Corrosion of Carbon Steel in Marine Environments: Role of the Corrosion Product Layer


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Exposure Sites
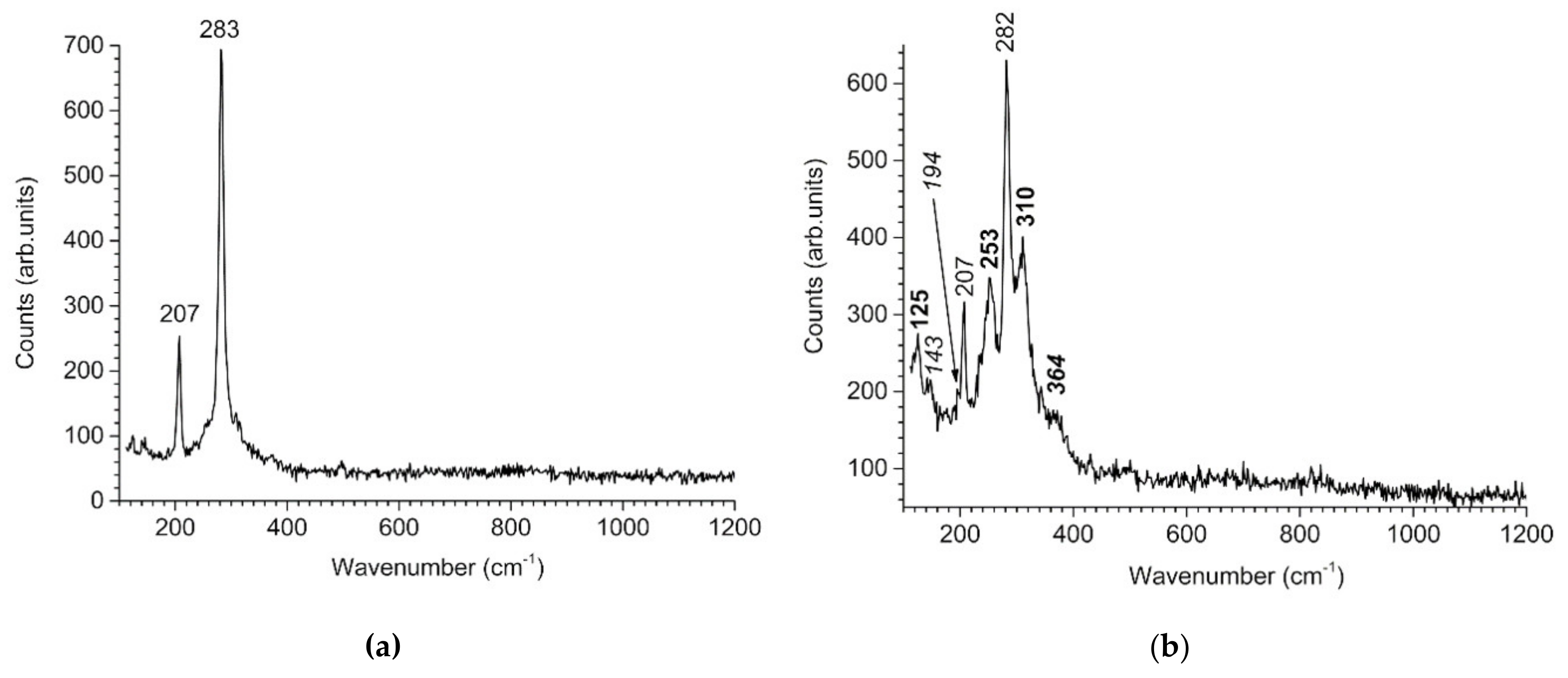
2.2. Characterization of the Corrosion Product Layers
3. General Features of the Corrosion Product Layer
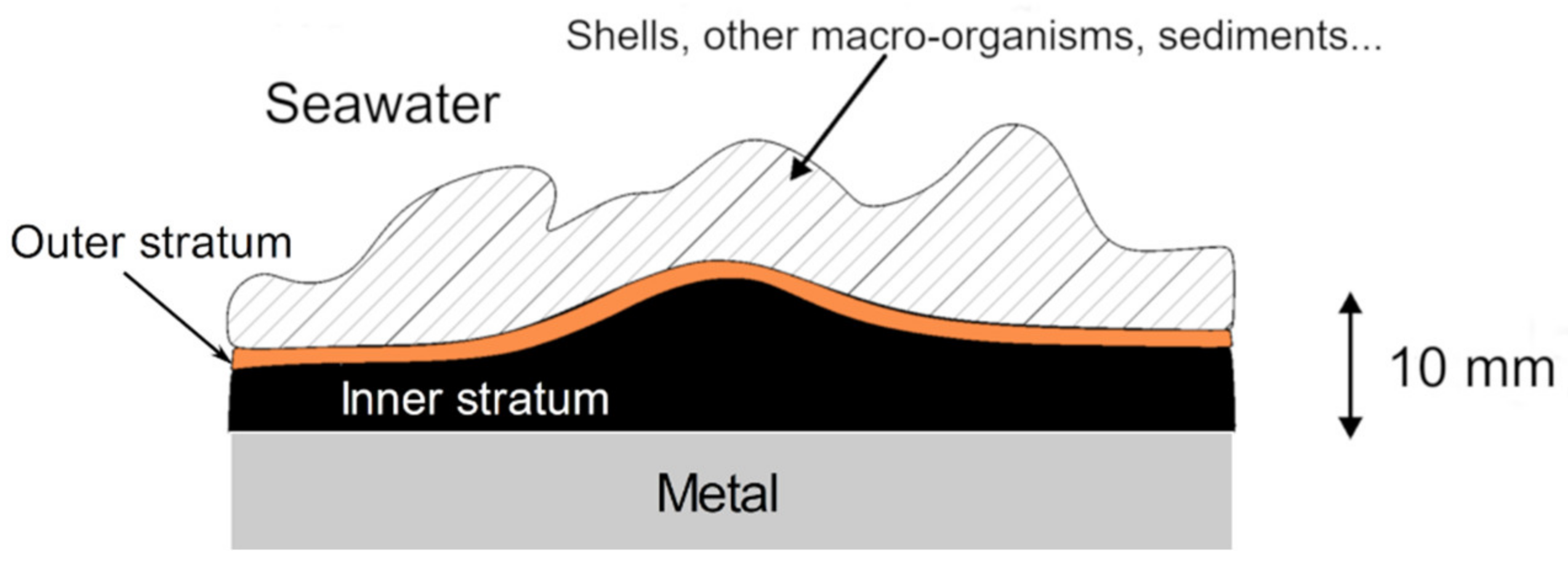
3.1. Stratification of the Corrosion Product Layer
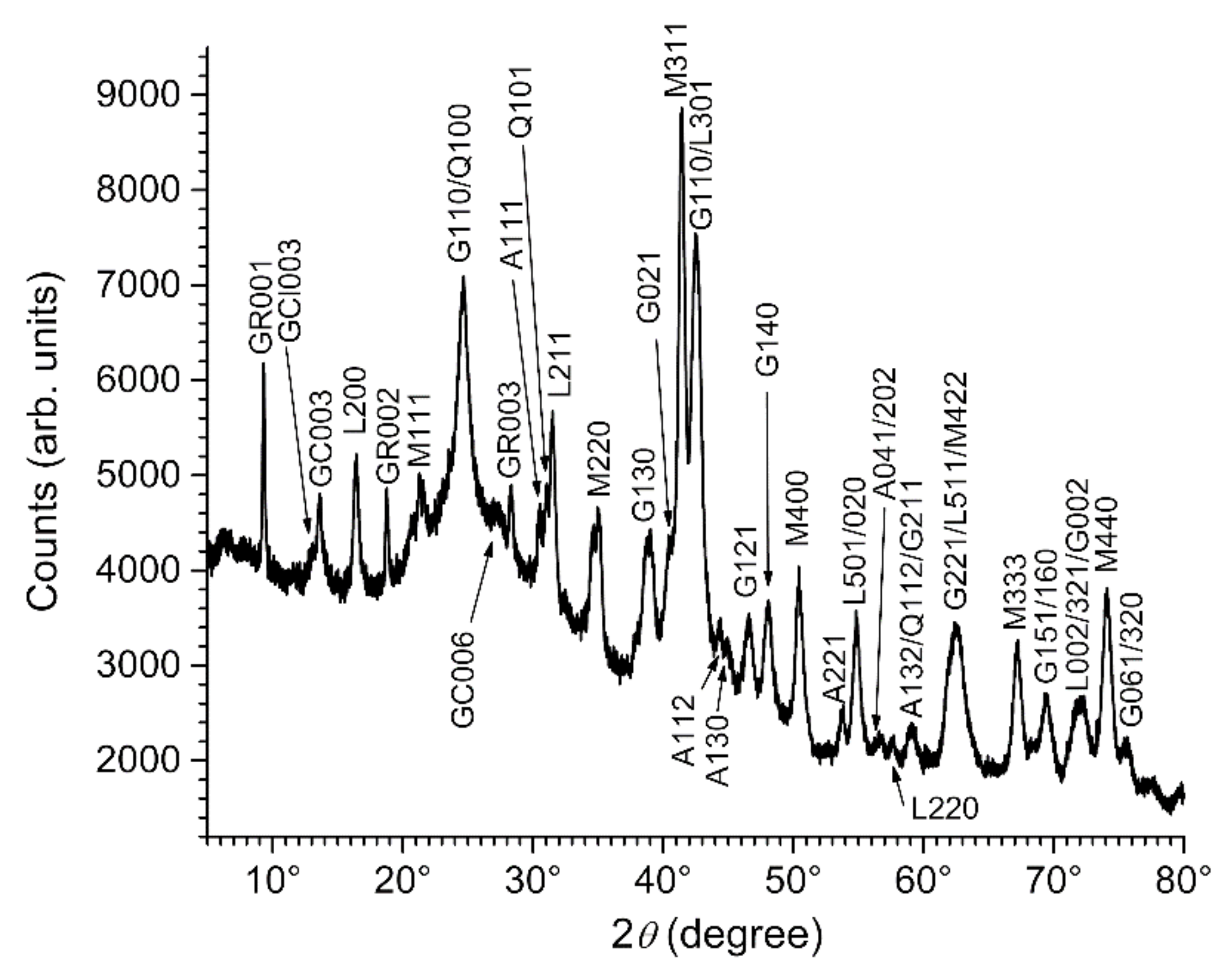
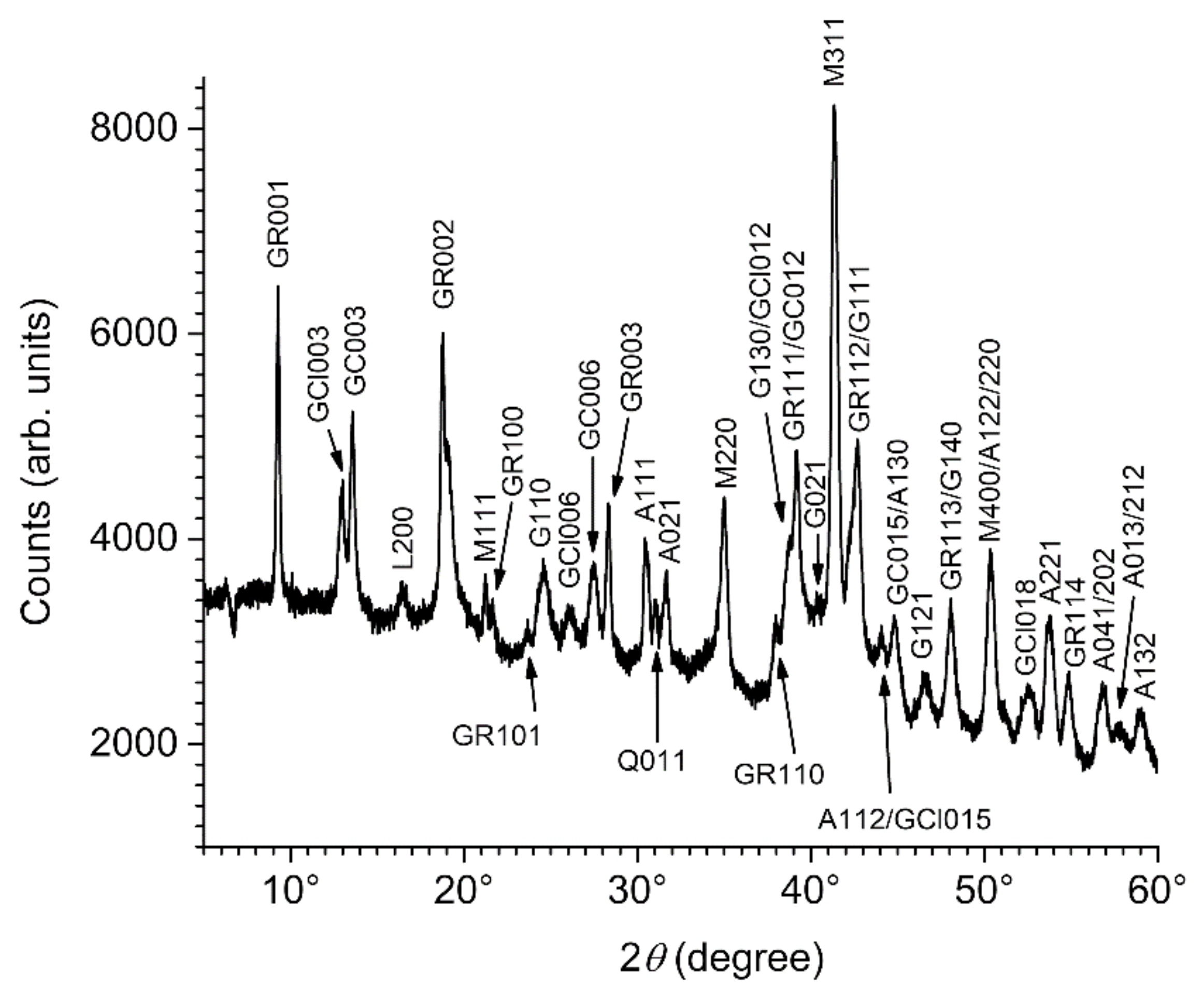
3.2. Composition of the Corrosion Product Layer
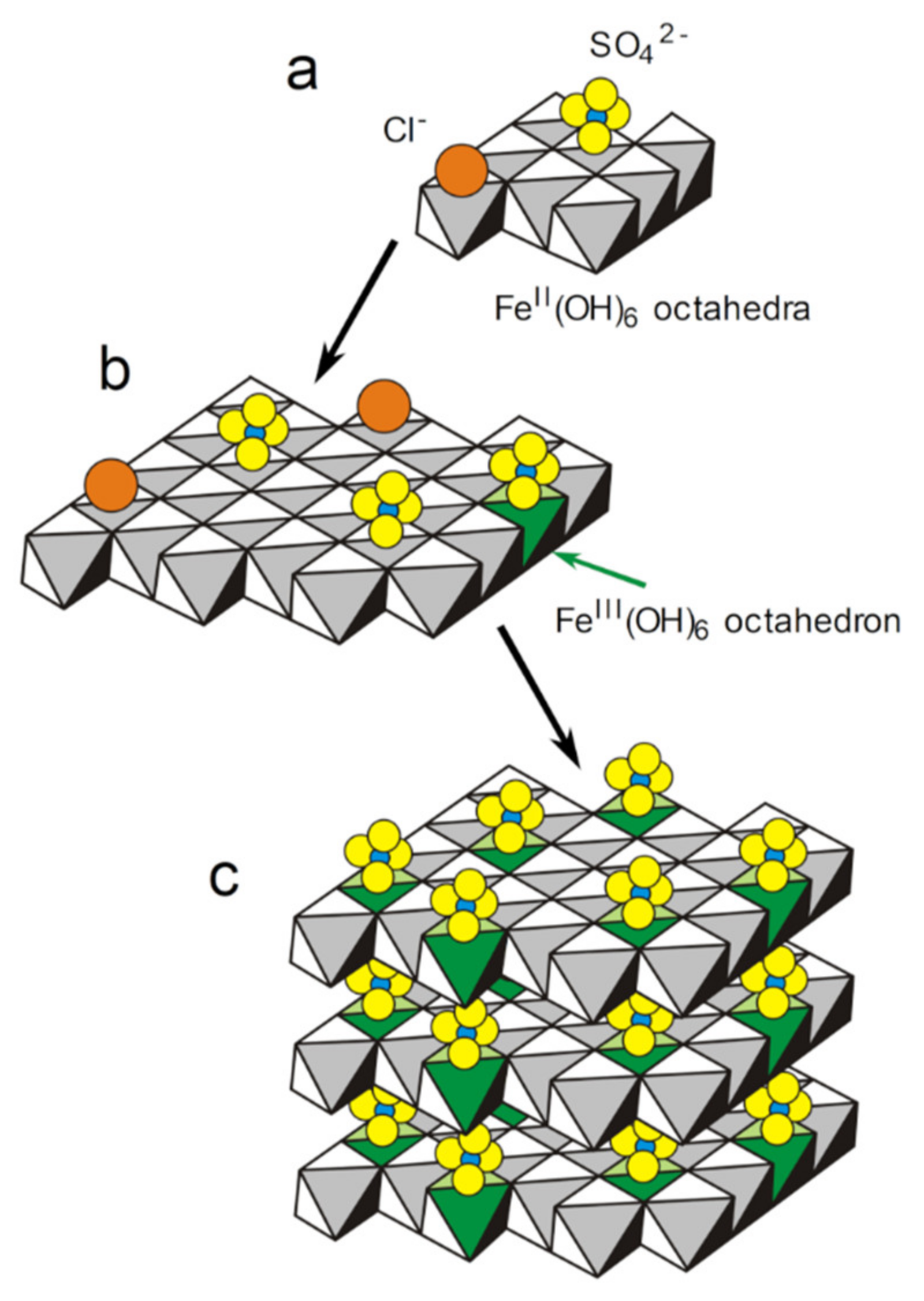
3.3. GR(SO42−), the First Compound Resulting of the Corrosion of Carbon Steel in Seawater
4. Characterization of Anodic and Cathodic Zones
4.1. Heterogeneity of the Corrosion Product Layer and Role of Interfacial pH
4.2. Equilibrium Conditions between GR(SO42−) and GR(CO32−)
4.3. Variations of the Magnetite Content
4.4. The Localized Corrosion Mechanism Associated with Magnetite-Rich Cathodic Zones
- (i)
- the layer covering the cathodic zone is covered by a thick layer of biofouling/calcareous deposit that prevents (as in anodic zones) dissolved O2 to reach magnetite particles,
- (ii)
- the corrosion product layer covering the cathodic zone becomes so thick that the network of interconnected Fe3O4/FeS particles is no more linked to the metal surface.
5. Oxidation of the Corrosion Product Layers and FORMATION of Akaganeite
6. Conclusions
- -
- It is generally admitted that, after a sufficiently long immersion time (variable but at least 6 months), anoxic conditions are met at the steel surface and inside the inner part of the corrosion product layer. Anaerobe microorganisms such as SRB can develop and be active, which leads to the formation of iron sulfides. Marine corrosion is then intrinsically a biologically influenced process and its complexity cannot be entirely mimicked by laboratory experiments and/or using artificial seawater. The biofilm itself and other microorganisms are also known to have an influence on corrosion processes.
- -
- The first solid phase to form on the steel surface is the sulfate GR. It is favored, with respect to magnetite and carbonate GR, in the anodic zones, by the decrease of the interfacial pH. A mechanism is proposed, involving (i) the adsorption of anions (mainly Cl− and SO42−) on the nuclei of Fe(OH)2 hydroxide sheets, (ii) the oxidation of part of the Fe(II) cations to Fe(III) in the hydroxide sheets and (iii) the stacking of the sheets leading to GR(SO42−) after the release of Cl− ions.
- -
- A particular mechanism can however involve oxygen even if anoxic conditions are established at the steel surface. The pH proved to have a significant influence on the composition of the corrosion product layer and this composition then depends on the anodic/cathodic character of the underlying metal surface. In cathodic zones, the increased interfacial pH favors the formation of magnetite (among other compounds) that is an electronic conductor. Associated with a low corrosion rate, the process then leads to a magnetite-rich layer that remains moderately thick (~2–5 mm) even after 6–8 years [41,42]. The reduction of dissolved O2 can then take place at the outer surface of this layer as long as the magnetite particles remain interconnected and connected to the steel surface.
- -
- The activity of SRB (and other sulfide-producing bacteria) in the cathodic zones may favor and reinforce this mechanism because it generates additional conductive corrosion products, i.e., iron sulfides.
- -
- Finally, it is necessary to preserve the samples from air to avoid the transformation of Fe(II)-based compounds. The nature of these compounds is directly related to the corrosion mechanisms. Moreover, the oxidation/transformation of the Fe(II)-based corrosion products may produce other compounds (e.g., akaganeite), thus possibly leading to an erroneous interpretation of the mechanisms.
Author Contributions
Funding
Conflicts of Interest
References
- Chaves, I.A.; Melchers, R.E. Long term localised corrosion of marine steel piling welds. Corros. Eng. Sci. Technol. 2013, 48, 469–474. [Google Scholar] [CrossRef]
- Melchers, R.E.; Jeffrey, R.J.; Usher, K.M. Localized corrosion of steel sheet piling. Corros. Sci. 2014, 79, 139–147. [Google Scholar] [CrossRef]
- Melchers, R.E.; Chaves, I.A.; Jeffrey, R. A conceptual model for the interaction between carbon content and manganese sulphide inclusions in the short-term seawater corrosion of low carbon steel. Metals 2016, 6, 132. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Cheng, X.; Dai, Z.; Liu, R.; Li, Z.; Cui, L.; Chen, M.; Ke, L. Synergistic effect of Al2O3 inclusion and pearlite on the localized corrosion evolution process of carbon steel in marine environment. Materials 2018, 11, 2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liduino, V.S.; Lutterbach, M.T.S.; Sérvulo, E.F.C. Biofilm activity on corrosion of API 5L X65 steel weld bead. Colloids Surf. B Biointerfaces 2018, 172, 43–50. [Google Scholar] [CrossRef]
- Evans, U.R. The Corrosion and Oxidation of Metals: Scientific Principles and Practical Applications; Edward Arnold (Publishers) Ltd.: London, UK, 1960. [Google Scholar]
- Jeffrey, R.; Melchers, R.E. Corrosion of vertical mild steel strips in seawater. Corros. Sci. 2009, 51, 2291–2297. [Google Scholar] [CrossRef]
- Zou, Y.; Wang, J.; Bai, Q.; Zhang, L.L.; Peng, X.; Kong, X.F. Potential distribution characteristics of mild steel in seawater. Corros. Sci. 2012, 57, 202–208. [Google Scholar] [CrossRef]
- Duboscq, J.; Sabot, R.; Jeannin, M.; Refait, P.H. Localized corrosion of carbon steel in seawater: Processes occurring in cathodic zones. Mater. Corros. 2019, 70, 973–984. [Google Scholar] [CrossRef]
- Lee, W.; Lewandowski, Z.; Nielsen, P.H.; Hamilton, W.A. Role of sulfate-reducing bacteria in corrosion of mild steel: A review. Biofouling 1995, 8, 165–194. [Google Scholar] [CrossRef]
- Starovetsky, D.; Khaselev, O.; Starovetsky, J.; Armon, R.; Yahalom, J. Effect of iron exposure in SRB media on pitting initiation. Corros. Sci. 2000, 42, 345–359. [Google Scholar]
- Huang, Y.; Duan, J.; Ma, S. Effects of anaerobe in sea bottom sediment on the corrosion of carbon steel. Mater. Corros. 2004, 55, 46–48. [Google Scholar] [CrossRef]
- Jeffrey, R.; Melchers, R.E. The changing topography of corroding mild steel surfaces in seawater. Corros. Sci. 2007, 49, 2270–2288. [Google Scholar] [CrossRef]
- Castaneda, H.; Benetton, X.D. SRB-biofilm influence in active corrosion sites formed at the steel-electrolyte interface when exposed to artificial seawater conditions. Corros. Sci. 2008, 50, 1169–1183. [Google Scholar] [CrossRef]
- Beech, I.B.; Campbell, S.A. Accelerated low water corrosion of carbon steel in the presence of a biofilm harbouring sulphate-reducing and sulphur-oxidising bacteria recovered from a marine sediment. Electrochim. Acta 2008, 54, 14–21. [Google Scholar] [CrossRef]
- Dong, Z.H.; Shi, W.; Ruan, H.M.; An Zhang, G. Heterogeneous corrosion of mild steel under SRB-biofilm characterised by electrochemical mapping technique. Corros. Sci. 2011, 53, 2978–2987. [Google Scholar] [CrossRef]
- Melchers, R.E.; Jeffrey, R. Corrosion of long vertical steel strips in the marine tidal zone and implications for ALWC. Corros. Sci. 2012, 65, 26–36. [Google Scholar] [CrossRef]
- Stipaničev, M.; Turcu, F.; Esnault, L.; Schweitzer, E.W.; Kilian, R.; Basseguy, R. Corrosion behavior of carbon steel in presence of sulfate-reducing bacteria in seawater environment. Electrochim. Acta 2013, 113, 390–406. [Google Scholar] [CrossRef] [Green Version]
- World Steel Association. “World Steel in Figures 2019”, Brussels, Belgium. 2019. Available online: https://www.worldsteel.org/publications/infographics.html (accessed on 2 June 2020).
- Melchers, R.E. Mathematical modelling of the diffusion controlled phase in marine immersion corrosion of mild steel. Corros. Sci. 2003, 45, 923–940. [Google Scholar] [CrossRef]
- Melchers, R.E.; Jeffrey, R. Early corrosion of mild steel in seawater. Corros. Sci. 2005, 47, 1678–1693. [Google Scholar] [CrossRef]
- Melchers, R.E.; Wells, T. Models for the anaerobic phases of marine immersion corrosion. Corros. Sci. 2006, 48, 1791–1811. [Google Scholar] [CrossRef]
- Enning, D.; Venzlaff, H.; Garrelfs, J.; Dinh, H.T.; Meyer, V.; Mayrhofer, K.J.J.; Hassel, A.W.; Stratmann, M.; Widdel, F. Marine sulfate-reducing bacteria cause serious corrosion of iron under electroconductive biogenic mineral crust. Environ. Microbiol. 2012, 14, 1772–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venzlaff, H.; Enning, D.; Srinivasan, J.; Mayrhofer, K.J.J.; Hassel, A.W.; Widdel, F.; Stratmann, M. Accelerated cathodic reaction in microbial corrosion of iron due to direct electron uptake by sulfate-reducing bacteria. Corros. Sci. 2013, 66, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Duan, J.; Du, X.; Huang, Y.; Hou, B. Accelerated anaerobic corrosion of electroactive sulfate-reducing bacteria by electrochemical impedance spectroscopy and chronoamperometry. Electrochem. Commun. 2013, 26, 101–104. [Google Scholar] [CrossRef]
- Pineau, S.; Sabot, R.; Quillet, L.; Jeannin, M.; Caplat, C.H.; Dupont-Morral, I.; Refait, P.H. Formation of the Fe(II-III) hydroxysulphate green rust during marine corrosion of steel associated to molecular detection of dissimilatory sulphite-reductase. Corros. Sci. 2008, 50, 1099–1111. [Google Scholar] [CrossRef]
- Duan, J.; Wu, S.; Zhang, X.; Huang, G.; Du, M.; Hou, B. Corrosion of carbon steel influenced by anaerobic biofilm in natural seawater. Electrochim. Acta 2008, 54, 22–28. [Google Scholar] [CrossRef]
- Boudaud, N.; Coton, M.; Coton, E.; Pineau, S.; Travert, J.; Amiel, C. Biodiversity analysis by polyphasic study of marine bacteria associated with biocorrosion phenomena. J. Appl. Microbiol. 2010, 109, 166–179. [Google Scholar] [CrossRef]
- Usher, K.M.; Kaksonen, A.H.; MacLeod, I.D. Marine rust tubercles harbour iron corroding archaea and sulphate reducing bacteria. Corros. Sci. 2014, 83, 189–197. [Google Scholar] [CrossRef]
- Lanneluc, I.; Langumier, M.; Sabot, R.; Jeannin, M.; Refait, P.; Sablé, S. On the bacterial communities associated with the corrosion product layer during the early stages of marine corrosion of carbon steel. Int. Biodeter. Biodegrad. 2015, 99, 55–65. [Google Scholar] [CrossRef]
- Stipanicev, M.; Turcu, F.; Esnault, L.; Rosas, O.; Basseguy, R.; Sztyler, M.; Beech, I.B. Corrosion of carbon steel by bacteria from North Sea offshore seawater injection systems: Laboratory investigation. Bioelectrochemistry 2014, 97, 76–88. [Google Scholar] [CrossRef] [Green Version]
- Rlinger, M.I.; Samokhvalov, A.A. Electron conduction in magnetite and ferrites. Phys. Stat. Sol. B 1977, 79, 9–48. [Google Scholar] [CrossRef]
- Ihle, D.; Lorenz, B. Small-polaron band versus hopping conduction in Fe3O4. J. Phys. C Solid State Phys. 1985, 18, L647–L650. [Google Scholar] [CrossRef]
- Lopez Maldonado, K.L.; de la Presa, P.; de la Rubia, M.A.; Crespo, P.; de Frutos, J.; Hernando, A.; Matutes-Aquino, J.A.; Elizalde Galindo, J. Effects of grain boundary width and crystallite size on conductivity and magnetic properties of magnetite nanoparticles. J. Nanopart. Res. 2014, 16, 2482. [Google Scholar] [CrossRef]
- Li, Y.; Hou, B.; Li, H.; Zhang, J. Corrosion behavior of steel in Chengdao offshore oil exploitation area. Mater. Corros. 2004, 55, 305–309. [Google Scholar] [CrossRef]
- Refait, P.; Nguyen, D.D.; Jeannin, M.; Sablé, S.; Langumier, M.; Sabot, R. Electrochemical formation of green rusts in deaerated seawater-like solutions. Electrochim. Acta 2011, 56, 6481–6488. [Google Scholar] [CrossRef]
- Refait, P.; Jeannin, M.; Sabot, R.; Antony, H.; Pineau, S. Corrosion and cathodic protection of carbon steel in the tidal zone: Products, mechanisms and kinetics. Corros. Sci. 2015, 90, 375–382. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; Alcantara, J.; Diaz, I.; Wolthuis, R.; de la Fuente, D. SEM/Micro-Raman characterization of the morphologies of marine atmospheric corrosion products formed on mild steel. J. Electrochem. Soc. 2016, 163, C426–C439. [Google Scholar] [CrossRef]
- Morcillo, M.; Chico, B.; de la Fuente, D.; Alcantara, J.; Odnevall Wallinder, I.; Leygraf, C. On the Mechanism of Rust Exfoliation in Marine Environment. J. Electrochem. Soc. 2017, 164, C8–C16. [Google Scholar] [CrossRef]
- Refait, P.; Grolleau, A.-M.; Jeannin, M.; François, E.; Sabot, R. Corrosion of carbon steel at the mud zone/seawater interface: Mechanisms and kinetics. Corros. Sci. 2018, 130, 76–84. [Google Scholar] [CrossRef]
- Refait, P.; Grolleau, A.-M.; Jeannin, M.; François, E.; Sabot, R. Localized corrosion of carbon steel in marine media: Galvanic coupling and heterogeneity of the corrosion product layer. Corros. Sci. 2016, 111, 583–595. [Google Scholar] [CrossRef]
- Refait, P.; Jeannin, M.; François, E.; Sabot, R.; Grolleau, A.-M. Galvanic corrosion in marine environments: Effects associated with the inversion of polarity of Zn/carbon steel couples. Mater. Corros. 2019, 70, 950–961. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Neff, D.; Kergourlay, F.; Foy, E.; Conforto, E.; Guilminot, E.; Reguer, S.; Refait, P.; Dillmann, P. Mechanisms of long-term anaerobic corrosion of iron archaeological artefacts in seawater. Corros. Sci. 2009, 51, 2932–2941. [Google Scholar] [CrossRef]
- Refait, P.; Jeannin, M.; Sabot, R.; Antony, H.; Pineau, S. Electrochemical formation and transformation of corrosion products on carbon steel under cathodic protection in seawater. Corros. Sci. 2013, 71, 32–36. [Google Scholar] [CrossRef]
- De Faria, D.L.A.; Silva, S.V.; Oliveira, M.T.D. Raman micro spectroscopy study of some iron oxides and oxyhydroxides. J. Raman Spectrosc. 1997, 28, 873–878. [Google Scholar] [CrossRef]
- Shebanova, O.N.; Lazor, P. Raman study of magnetite (Fe3O4): Laser-induced thermal effects and oxidation. J. Raman Spectrosc. 2003, 34, 845–852. [Google Scholar] [CrossRef]
- Hansen, H.C.B. Composition, stabilisation, and light absorption of Fe(II)-Fe(III) hydroxycarbonate (green rust). Clay Miner. 1989, 24, 663–669. [Google Scholar] [CrossRef]
- Duboscq, J.; Abdelmoula, M.; Jeannin, M.; Sabot, R.; Refait, P. On the formation and transformation of Fe(III)-containing chukanovite, FeII2−xFeIIIx(OH)2−xOxCO3. J. Phys. Chem. Solids 2020, 138, 109310. [Google Scholar] [CrossRef]
- Pourbaix, M. Thermodynamics and corrosion. Corros. Sci. 1990, 30, 963–988. [Google Scholar] [CrossRef]
- Refait, P.; Géhin, A.; Abdelmoula, M.; Génin, J.-M.R. Coprecipitation thermodynamics of iron(II-III) hydroxysulphate green rust from Fe(II) and Fe(III) salts. Corros. Sci. 2003, 45, 656–676. [Google Scholar] [CrossRef]
- Ruby, C.; Abdelmoula, M.; Naille, S.; Renard, A.; Khare, V.; Ona-Nguema, G.; Morin, G.; Génin, J.-M.R. Oxidation modes and thermodynamics of FeII-III oxyhydroxycarbonate green rust: Dissolution-precipitation versus in situ deprotonation. Geochim. Cosmochim. Acta 2010, 74, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Stampfl, P.P. Ein basisches eisen-II-III-karbonat in rost. Corros. Sci. 1969, 9, 185–187. [Google Scholar] [CrossRef]
- Allmann, R. Doppelschichtstrukturen mit brucitähnlichen Schichtionen [Me(II)1−xMe(III)x(OH)2]x+. Chimia 1970, 24, 99–108. [Google Scholar]
- Refait, P.; Abdelmoula, M.; Génin, J.-M.R. Mechanisms of formation and structure of green rust one in aqueous corrosion of iron in the presence of chloride ions. Corros. Sci. 1998, 40, 1547–1560. [Google Scholar] [CrossRef]
- Simon, L.; François, M.; Refait, P.; Renaudin, G.; Lelaurain, M.; Génin, J.-M.R. Structure of the Fe(II-III) layered double hydroxysulphate green rust two from Rietveld analysis. Sol. State Sci. 2003, 5, 327–334. [Google Scholar] [CrossRef]
- Miyata, S. Anion-exchange properties of hydrotalcite-like compounds. Clays Clay Miner. 1983, 31, 305–311. [Google Scholar] [CrossRef]
- Mendiboure, A.; Schöllhorn, R. Formation and anion exchange reactions of layered transition metal hydroxides [Ni1−xMx](OH)2(CO3)x/2(H2O)z (M=Fe,Co). Rev. Chim. Miner. 1986, 23, 819–827. [Google Scholar] [CrossRef]
- Refait, P.; Memet, J.B.; Bon, C.; Sabot, S.; Génin, J.-M.R. Formation of the Fe(II)-Fe(III) hydroxysulphate green rust during marine corrosion of steel. Corros. Sci. 2003, 45, 833–845. [Google Scholar] [CrossRef]
- Bourdoiseau, J.A.; Jeannin, M.; Sabot, R.; Rémazeilles, C.; Refait, P. Characterisation of mackinawite by Raman spectroscopy: Effects of crystallisation, drying and oxidation. Corros. Sci. 2008, 50, 3247–3255. [Google Scholar] [CrossRef]
- Bocher, F.; Géhin, A.; Ruby, C.; Ghanbaja, J.; Abdelmoula, M.; Génin, J.-M.R. Coprecipitation of Fe(II-III) hydroxycarbonate green rust stabilised by phosphate adsorption. Sol. State Sci. 2004, 6, 117–124. [Google Scholar] [CrossRef]
- Barthélémy, K.; Naille, S.; Despas, C.; Ruby, C.; Mallet, M. Carbonated ferric green rust as a new material for efficient phosphate removal. J. Colloid Interface Sci. 2012, 384, 121–127. [Google Scholar] [CrossRef]
- Usman, M.; Hanna, K.; Abdelmoula, M.; Zegeye, A.; Faure, P.; Ruby, C. Formation of green rust via mineralogical transformation of ferric oxides (ferrihydrite, goethite and hematite). Appl. Clay Sci. 2012, 64, 38–43. [Google Scholar] [CrossRef]
- Jeong, H.Y.; Lee, J.H.; Hayes, K.F. Characterization of nanocrystalline mackinawite: Crystal structure, particle size, and specific surface area. Geochim. Cosmochim. Acta 2008, 72, 493–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohfuji, H.; Rickard, D. High resolution transmission electron microscopic study of synthetic nanocrystalline mackinawite. Earth Planet. Sci. Lett. 2006, 241, 227–233. [Google Scholar] [CrossRef]
- Bourdoiseau, J.-A.; Jeannin, M.; Rémazeilles, C.; Sabot, R.; Refait, P. The transformation of mackinawite into greigite studied by Raman spectroscopy. J. Raman Spectrosc. 2011, 42, 496–504. [Google Scholar] [CrossRef]
- Mullet, M.; Boursiquot, S.; Abdelmoula, M.; Génin, J.-M.R.; Ehrhardt, J.J. Surface chemistry and structural properties of mackinawite prepared by reaction of sulfide with metallic iron. Geochim. Cosmochim. Acta 2002, 66, 829–836. [Google Scholar] [CrossRef]
- Lennie, A.R.; Redfern, S.A.T.; Champness, P.E.; Stoddart, C.P.; Schofield, P.F.; Vaughan, D.J. Transformation of mackinawite to greigite: An in situ X-ray powder diffraction and transmission electron microscope study. Am. Min. 1997, 82, 302–309. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Saheb, M.; Neff, D.; Guilminot, E.; Tran, K.; Bourdoiseau, J.-A.; Sabot, R.; Jeannin, M.; Matthiesen, H.; Dillmann, P.; et al. Microbiologically influenced corrosion of archaeological artefacts; characterisation of iron(II) sulphides by Raman spectroscopy. J. Raman Spectrosc. 2010, 41, 1135–1143. [Google Scholar] [CrossRef]
- Malard, E.; Kervadec, D.; Gil, O.; Lefevre, Y.; Malard, S. Interactions between steels and sulphide-producing bacteria-Corrosion of carbon steels and low-alloy steels in natural seawater. Electrochim. Acta 2008, 54, 8–13. [Google Scholar] [CrossRef]
- Refait, P.; Sabot, R.; Jeannin, M. Role of Al(III) and Cr(III) on the formation and oxidation of the Fe(II-III) hydroxysulfate Green Rust. Colloids Surf. A Phys. Eng. Asp. 2017, 531, 203–212. [Google Scholar] [CrossRef]
- Hartt, W.H.; Culberson, C.H.; Smith, S.W. Calcareous deposits on metal surfaces in seawater- A critical review. Corrosion 1984, 40, 609–618. [Google Scholar] [CrossRef]
- Lee, R.U.; Ambrose, J.R. Influence of cathodic protection parameters on calcareous deposit formation. Corrosion 1986, 44, 887–891. [Google Scholar] [CrossRef]
- Yan, J.F.; White, R.E.; Griffin, R.B. Parametric studies of the formation of calcareous deposits on cathodically protected steel in seawater. J. Electrochem. Soc. 1993, 141, 1275–1280. [Google Scholar] [CrossRef] [Green Version]
- Barchiche, C.; Deslouis, C.; Festy, D.; Gil, O.; Refait, P.; Touzain, S.; Tribollet, B. Characterization of calcareous deposits in artificial sea water by impedance techniques. 3- Deposit of CaCO3 in the presence of Mg(II). Electrochim. Acta 2003, 48, 1645–1654. [Google Scholar] [CrossRef]
- Cook, D.C.; van Orden, A.C.; Carpio, J.J.; Oh, S.J. Atmospheric corrosion in the Gulf of Mexico. Hyperf. Interact. 1998, 113, 319–329. [Google Scholar] [CrossRef]
- Li, S.; Hihara, L.H. A micro-Raman spectroscopic study of marine atmospheric corrosion of carbon steel: The effect of akaganeite. J. Electrochem. Soc. 2015, 162, C495–C502. [Google Scholar] [CrossRef]
- Stahl, K.; Nielsen, K.; Jiang, J.; Lebech, B.; Hanson, J.C.; Norby, P.; van Lanschot, J. On the akaganeite crystal structure, phase transformations and possible role in post-excavational corrosion of iron artifacts. Corros. Sci. 2003, 45, 2563–2575. [Google Scholar] [CrossRef]
- Rémazeilles, C.; Refait, P. On the formation of β-FeOOH (Akaganéite) in chloride-containing environments. Corros. Sci. 2007, 49, 844–857. [Google Scholar] [CrossRef]
- Refait, P.; Génin, J.-M.R. The mechanisms of oxidation of ferrous hydroxychloride β-Fe2(OH)3Cl in chloride-containing aqueous solution: The formation of β-FeOOH akaganeite; an X-ray diffraction, Mössbauer spectroscopy and electrochemical study. Corros. Sci. 1997, 39, 539–553. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon Steel | C | Mn | P | S | Si | Al | Cr | Cu | Ni |
|---|---|---|---|---|---|---|---|---|---|
| S355GP | ≤0.27 | ≤1.7 | ≤0.055 | ≤0.055 | ≤0.6 | 0.02 | - | - | - |
| S355NL | 0.17 | 1.4 | 0.015 | 0.005 | 0.21 | 0.02 | 0.02 | 0.01 | 0.02 |
| TU250B | 0.12 | 0.6 | 0.017 | 0.006 | 0.22 | - | 0.08 | 0.19 | 0.09 |
| Compound Name: Chemical Formula | Importance/Abundance and Localization | Link to Anodic and Cathodic Zones |
|---|---|---|
| Sulfate green rust, GR(SO42−): FeII4FeIII2(OH)12SO4·8H2O | Main component of the inner layer | Favored in anodic zones |
| Carbonate green rust, GR(CO32−): FeII4FeIII2(OH)12CO3·2H2O | Minor component of the inner layer | Favored in cathodic zones |
| Chloride green rust, GR(Cl−): FeII3FeIII(OH)8Cl·2H2O | Minor component of the inner layer | Not known |
| Chukanovite: Fe2(OH)2CO3 | Rare | Cathodic zones |
| Mackinawite/Fe(III)-containing mackinawite: FeIIS/FeII1-3xFeIII2xS | Main component of the inner layer, due to SRB | Not known |
| Greigite: Fe3S4 | Rare | Not known |
| Magnetite: Fe3O4 | Main component of the inner layer | Favored in cathodic zones |
| Lepidocrocite: γ-FeOOH | Main component of the outer layer | Anodic zones or uniform corrosion |
| Goethite: α-FeOOH | Main component of the outer layer | Anodic zones or uniform corrosion |
| Akaganeite: β-FeOOH | Rare, minor component | Not known |
| Aragonite: CaCO3 | Outer layer | Cathodic zones |
| Zone | Corrosion Rate (mm/Year) | I(M311)/I(GR003) |
|---|---|---|
| Strongly anodic | 0.50 ± 0.01 | 0 |
| Anodic | 0.20 ± 0.03 | 0.12 |
| Anodic | 0.15 ± 0.04 | 0.5 |
| Anodic | 0.14 ± 0.02 | 0.03 |
| Slightly anodic | 0.08 ± 0.02 | 0.51 |
| Uniform corrosion | 0.07 ± 0.02 | 0.77 to 1.05 |
| Slightly cathodic | 0.06 ± 0.01 | 0.5 |
| Slightly cathodic | 0.06 ± 0.01 | 3.1 |
| Cathodic | 0.05 ± 0.01 | 1.9 to 3.3 |
| Cathodic | 0.040 ± 0.005 | 8.9 to 10 |
| Cathodic | 0.035 ± 0.005 | 2.6 |
| Strongly cathodic | ~ 0.01 | 4.9 to 14.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Refait, P.; Grolleau, A.-M.; Jeannin, M.; Rémazeilles, C.; Sabot, R. Corrosion of Carbon Steel in Marine Environments: Role of the Corrosion Product Layer. Corros. Mater. Degrad. 2020, 1, 198-218. https://0-doi-org.brum.beds.ac.uk/10.3390/cmd1010010
Refait P, Grolleau A-M, Jeannin M, Rémazeilles C, Sabot R. Corrosion of Carbon Steel in Marine Environments: Role of the Corrosion Product Layer. Corrosion and Materials Degradation. 2020; 1(1):198-218. https://0-doi-org.brum.beds.ac.uk/10.3390/cmd1010010
Chicago/Turabian StyleRefait, Philippe, Anne-Marie Grolleau, Marc Jeannin, Celine Rémazeilles, and René Sabot. 2020. "Corrosion of Carbon Steel in Marine Environments: Role of the Corrosion Product Layer" Corrosion and Materials Degradation 1, no. 1: 198-218. https://0-doi-org.brum.beds.ac.uk/10.3390/cmd1010010