Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study


1
Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, China
2
School of Physics, University of Chinese Academy of Sciences, Beijing 100049, China
3
CAS Center for Excellence in Topological Quantum Computation, Beijing 100190, China
4
Songshan Lake Materials Laboratory, Dongguan 523803, China
*
Author to whom correspondence should be addressed.
Surfaces 2021, 4(1), 31-38; https://0-doi-org.brum.beds.ac.uk/10.3390/surfaces4010005
Submission received: 30 December 2020
/
Revised: 16 January 2021
/
Accepted: 19 January 2021
/
Published: 22 January 2021
Abstract
:Single-molecular devices show remarkable potential for applications in downscale electronic devices. The adsorption behavior of a molecule on a metal surface is of great importance from both fundamental and technological points of view. Herein, based on first-principles calculations, the adsorption of a 4,4″-diamino-p-terphenyl (DAT) molecule on a Cu(001) surface has been systematically explored. The most stable configuration is the DAT molecule lying flat with a rotation angle of 13° relative to the [100] surface direction. It was found that the adsorption sites of benzene rings and nitrogen atoms in the DAT molecule have important influences on the stability of the adsorption configuration. Electron density differences analysis shows that the electrons accumulate at the DAT-Cu(001) interface. The density of states projected on a DAT molecule of DAT/Cu(001) exhibits a metallic character, while the freestanding ones are semiconducting, indicating a strong interaction between the DAT molecule and the Cu(001) surface in the most stable adsorption configuration. These results provide useful information for tuning the properties and functions of DAT molecules, and may offer useful insights for other organic molecule/surface systems.
1. Introduction
Molecule–metal hybrid systems have attracted much attention in modern surface science and technology for their potential applications in heterogeneous catalysis, molecular electronics, photovoltaics, and light-emitting diodes [1,2,3,4,5,6,7,8]. The hybrid systems are very sensitive to the adsorption configuration, the stability of the molecules on surfaces, and the physical or chemical interactions between the molecules and the substrate. Hence, it is key to figure out the adsorption behavior in order to design novel nanodevices at the single-molecule scale. However, the precise chemical structure and orientation of 3D twisted/bending molecules on a surface are difficult to be visualized by using standard experimental techniques, such as atomic force microscopy and scanning tunneling microscopy [9,10]. First principles calculations based on density functional theory are playing an increasingly important role in describing molecule–metal hybrid systems—not only the adsorption configurations and the interactions between molecules and surfaces, but also potential reaction pathways [6,11,12].
Among various metal surfaces, the Cu(001) surface is very active with great potential for strong interactions with molecules [13,14,15,16,17,18,19,20,21,22,23]. The simple yet elegant 4,4″-diamino-p-terphenyl (DAT) molecule is composed of three non-planar benzene rings and two amino groups, which is small but complete for evaluating the absorption mechanism of a 3D amino-functionalized organic molecule. In addition, it represents one of the most promising and versatile classes owing to its unique properties as a monomer of a conjugated polymer, polyazomethine, and has been applied to polymer light-emitting diodes by vapor deposition polymerization [24].
The adsorption of a single DAT molecule and the DAT molecules’ assembly behavior on substrates have received extensive research attention for a few decades [25,26,27,28,29,30,31,32,33]. A DAT molecule exhibits symmetric adsorption geometries on a Au(111) surface, and asymmetric on a Cu(111) surface resulting from the lattice mismatch and the interaction between the DAT molecule and the Cu(111)surface [25]. The amino group in the DAT molecule prefers to chemically bond with a Si(111) surface or a Si(001) surface [26,27,28]. The self-assembly of the DAT molecule and its co-assembly with other organic molecules are temperature-dependent [30,31,32,33]. However, to our knowledge, studies focused on a DAT/Cu(001) system are rare. Therefore, it is worthwhile to investigate the DAT/Cu(001) system by using theoretical methods, which could provide a better understanding of the mechanisms and features of 3D amino-functionalized organic molecules on metal surfaces. This work was designed to explore the adsorption mechanism of a DAT molecule on a Cu(001) surface.
In this article, based on first-principles calculations, the adsorption behavior of a DAT molecule on a Cu(001) surface has been investigated extensively, considering different adsorption sites and various rotation angles relative to the [100] direction. It was found that the adsorption site of the DAT molecule plays an important role in the adsorption configuration and its stability. It turns out that the DAT molecule prefers lying flat on the Cu(001) surface with a rotation angle around 13° relative to the [100] direction. Moreover, the calculated electron density difference and density of states show a strong interaction with a large electron accumulation between the DAT molecule and the Cu(001) surface. These results provide a comprehensive understanding for molecular adsorptions on metal surfaces.
2. Computational Methods
The first-principles calculations are based on the density functional theory (DFT) with the projector augmented wave (PAW) method [34] implemented in the Vienna ab initio simulation package (VASP) code [35]. The generalized gradient approximation (GGA) developed by Perdew–Burke–Ernzerhof (PBE) [36] was adopted for the exchange-correlation function for the structural relaxation. A plane-wave basis set with an energy cutoff of 400 eV is used. The Brillouin zone is sampled using a 1 × 1 × 1 Gamma-centered Monkhorst-Pack k-point grid. The Cu(001) surfaces are modeled by a periodic 3-layer 7 × 5 slab. The lattice constants are 24.94 Å, 17.82 Å, and 22.03 Å, respectively. To eliminate spurious interaction between two adjacent slabs, a vacuum layer of thickness larger than 15 Å is applied. Throughout the calculations, the bottom Cu layer is fixed, while the two top Cu layers and the DAT molecule are fully relaxed until the force for each atom is less than 0.02 eV/Å and the total energy is converged to 10−5 eV. All the calculations reported here were performed with van der Waals (vdW) corrections based on the DFT-D2 method of Grimme [37].
In order to find the most stable adsorption configuration of a DAT molecule on the Cu(001) surface, the center of the DAT molecule is initially located on different adsorption sites with different rotation angles. The typical adsorption sites are considered, namely, top, hollow, and bridge sites, as shown in Figure 1a. The initial rotation angle of a DAT molecule relative to the [100] direction is also considered as 0°, 15°, 30°, or 45°, respectively. In each case, a freestanding DAT molecule is set with an initial vertical distance of 3.00 Å from the Cu surface. The adsorption energy of a DAT molecule on a Cu(001) surface is estimated using the following equation:
where , , and are the total energies of the DAT/Cu(001) system, the isolated freestanding DAT molecule, and the Cu(001) surface, respectively.
3. Results and Discussion
3.1. Adsorption Configurations
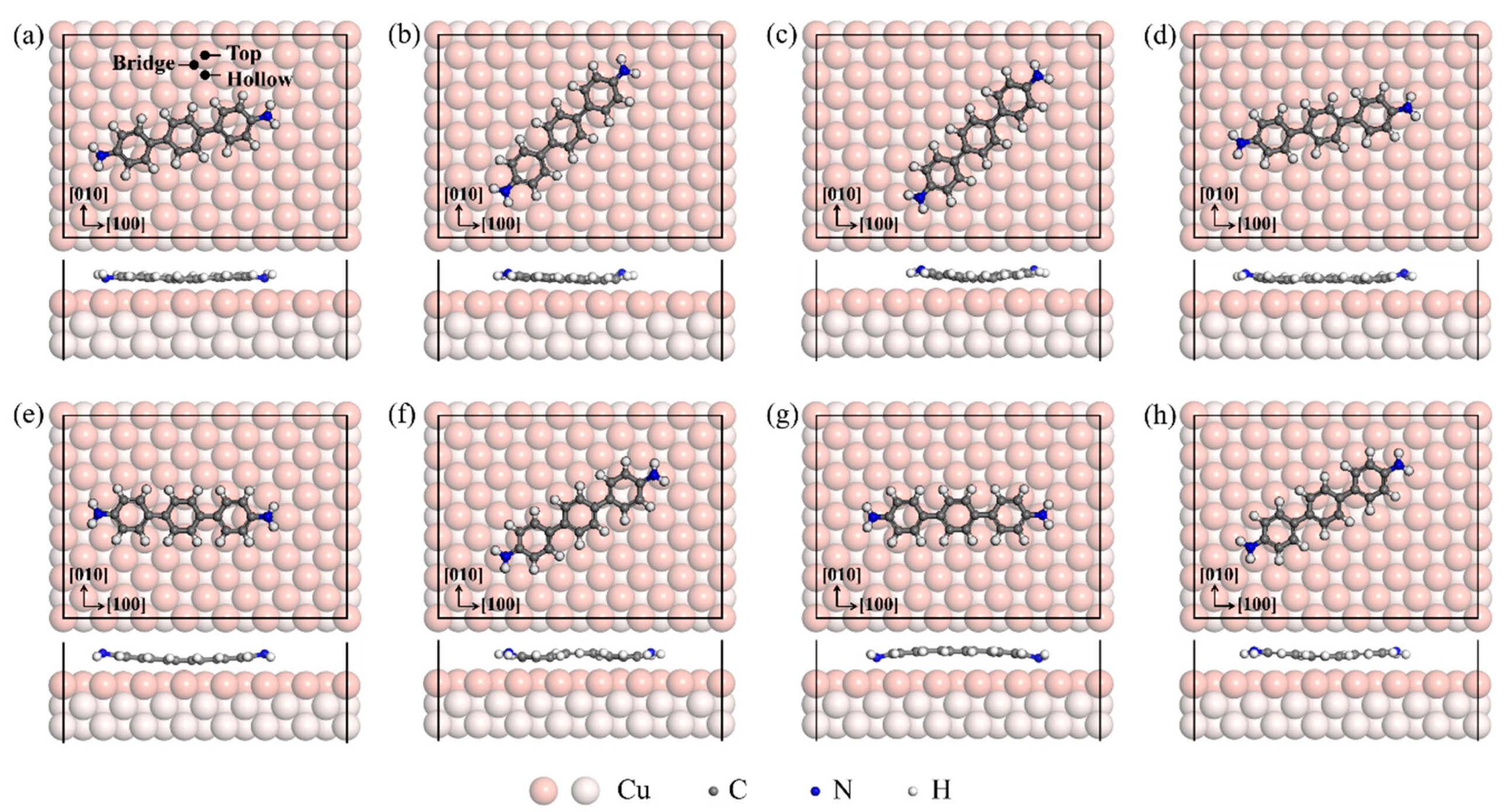
The DAT molecule shows various possible adsorption configurations on a Cu(001) surface, presumably due to adsorption-induced conformational changes. The optimized configurations with different rotation angles from the most stable configuration to the least stable one are given in Figure 1, which are named configurations a–h respectively. The corresponding adsorption energies and rotation angles are summarized in Table 1. We found that the benzene ring position relative to the surface is a key factor that influences the adsorption configuration. Figure 1 shows clearly that the benzene ring in the DAT molecule prefers to approach to the surface when its center adsorbs on the hollow site, whereas the benzene ring opts to stay away from the surface when located on the top site. Another key factor affecting the adsorption configuration of the DAT/Cu(001) system is the position of the nitrogen atom. The N atom on a top site tends to be close to the surface, while those at other sites relate to a further distance. The competition for the interactions between the three benzene rings and the substrate, together with that between the two nitrogen atoms and the substrate, results in a DAT/Cu(001) system with abundant adsorption configurations (planar (Figure 1a), upward-arched (Figure 1f,g), counter-arched (Figure 1c–e), and asymmetric (Figure 1b)).
The most stable (i.e., lowest-energy) adsorption configuration of the DAT/Cu(001) system is shown in Figure 1a, configuration a. For configuration a, the three benzene rings are adsorbed on one hollow and two bridge sites, and two N atoms both sit on the top sites. All atoms in the DAT molecule are almost in a horizontal plane. The DAT molecule has a rotation angle of 13.2° relative to the [100] direction of the substrate. The shortest distance between C and Cu atoms of configuration a is 2.19 Å, while that between N and Cu atoms is 2.20 Å. The adsorption energy of this configuration is −3.871 eV, showing a high stability (Table 1).
Figure 1b shows that the DAT molecule is asymmetrically adsorbed on the Cu(001) surface. Although there are almost two benzene rings locating on hollow sites, configuration b is the second-lowest-energy adsorption configuration since the two N atoms are away from the top sites. The DAT molecule has a rotation angle of 45.5° relative to the [100] direction. The shortest distance between C and Cu atoms of configuration b is 2.20 Å. Configuration c is a symmetric structure, with the center benzene ring adsorbed on the hollow site and the two N atoms close to the hollow sites (see Figure 1c). By comparing configurations b and c, it can be found that the asymmetric adsorption offers a more stable configuration.
Unlike the planar configuration of the configuration a, there are some other arched configurations due to the different adsorption sites for the benzene rings and the nitrogen atoms, such as a counter-arched configuration in configuration e and an upward-arched configuration in configuration g. The DAT molecules are largely arched and thus relatively metastable with relatively small adsorption energies of −3.364 and −3.587 eV on a Cu(001) surface (Table 1).
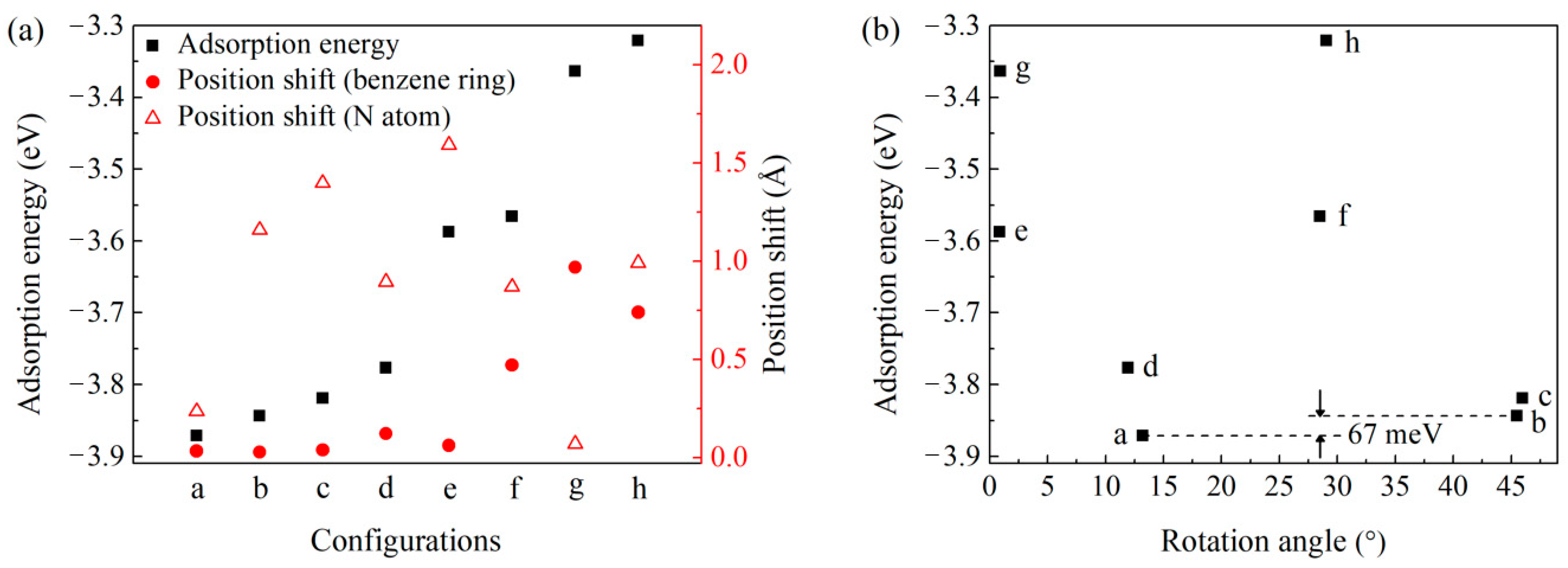
Figure 2a gives the correlations between the adsorption energies and the position shift between the DAT molecule and the Cu(001) surface. The adsorption energies (the black squares) and the smallest position shift between the center of a benzene ring and hollow sites (the red circle) exhibit a positive correlation, indicating an important effect on the stability of the adsorption configurations induced by the adsorption site of the benzene ring. Besides, an inverse correlation is observed for configurations e and h, due to the nonnegligible contribution from N atoms (the red triangles).
Based on the analysis of all the adsorption configurations, we conclude a simple rule to estimate the stability of a DAT molecule on a Cu(001) surface. The greater the number of benzene rings on hollow sites and that of nitrogen atoms on top sites, the more stable the adsorption configuration. When the rotation angle is around 13°, configuration a could satisfy the most points in the rule with one benzene ring on a hollow site and two nitrogen atoms on top sites. When the rotation angle is around 0° or 30°, most of the benzene rings and the N atoms are away from their favored adsorption sites (Figure 1e–h), resulting in adsorption configurations with higher adsorption energies (Figure 2b). It is worth noting that the energy of the configuration b with a rotation angle around 45° is only 67 meV higher than that of the most stable configuration (configuration a). Therefore, it can be expected that adsorption configurations with rotation angles around 15° and 45° coexist in the laboratory, and the rotation angles can be used as visual features in experiments by using surface characterization techniques.
3.2. Electronic Properties
Adsorption of the DAT molecule on the Cu(001) surface should induce an electron redistribution due to the interaction between the molecule and the substrate [25,26,27,28,29,30,31,32,33]. To quantitatively understand the adsorption behavior in the DAT/Cu(001) system, the electron density difference (EDD) has been calculated with the following equation:
where , and are the electron densities of the DAT/Cu(001) system, a DAT molecule, and a Cu(001) surface, respectively. Negative Δρ means electron loss, while positive Δρ means electron accumulation.
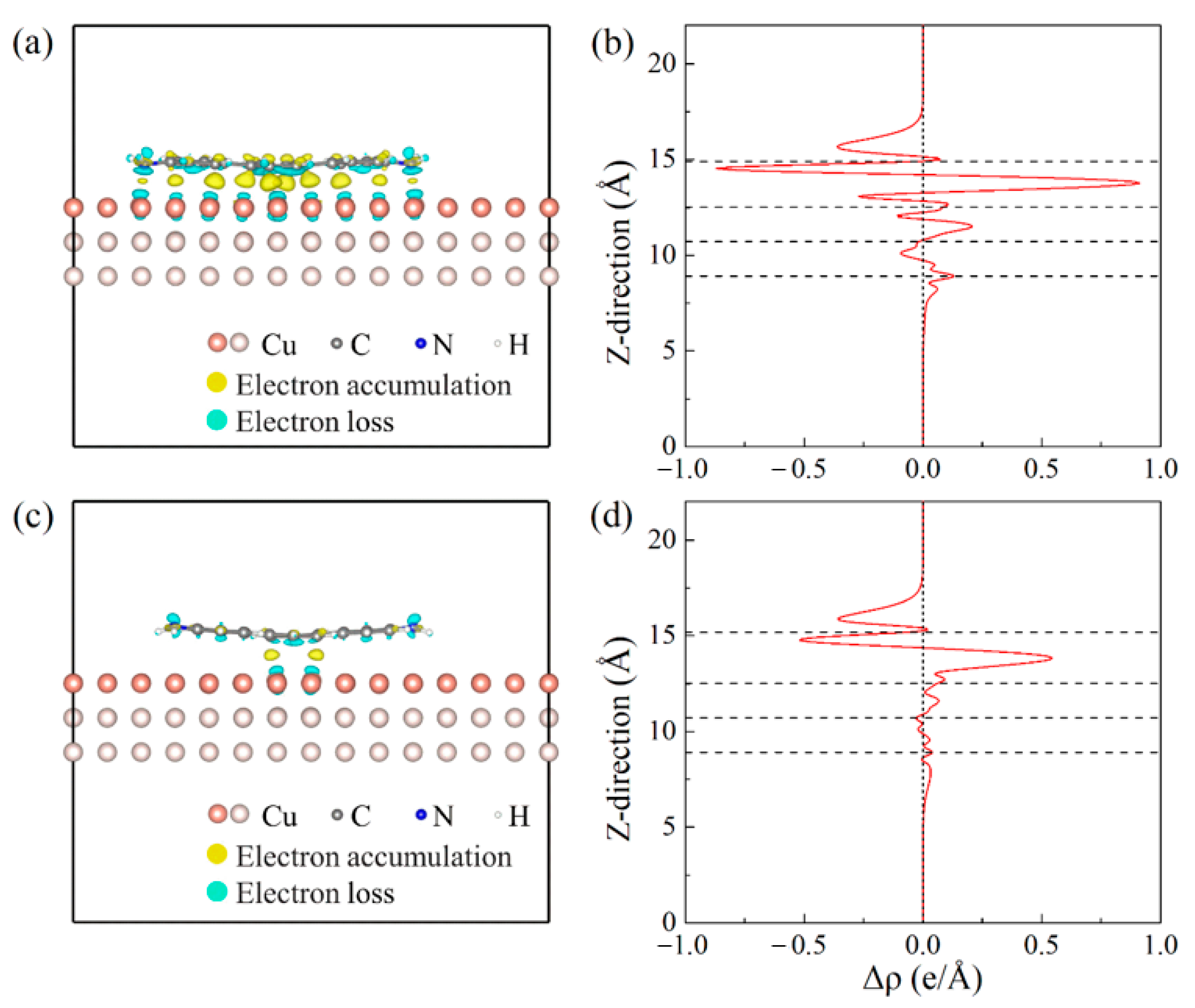
Configurations a and h are used as two examples here to analyze the interaction between the DAT molecule and the Cu(001) surface. One represents relatively strong interaction and the other one is for relatively weak interaction. The EDD maps of these two configurations have been plotted in the Figure 3a,c. There is an obvious electron accumulation at the interface between the DAT molecule and the Cu(001) surface in configuration a (Figure 3a). The corresponding integrated EDD in the z direction has been plotted in Figure 3b, showing a much obvious EDD oscillation from surface toward the inner Cu layers in configuration a (Figure 3b). Comparing the EDD and integrated EDD of configurations a and h (Figure 3), the interaction between the DAT molecule and the substrate in configuration a is stronger than that in configuration h. It also shows clearly that the electron transfer in the configuration a is larger than that in the configuration h.
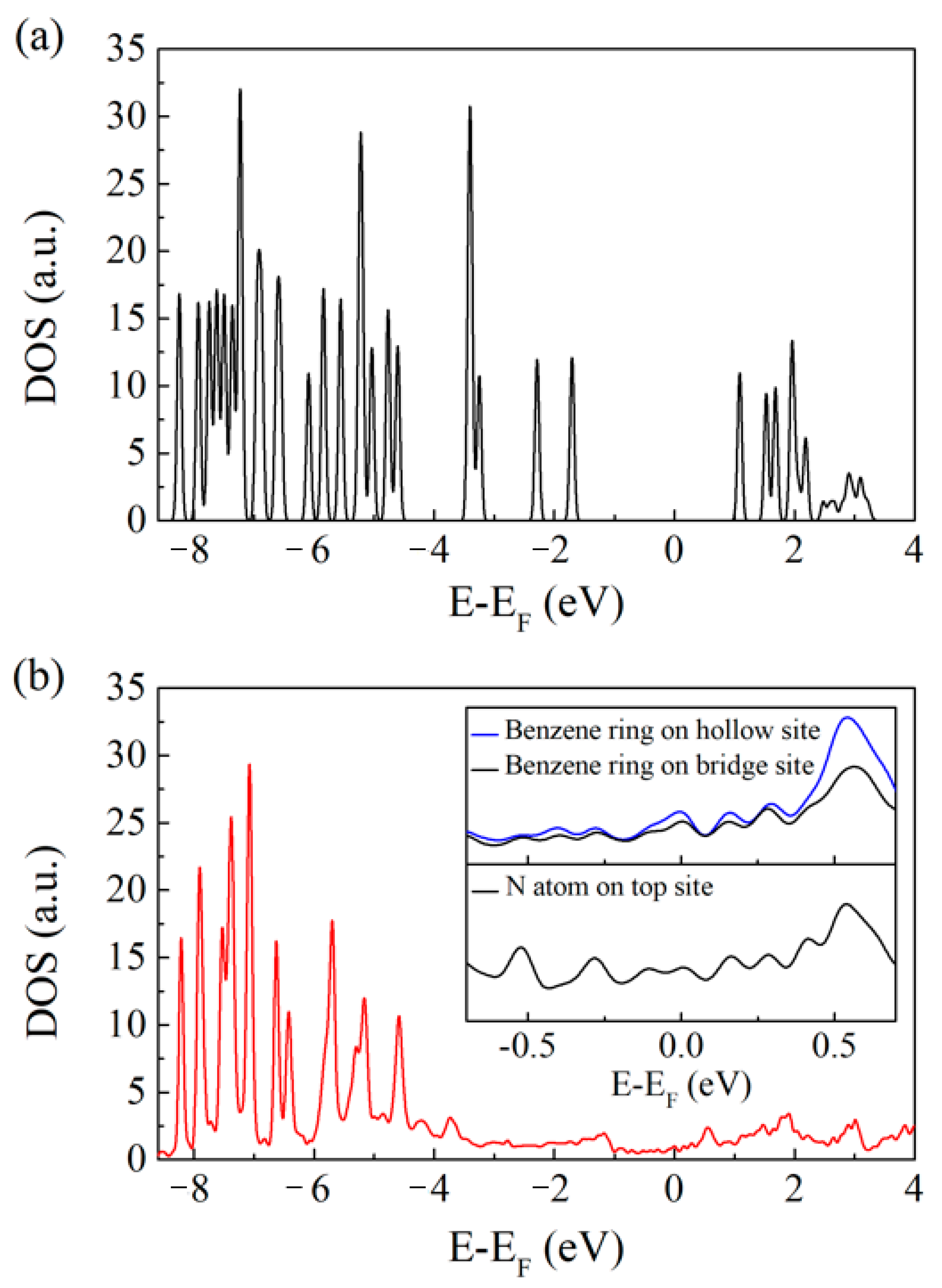
The electronic density of states (DOS) of a freestanding DAT molecule and the projected DOS of the configuration a have been plotted in Figure 4 to obtain further insights into the DAT/Cu(001) system. The DAT molecule on a Cu(001) surface has similar electronic states with a freestanding one at low energy levels. However, there is no electronic state around the Fermi level for a freestanding DAT molecule (Figure 4a), while the DOS projected on the DAT molecule adsorbed on a Cu(001) surface (see Figure 4b) exhibits a metallic character, showing a strong interaction between the DAT molecule and the Cu(001) surface. The inset figures in Figure 4b give the projected DOS around the Fermi level on benzene rings and a N atom of the configuration a. Comparing the projected DOS on benzene rings on different adsorption sites (the upper inset figure), the benzene ring on a hollow site has larger states than that on a bridge site around the Fermi level, indicating a stronger interaction with the Cu(001) surface. The lower inset figure shows the interaction between the N atom and the surface.
4. Conclusions
In conclusion, the adsorption of a 4,4″-diamino-p-terphenyl molecule on the Cu(001) surface has been investigated by first-principles calculations. A simple rule to check whether an adsorption configuration of the DAT/Cu(001) system is favored or not has been summarized. We found that the structural geometry and stability are strongly dependent on the adsorption sites of the benzene rings and the nitrogen atoms in the DAT molecule. The most stable configuration is the DAT molecule lying flat with a rotation angle of 13° relative to the [100] surface direction. Detailed electron density difference analysis shows that a large electron accumulation occurs between the DAT molecule and the Cu(001) surface in the most stable adsorption configuration. In addition, the calculated electronic density of states confirms the strong interaction between the DAT molecule and the Cu surface. These results provide useful information for molecules on Cu(001) surfaces, and may offer useful guidance for the stability of other organic molecules on metal surfaces.
Author Contributions
C.-T.W. performed the calculations under the guidance of S.D. and Y.-F.Z. The manuscript was written via contributions by all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (number 61888102).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available free of charge from the corresponding author.
Acknowledgments
All authors would like to thank De-Liang Bao for critical discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eremtchenko, M.; Schaefer, J.A.; Tautz, F.S. Understanding and tuning the epitaxy of large aromatic adsorbates by molecular design. Nature 2003, 425, 602–605. [Google Scholar] [CrossRef] [PubMed]
- Du, S.X.; Gao, H.J.; Seidel, C.; Tsetseris, L.; Ji, W.; Kopf, H.; Chi, L.F.; Fuchs, H.; Pennycook, S.J.; Pantelides, S.T. Selective Nontemplated Adsorption of Organic Molecules on Nanofacets and the Role of Bonding Patterns. Phys. Rev. Lett. 2006, 97, 156105. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.J.; Gao, L. Scanning tunneling microscopy of functional nanostructures on solid surfaces: Manipulation, self-assembly, and applications. Prog. Surf. Sci. 2010, 85, 28–91. [Google Scholar] [CrossRef]
- Vuillaume, D. Molecular Nanoelectronics. Proc. IEEE 2010, 98, 2111–2123. [Google Scholar]
- Gao, L.; Liu, Q.; Zhang, Y.Y.; Jiang, N.; Zhang, H.G.; Cheng, Z.H.; Qiu, W.F.; Du, S.X.; Liu, Y.Q.; Hofer, W.A.; et al. Constructing an Array of Anchored Single-Molecule Rotors on Gold Surfaces. Phys. Rev. Lett. 2008, 101, 197209. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Zhang, Y.-Y.; Pantelides, S.T.; Du, S. Tuning the Catalytic Activity of a Quantum Nutcracker for Hydrogen Dissociation. Surfaces 2020, 3, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Yan, L.; Bao, D.-L.; Ren, J.; Du, S.; Wang, Y.; Huan, Q.; Gao, H.-J. Self-Assembly Evolution of Metal-Free Naphthalocyanine Molecules on Ag(111) at the Submonolayer Coverage. J. Phys. Chem. C 2019, 123, 7202–7208. [Google Scholar] [CrossRef]
- Lu, H.; Wenlong, E.; Ma, Z.; Yang, X. Organometallic polymers synthesized from prochiral molecules by a surface-assisted synthesis on Ag(111). Phys. Chem. Chem. Phys. 2020, 22, 8141–8145. [Google Scholar] [CrossRef]
- Martin-Jimenez, D.; Ahles, S.; Mollenhauer, D.; Wegner, H.A.; Schirmeisen, A.; Ebeling, D. Bond-Level Imaging of the 3D Conformation of Adsorbed Organic Molecules Using Atomic Force Microscopy with Simultaneous Tunneling Feedback. Phys. Rev. Lett. 2019, 122, 196101. [Google Scholar] [CrossRef] [Green Version]
- Ebeling, D.; Zhong, Q.; Schlöder, T.; Tschakert, J.; Henkel, P.; Ahles, S.; Chi, L.; Mollenhauer, D.; Wegner, H.A.; Schirmeisen, A. Adsorption Structure of Mono- and Diradicals on a Cu(111) Surface: Chemoselective Dehalogenation of 4-Bromo-3″-iodo-p-terphenyl. ACS Nano 2019, 13, 324–336. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Q.; Cai, K.; Li, J.; Li, Y.; Yang, X.; Zhang, Y.; Wang, Y.; Tang, H.; Zhao, D.; et al. Stepwise on-surface dissymmetric reaction to construct binodal organometallic network. Nat. Commun. 2019, 10, 2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telychko, M.; Su, J.; Gallardo, A.; Gu, Y.; Mendieta-Moreno, J.I.; Qi, D.; Tadich, A.; Song, S.; Lyu, P.; Qiu, Z.; et al. Strain-Induced Isomerization in One-Dimensional Metal–Organic Chains. Angew. Chem. Int. Ed. 2019, 58, 18591–18597. [Google Scholar] [CrossRef] [PubMed]
- Oreshkin, A.I.; Muzychenko, D.A.; Oreshkin, S.I.; Yakovlev, V.A.; Murugan, P.; Chandrasekaran, S.S.; Kumar, V.; Bakhtizin, R.Z. Real-Time decay of fluorinated fullerene molecules on Cu(001) surface controlled by initial coverage. Nano Res. 2018, 11, 2069–2082. [Google Scholar] [CrossRef]
- Ienaga, K.; Miyamachi, T.; Takahashi, Y.; Kawamura, N.; Komori, F. Enhanced periodic modulation of electronic states in a hexagonal iron-nitride monolayer on Cu(001) via interfacial interaction. Phys. Rev. B 2017, 96, 085439. [Google Scholar] [CrossRef]
- Bahlke, M.P.; Karolak, M.; Herrmann, C. Interplay between strong correlation and adsorption distances: Co on Cu(001). Phys. Rev. B 2018, 97, 035119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhao, Y.; Wang, C.; Wei, Z.; Yang, J.; Ma, J. Zn-Doped Cu(100) facet with efficient catalytic ability for the CO2 electroreduction to ethylene. Phys. Chem. Chem. Phys. 2019, 21, 21341–21348. [Google Scholar] [CrossRef]
- Dokukin, S.A.; Kolesnikov, S.V.; Saletsky, A.M.; Klavsyuk, A.L. Diffusion-Mediated processes in Pt/Cu(001) surface alloy. Surf. Sci. 2020, 692, 121515. [Google Scholar] [CrossRef]
- Dou, W.-D.; Zhang, H.-J.; Bao, S.-N. Scanning tunneling microscopy study of surface reconstruction induced by N adsorption on Cu (100) surface. Chin. Phys. B 2010, 19, 026803. [Google Scholar]
- Benlattar, M.; Elkoraychy, E.; Sbiaai, K.; Mazroui, M.; Boughaleb, Y. Ehrlich-Schwöbel barriers and adsorption of Au, Cu and Ag stepped (100) surfaces. Mod. Phys. Lett. B 2017, 31, 1750037. [Google Scholar] [CrossRef]
- Chen, S.; Sun, S.; Lian, B.; Ma, Y.; Yan, Y.; Hu, S. The adsorption and dissociation of H2S on Cu(100) surface: A DTF study. Surf. Sci. 2014, 620, 51–58. [Google Scholar] [CrossRef]
- Robledo, M.; Díaz-Tendero, S. Exploring the Adsorption and the Potential Energy Surface of Acrylonitrile on Cu(100) and Cu(100) Coated with NaCl Layers. J. Phys. Chem. C 2015, 119, 15125–15136. [Google Scholar] [CrossRef]
- Guo, Q.; Qin, Z.; Huang, M.; Mantsevich, V.N.; Cao, G. Image potential states mediated STM imaging of cobalt phthalocyanine on NaCl/Cu(100). Chin. Phys. B 2016, 25, 036801. [Google Scholar] [CrossRef]
- Eren, B.; Weatherup, R.S.; Liakakos, N.; Somorjai, G.A.; Salmeron, M. Dissociative Carbon Dioxide Adsorption and Morphological Changes on Cu(100) and Cu(111) at Ambient Pressures. J. Am. Chem. Soc. 2016, 138, 8207–8211. [Google Scholar] [CrossRef] [PubMed]
- Itabashi, A.; Fukushima, M.; Murata, H. Multi-Layer Polymer Light-Emitting Diodes Prepared by Vapor Deposition Polymerization of Polyazomethine Thin Film. Jpn. J. Appl. Phys. 2008, 47, 1271–1275. [Google Scholar] [CrossRef]
- Zhong, Q.; Ebeling, D.; Tschakert, J.; Gao, Y.; Bao, D.; Du, S.; Li, C.; Chi, L.; Schirmeisen, A. Symmetry breakdown of 4,4″-diamino-p-terphenyl on a Cu(111) surface by lattice mismatch. Nat. Commun. 2018, 9, 3277. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Itabashi, A.; Sasahara, A.; Murata, H.; Arai, T.; Tomitori, M. Adsorption State of 4,4″-Diamino-p-terphenyl through an Amino Group Bound to Si(111)-7 × 7 Surface Examined by X-ray Photoelectron Spectroscopy and Scanning Tunneling Microscopy. J. Phys. Chem. C 2010, 114, 11109–11114. [Google Scholar] [CrossRef]
- Nishimura, T.; Sasahara, A.; Murata, H.; Arai, T.; Tomitori, M. Thermal Transformation of 4,4″-Diamino-p-terphenyl on a Si(111)-7 × 7 Surface Analyzed by X-ray Photoemission Spectroscopy and Scanning Tunneling Microscopy. J. Phys. Chem. C 2014, 118, 25104–25109. [Google Scholar] [CrossRef]
- Hassan, A.M.A.; Nishimura, T.; Sasahara, A.; Murata, H.; Tomitori, M. Stable alignment of 4,4″-diamino-p-terphenyl chemically adsorbed on a Si(001)-(2 × 1) surface observed by scanning tunneling microscopy. Surf. Sci. 2014, 630, 96–100. [Google Scholar] [CrossRef]
- Ren, J.; Bao, D.-L.; Dong, L.; Gao, L.; Wu, R.; Yan, L.; Wang, A.; Yan, J.; Wang, Y.; Du, S.-X.; et al. Thermo-controllable self-assembled structures of single-layer 4, 4″-diamino-p-terphenyl molecules on Au(110). Chin. Phys. B 2017, 26, 086801. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.-T.; Pignedoli, C.A.; Treier, M.; Fasel, R.; Passerone, D. The role of van der Waals interactions in surface-supported supramolecular networks. Phys. Chem. Chem. Phys. 2010, 12, 992–999. [Google Scholar] [CrossRef]
- Treier, M.; Nguyen, M.-T.; Richardson, N.V.; Pignedoli, C.; Passerone, D.; Fasel, R. Tailoring Low-Dimensional Organic Semiconductor Nanostructures. Nano Lett. 2009, 9, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Ding, J.; Yang, B.; Zhang, J.; Peng, C.; Lin, H.; Zhang, H.; Li, Q.; Chi, L. Deprotonation-Induced Phase Evolutions in Co-Assembled Molecular Structures. Langmuir 2018, 34, 7852–7858. [Google Scholar] [CrossRef] [PubMed]
- Rauls, E.; Blankenburg, S.; Schmidt, W.G. Chemical reactivity on surfaces: Modeling the imide synthesis from DATP and PTCDA on Au(111). Phys. Rev. B 2010, 81, 125401. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
Figure 1.
The top and side views of the optimized configurations for DAT/Cu(001) systems from the most stable configuration (a) to the least stable one (h). The rotation angles are (a) 13.2°, (b) 45.5°, (c) 46.0°, (d) 11.9°, (e) 0.9°, (f) 28.5°, (g) 0.9°, (h) 29.0°, relative to the [100] direction, respectively. The unit cell is marked by black lines. The [100] and [010] directions of the Cu slab are marked as [100] and [010]. The orange and light orange balls represent the top and two bottom layers of Cu atoms, respectively. The gray, blue, and white balls represent the C, N, and H atoms, respectively. The top, hollow, and bridge sites are shown in Figure 1a.
Figure 1.
The top and side views of the optimized configurations for DAT/Cu(001) systems from the most stable configuration (a) to the least stable one (h). The rotation angles are (a) 13.2°, (b) 45.5°, (c) 46.0°, (d) 11.9°, (e) 0.9°, (f) 28.5°, (g) 0.9°, (h) 29.0°, relative to the [100] direction, respectively. The unit cell is marked by black lines. The [100] and [010] directions of the Cu slab are marked as [100] and [010]. The orange and light orange balls represent the top and two bottom layers of Cu atoms, respectively. The gray, blue, and white balls represent the C, N, and H atoms, respectively. The top, hollow, and bridge sites are shown in Figure 1a.

Figure 2.
The adsorption energies of configurations a to h. (a) The adsorption energies and the position shifts between the DAT molecule and the Cu(001) surface. The black square is the adsorption energy. The red circle and the red triangle represent the smallest position shift between the center of a benzene ring and hollow sites, and that between a N atom and top sites, respectively. (b) The adsorption energies distribution at different rotation angles ranging from 0° to 45°. The detailed structural configurations of a to h are given in Figure 1.
Figure 2.
The adsorption energies of configurations a to h. (a) The adsorption energies and the position shifts between the DAT molecule and the Cu(001) surface. The black square is the adsorption energy. The red circle and the red triangle represent the smallest position shift between the center of a benzene ring and hollow sites, and that between a N atom and top sites, respectively. (b) The adsorption energies distribution at different rotation angles ranging from 0° to 45°. The detailed structural configurations of a to h are given in Figure 1.

Figure 3.
(a,c) The EDD maps of configurations a and h with iso-surfaces of Δρ = ±0.027 e/Å3. The yellow regions indicate the electron accumulation, while blue regions indicate the electron loss. (b,d) The integrated electron density difference figures of configurations a and h in z direction. The electron transfer in the configuration a is larger than in the configuration h. The black dashed lines represent the relative positions of the DAT molecule and three Cu layers in the z direction. Negative Δρ means electron loss, while positive Δρ means electron accumulation.
Figure 3.
(a,c) The EDD maps of configurations a and h with iso-surfaces of Δρ = ±0.027 e/Å3. The yellow regions indicate the electron accumulation, while blue regions indicate the electron loss. (b,d) The integrated electron density difference figures of configurations a and h in z direction. The electron transfer in the configuration a is larger than in the configuration h. The black dashed lines represent the relative positions of the DAT molecule and three Cu layers in the z direction. Negative Δρ means electron loss, while positive Δρ means electron accumulation.

Figure 4.
(a) Electronic density of states (DOS) for the freestanding DAT molecule. (b) Projected electronic density of states for the DAT molecule adsorbed on the Cu(001) surface. The insets show expanded views of the projected DOS on benzene rings and a N atom of the DAT/Cu(001) system around the Fermi level, respectively. E-EF means the Fermi level is set to zero eV.
Figure 4.
(a) Electronic density of states (DOS) for the freestanding DAT molecule. (b) Projected electronic density of states for the DAT molecule adsorbed on the Cu(001) surface. The insets show expanded views of the projected DOS on benzene rings and a N atom of the DAT/Cu(001) system around the Fermi level, respectively. E-EF means the Fermi level is set to zero eV.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The adsorption energies and rotation angles of a DAT molecule on a Cu(001) surface in different adsorption configurations. The detailed structural configurations are given in Figure 1.
Table 1.
The adsorption energies and rotation angles of a DAT molecule on a Cu(001) surface in different adsorption configurations. The detailed structural configurations are given in Figure 1.
| Structure | a | b | c | d | e | f | g | h |
|---|---|---|---|---|---|---|---|---|
| Eads (eV/molecule) | −3.871 | −3.844 | −3.819 | −3.777 | −3.587 | −3.566 | −3.364 | −3.321 |
| Angle (°) | 13.2 | 45.5 | 46.0 | 11.9 | 0.9 | 28.5 | 0.9 | 29.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, C.-T.; Zhang, Y.-F.; Du, S. Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study. Surfaces 2021, 4, 31-38. https://0-doi-org.brum.beds.ac.uk/10.3390/surfaces4010005
AMA Style
Wang C-T, Zhang Y-F, Du S. Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study. Surfaces. 2021; 4(1):31-38. https://0-doi-org.brum.beds.ac.uk/10.3390/surfaces4010005
Chicago/Turabian StyleWang, Chang-Tian, Yan-Fang Zhang, and Shixuan Du. 2021. "Adsorption of 4,4″-Diamino-p-Terphenyl on Cu(001): A First-Principles Study" Surfaces 4, no. 1: 31-38. https://0-doi-org.brum.beds.ac.uk/10.3390/surfaces4010005