wang, W.; Li, S.P.; Dai, L.; Zhang, Z.R. Design, synthesis and preliminary evaluation of the anti-inflammatory of the specific selective targeting druggable enzymome cyclooxygenase-2 (COX-2) small molecule. Pharm. Biol. 2016, 54, 2505–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parenti, A.; De Logu, F.; Geppetti, P.; Benemei, S. What is the evidence for the role of TRP channels in inflammatory and immune cells? Br. J. Pharmacol. 2016, 173, 953–969. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Mickle, A.D.; Shepherd, A.J.; Mohapatra, D.P. Sensory TRP channels: The key transducers of nociception and pain. Prog. Mol. Biol. Transl. Sci. 2015, 131, 73–118. [Google Scholar]
- Southall, M.D.; Li, T.; Gharibova, L.S.; Pei, Y.; Nicol, G.D.; Travers, J.B. Activation of epidermal vanilloid receptor-1 induces release of proinflammatory mediators in human keratinocytes. J. Pharmacol. Exp. Ther. 2003, 304, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Biro, T.; Acs, G.; Acs, P.; Modarres, S.; Blumberg, P.M. Recent advances in understanding of vanilloid receptors: A therapeutic target for treatment of pain and inflammation in skin. J. Investig. Dermatol. Symp. Proc. 1997, 2, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, C.M.; Ganev, E.G.; Mazzardo-Martins, L.; Martins, D.F.; Rocha, L.R.; Santos, A.R.; Hiruma-Lima, C.A. Citral: A monoterpene with prophylactic and therapeutic anti-nociceptive effects in experimental models of acute and chronic pain. Eur. J. Pharmacol. 2014, 736, 16–25. [Google Scholar] [CrossRef]
- Hamidpour, R.; Hamidpour, S.; Hamidpour, M.; Shahlari, M. Camphor (Cinnamomum camphora), a traditional remedy with the history of treating several diseases. IJCRI 2013, 4, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Sherkheli, M.A.; Vogt-Eisele, A.K.; Weber, K.; Hatt, H. Camphor modulates TRPV3 cation channels activity by interacting with critical pore-region cysteine residues. Pak. J. Pharm. Sci. 2013, 26, 431–438. [Google Scholar] [PubMed]
- Vogt-Eisele, A.K.; Weber, K.; Sherkheli, M.A.; Vielhaber, G.; Panten, J.; Gisselmann, G.; Hatt, H. Monoterpenoid agonists of TRPV3. Br. J. Pharmacol. 2007, 151, 530–540. [Google Scholar] [CrossRef] [Green Version]
- Caceres, A.I.; Liu, B.; Jabba, S.V.; Achanta, S.; Morris, J.B.; Jordt, S.E. Transient Receptor Potential Cation Channel Subfamily M Member 8 channels mediate the anti-inflammatory effects of eucalyptol. Br. J. Pharmacol. 2017, 174, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Rahman, L.; Verma, R.; Chauhan, A.; Yadav, A.; Singh, A. Essential Oil Composition of Menthol Mint (Mentha arvensis) and Peppermint (Mentha piperita) Cultivars at Different Stages of Plant Growth from Kumaon Region of Western Himalaya. Open Access J. Med. Aromatic Plants. 2010, 1, 13–18. [Google Scholar]
- Kamatou, G.P.; Vermaak, I.; Viljoen, A.M.; Lawrence, B.M. Menthol: A simple monoterpene with remarkable biological properties. Phytochemistry 2013, 96, 15–25. [Google Scholar] [CrossRef]
- Amaral, R.G.; Fonseca, C.S.; Silva, T.K.; Andrade, L.N.; França, M.E.; Barbosa-Filho, J.M.; de Sousa, D.P.; Moraes, M.O.; Pessoa, C.; Carvalho, A.A.; et al. Evaluation of the cytotoxic and antitumour effects of the essential oil from Mentha x villosa and its main compound, rotundifolone. J. Pharm. Pharmacol. 2015, 67, 1100–1106. [Google Scholar] [CrossRef]
- Silva, D.F.; de Almeida, M.M.; Chaves, C.G.; Braz, A.L.; Gomes, M.A.; Pinho-da-Silva, L.; Pesquero, J.L.; Andrade, V.A.; Leite, M.e.F.; de Albuquerque, J.G.; et al. TRPM8 Channel Activation Induced by Monoterpenoid Rotundifolone Underlies Mesenteric Artery Relaxation. PLoS ONE 2015, 10, e0143171. [Google Scholar] [CrossRef] [Green Version]
- Sherkheli, M.A.; Vogt-Eisele, A.K.; Bura, D.; Beltrán Márques, L.R.; Gisselmann, G.; Hatt, H. Characterization of selective TRPM8 ligands and their structure activity response (S.A.R) relationship. J. Pharm. Pharm. Sci. 2010, 13, 242–253. [Google Scholar] [CrossRef] [Green Version]
- Skerratt, S. Recent Progress in the Discovery and Development of TRPA1 Modulators. Prog. Med. Chem. 2017, 56, 81–115. [Google Scholar]
- Meseguer, V.; Alpizar, Y.A.; Luis, E.; Tajada, S.; Denliger, B.; Fajardo, O.; Manenschijn, F.P.; Talavera, A.; Kichko, T.; Navia, B.; et al. TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, G.; Gershenzon, J.; Nielson, E.E.; Froehlich, J.E.; Croteau, R. Limonene synthase, the enzyme responsible for monoterpene biosynthesis in peppermint, is localized to leucoplasts of oil gland secretory cells. Plant Physiol. 1999, 120, 879–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, B.; Dubin, A.; Bursulaya, B.; Viswanath, V.; Jegla, T.; Patapoutian, A. Identification of the Transmembrane Domain Five as a Critical Molecular Determinant of Menthol Sensitivity in Mammalian TRPA1 Channels. J. Neurosci. 2008, 24, 9640–9651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpizar, Y.; Gees, M.; Sanchez, A.; Apetrei, A.; Voets, T.; Nilius, B.; Talavera, K. Bimodal effects of cinnamaldehyde and camphor on mouse TRPA1. Eur. J. Physiol. 2013, 465, 853–864. [Google Scholar] [CrossRef]
- Kaimoto, T.; Hatakeyama, Y.; Tkahashi, K.; Imagawa, T.; Tominaga, M.; Ohta, T. Involvement of transient receptor potential A1 channel in algesic and analgesic actions of the organic compound limonene. Eur. J. Pain 2016, 20, 1155–1165. [Google Scholar] [CrossRef]
- Oz, M.; Lozon, Y.; Sultan, A.; Yang, K.H.; Galadari, S. Effects of monoterpenes on ion channels of excitable cells. Pharmacol. Ther. 2015, 152, 83–97. [Google Scholar] [CrossRef]
- Mélik Parsadaniantz, S.; Rivat, C.; Rostène, W.; Réaux-Le Goazigo, A. Opioid and chemokine receptor crosstalk: A promising target for pain therapy? Nat. Rev. Neurosci. 2015, 16, 69–78. [Google Scholar] [CrossRef]
- Kumamoto, E.; Fujita, T. Differential Activation of TRP Channels in the Adult Rat Spinal Substantia Gelatinosa by Stereoisomers of Plant-Derived Chemicals. Pharmaceuticals 2016, 9, 46. [Google Scholar] [CrossRef]
- Mastrangelo, F.; Frydas, I.; Ronconi, G.; Kritas, S.K.; Tettamanti, L.; Caraffa, A.; Ovidio, D.C.; Younes, A.; Gallenga, C.E.; Conti, P. Low-grade chronic inflammation mediated by mast cells in fibromyalgia: Role of IL-37. J. Biol. Regul. Homeost Agents. 2018, 32, 195–198. [Google Scholar]
Figure 1.
Molecular mechanism of nociception and peripheral sensitization mediated by prostaglandins. Prostaglandins (PGs) (purple circles) from induced COX-2 in macrophages and constitutive COX-1 activate PGs receptors (purple tetragons), triggering the pain stimulus (blue arrows). The activation of the PG-receptor-dependent cascade (black arrow) also results in the activation of PKA and PKC, which modulate (gray arrows) the permeability of ion channels (TRPV1: Red; TTX-R Na+: Green; K+: Magenta) and the sensitivity of Bradykinin receptors (blue tetragons).
Figure 1.
Molecular mechanism of nociception and peripheral sensitization mediated by prostaglandins. Prostaglandins (PGs) (purple circles) from induced COX-2 in macrophages and constitutive COX-1 activate PGs receptors (purple tetragons), triggering the pain stimulus (blue arrows). The activation of the PG-receptor-dependent cascade (black arrow) also results in the activation of PKA and PKC, which modulate (gray arrows) the permeability of ion channels (TRPV1: Red; TTX-R Na+: Green; K+: Magenta) and the sensitivity of Bradykinin receptors (blue tetragons).

Figure 2.
Th17 cell (green)–macrophage (purple) proinflammatory loop.

Figure 3.
Schematic representation of cyclooxygenase involvement in physiological and inflammatory pathways. Inflammatory stimuli induce the up-regulation of COX-2 via the NF-κB pathway in injured tissues. The increase of prostaglandins above their constitutive level produces inflammation and pain.
Figure 3.
Schematic representation of cyclooxygenase involvement in physiological and inflammatory pathways. Inflammatory stimuli induce the up-regulation of COX-2 via the NF-κB pathway in injured tissues. The increase of prostaglandins above their constitutive level produces inflammation and pain.

Figure 4.
Structures of the described monoterpenoids.


Figure 5.
Schematic summary of the discussed substances and temperature spectrum effective in the activation/desensitization of transient receptor potential (TRP) cation channels involved in the mechanisms of nociception and pain.
Figure 5.
Schematic summary of the discussed substances and temperature spectrum effective in the activation/desensitization of transient receptor potential (TRP) cation channels involved in the mechanisms of nociception and pain.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Perri, F.; Coricello, A.; Adams, J.D. Monoterpenoids: The Next Frontier in the Treatment of Chronic Pain? J 2020, 3, 195-214. https://0-doi-org.brum.beds.ac.uk/10.3390/j3020016
AMA Style
Perri F, Coricello A, Adams JD. Monoterpenoids: The Next Frontier in the Treatment of Chronic Pain? J. 2020; 3(2):195-214. https://0-doi-org.brum.beds.ac.uk/10.3390/j3020016
Chicago/Turabian StylePerri, Filomena, Adriana Coricello, and James D. Adams. 2020. "Monoterpenoids: The Next Frontier in the Treatment of Chronic Pain?" J 3, no. 2: 195-214. https://0-doi-org.brum.beds.ac.uk/10.3390/j3020016