
Atmospheric Corrosion of Silver and Silver Nanoparticles

School of Information and Physical Science, The University of Newcastle, Callaghan NSW 2308, Australia
Corros. Mater. Degrad. 2022, 3(2), 221-234; https://0-doi-org.brum.beds.ac.uk/10.3390/cmd3020013
Submission received: 11 April 2022
/
Revised: 26 April 2022
/
Accepted: 20 May 2022
/
Published: 24 May 2022
(This article belongs to the Special Issue Atmospheric Corrosion of Materials)
Abstract
:Even though it is a noble metal, silver will corrode in ambient atmospheres, predominantly by reacting with sulfur-containing gases such as hydrogen sulfide (H2S) and carbonyl sulfide (OCS) to form the silver sulfide (Ag2S) acanthite. Other aspects of the environment, such as relative humidity and the presence of oxidizing species, also play a critical role. With the emergence of silver nanoparticles for a range of technological and medical applications, there has been a revival of interest in the corrosion behavior of this important metal. This article reviews the current understanding of the atmospheric corrosion of silver in both the bulk and nanoparticle forms. Gaps in our current understanding and areas for future investigation are identified.
1. Introduction
For centuries, silver metal has played an integral role in human society, where it has been widely used for tableware, coinage, jewellery, and decorative purposes. Silver is an outstanding thermal and electrical conductor with high reflectivity, high malleability, and low tarnishing rates when compared to many other metals. These beneficial properties mean that silver technology remains relevant today for a diverse range of modern applications, such as in mirrors, as an electrical contact, as a coating for engine bearings, in brazing and soldering, and many others [1]. Although silver is one of the more corrosion-resistant metals, degradation upon exposure to the ambient environment will occur and needs to be considered as a factor in many applications.
Recently, nanostructured and nanoparticle silver have been identified as the materials of choice for the new field of plasmonics [2], in which they have diverse applications, including chemical and bio-sensors, nano-photonic circuitry, tumour therapy, and photo-catalytic and photovoltaic enhancement [3]. Silver nanoparticles have also been suggested for applications in catalysis [4]. The large surface area of nanostructured silver means that the deleterious effects of corrosion can occur rapidly and limit practical applications. Accordingly, there is an ongoing search for materials with a plasmonic response equivalent to silver but with superior stability [5,6].
The most widespread use of nanoparticle silver is as an anti-bacterial and sterilizing agent [7]. In aqueous environments, silver releases Ag+ ions, and the interaction of these ions with biological thiol groups is linked to antibacterial activity [8,9]. There is a long tradition of the use of silver and colloidal silver for health-related applications [10,11], but this has accelerated with the greater need for wide-spectrum antibacterial materials in an era of increasing antibacterial resistance [12,13]. Silver in nanoparticle form has been demonstrated to be effective for applications in wound and burn dressings, in fabrics for odour reduction, in water treatment, and in medical instruments and implants [12,14,15]. However, the effectiveness observed in in vitro laboratory experiments has not always translated to improved outcomes in real world applications [13,14,15,16]. Furthermore, with their growing use in a wide variety of antibacterial consumer products, there are concerns about the ecotoxic effects of silver nanoparticles upon their later release into the environment [12,17].
Any Ag+ ions released from bulk or nanoparticle silver can also react with other chemical species in the environment to form other silver-based compounds. These processes are important for both the effectiveness of the antibacterial products and the eventual fate of silver nanoparticles in the environment. Alternatively, Ag+ ions can undergo a reduction reaction back to silver metal, for example, by exposure to light. This last reaction is related to the processes of photography using conventional film, although it has now been generally superseded by digital imaging technologies. The release of Ag+ ions can also occur within the adsorbed water layer found on surfaces in ambient atmospheres. The released ions may then react with any other dissolved species in this layer, which can include dissolved atmospheric gases. Thus, relative humidity (RH) is a critical aspect of the corrosion and tarnishing of silver in air, as it controls the thickness and extent of the surface water layer.
Given the ubiquity of silver and silver nanoparticle applications, understanding the corrosion processes is important for several reasons.
- A thorough understanding of the corrosion mechanism can guide materials design to slow or prevent the corrosion process and expand the options for technological, medical, practical, or decorative applications.
- Knowledge about the speed and mechanism of the corrosion of silver nanoparticles can inform the extent to which we need to be concerned about their ecotoxic effect upon release to the environment.
- As both corrosion and antibacterial activity have a common link to the release of Ag+ ions, understanding the corrosion process can provide insight into antibacterial effectiveness.
- An understanding of the corrosion of silver has importance in the conservation and restoration of historical and cultural objects.
Silver corrosion has been the subject of extensive research over many decades. The recent interest in new applications using silver in nanostructured forms has reinvigorated this topic. Investigators studying the degradation of nanoparticle silver may not be familiar with details of the literature on bulk corrosion, most of which is over 30 years old. Surprisingly, for such a well-known and studied phenomenon, there remain significant questions yet to be answered. Although there are many commonalities found in the corrosion of the bulk and nanostructured forms, there are some important distinctions. This review paper will synthesise and summarise the current knowledge on the atmospheric corrosion of silver in both bulk and nanoparticle form and will identify important areas requiring further investigation.
2. Corrosion of Bulk Silver
Anyone who has owned a piece of silver tableware or jewellery will be familiar with the dark grey or black tarnish that develops over time and must be polished off if the silverware is to retain its bright lustre. However, as silver is a very soft metal, these everyday objects are usually made from sterling silver, in which silver has been alloyed with other metals, most often copper. The corrosion of these other components will be a dominant contribution to the tarnish [18,19]. Nevertheless, even pure silver will eventually tarnish when exposed to corroding gas species found in the ambient atmosphere, as illustrated in Figure 1.
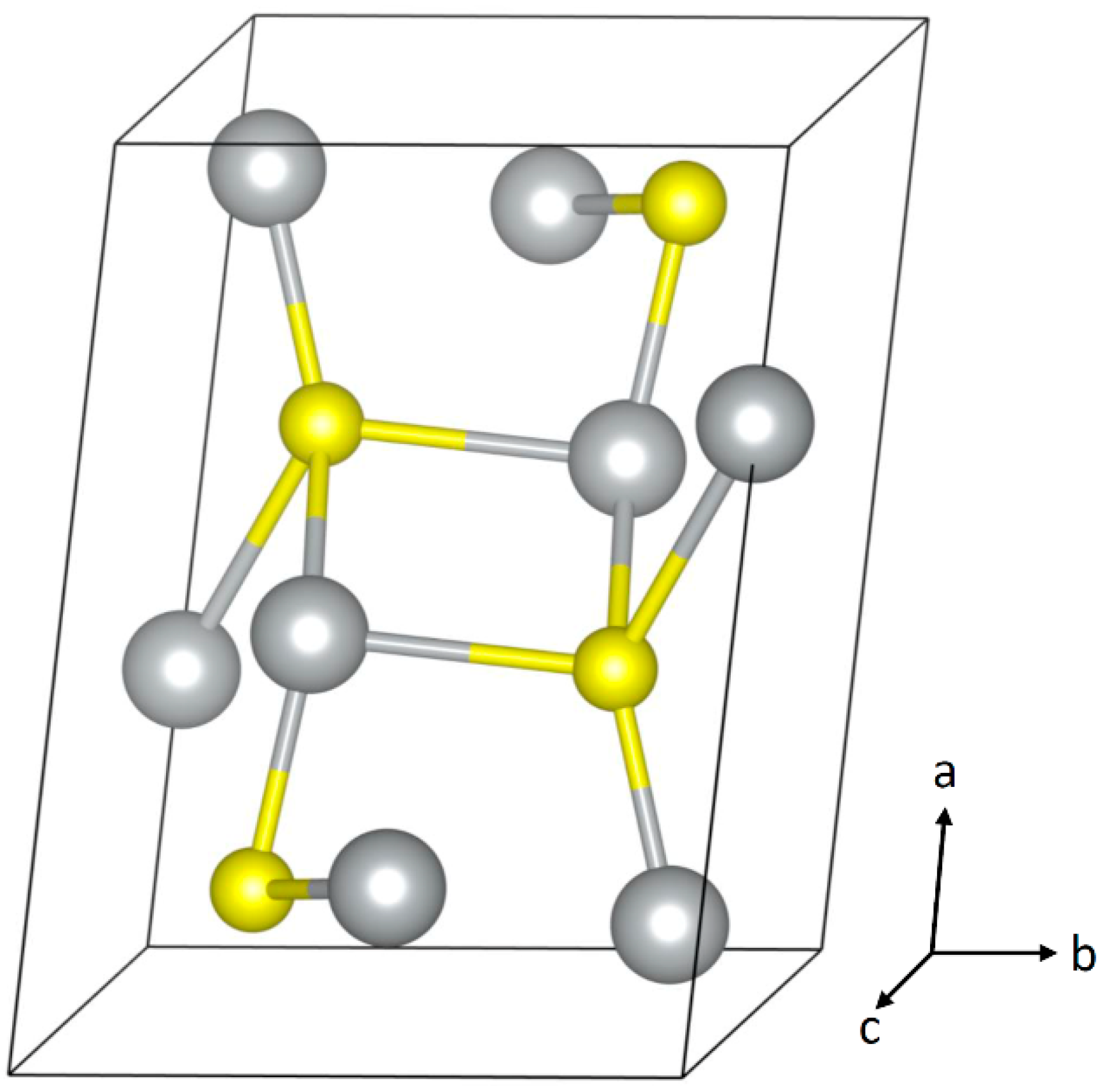
As a noble metal, silver has a low affinity for oxygen, so, in contrast to most metals, the corrosion product in indoor ambient air is usually not an oxide but is instead the sulfide Ag2S [20]. Below about 180 °C, the stable phase of Ag2S is the orthorhombic acanthite phase, and this is what is usually observed as the corrosion product. The crystal structure of acanthite is described in Table 1 and shown in Figure 2. There have been reports of the cubic argentite structure of Ag2S as well as Ag8S as corrosion products [21,22,23], but these are inconsistent with the binary phase diagram and with most other studies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Crystal structure of acanthite Ag2S [24].
Table 1.
Crystal structure of acanthite Ag2S [24].
| Ag2S | Space Group: P21/c (14) | |||
|---|---|---|---|---|
| Unit Cell Parameters | ||||
| a = 4.231 Å | b = 6.93 Å | c = 9.526 Å | ||
| α = 90° | β = 125.48° | γ = 90° | ||
| Fractional Atom Coordinates (Wykoff site) | ||||
| Ag | 0.0438 | 0.0169 | 0.3075 | (4e) |
| Ag | 0.6465 | 0.3213 | 0.4362 | (4e) |
| S | 0.2612 | 0.2383 | 0.1306 | (4e) |
The corrosion in outdoor environments is more complex, with a greater variety of products observed, including chlorides, sulfides, sulfates, oxides, and carbonates, depending on the location [26,27,28]. As some of these corrosion products have high solubility, whether they will be retained and observed will depend on the exposure conditions. The formation of AgCl has been associated with the presence of chloride-containing species, particularly NaCl, and is usually the dominant corrosion product outdoors [22,28,29,30,31]. Ultraviolet (UV) light, ozone, and RH have been found to play a role in the corrosion to AgCl [32,33]. As most applications of silver are intended for indoor use, the question of outdoor corrosion has not received as much attention in the literature, as has been the case for some other metals.
Indoor corrosion is usually attributed to a rapid reaction with atmospheric H2S, although carbonyl sulfide (OCS) has also been identified as being involved [34,35]. It is believed that the OCS molecule is first hydrolysed to H2S, so in both cases, it is the HS− ion species involved in the conversion to Ag2S [36]. The presence of SO2 has a much slower effect on silver tarnishing [34,37,38,39]. Corrosion by organic sulfur species has been observed, and this occurred more rapidly in the presence of light [40]. Most studies have demonstrated that RH plays a critical role in corrosion rates [37,39,41,42]. Silver corrosion ceases when samples are transferred to a dry nitrogen environment [43]. However, at least one study did not observe a dependence on RH [38]. As silver carbonate is highly soluble, the role of atmospheric CO2 is expected to be to a small decrease in the pH of the surface aqueous layer [36].
Even though the corrosion product is not an oxide, corrosion is enhanced by the presence of an oxidising agent such as ozone, NO2 or Cl2 [38,44,45], or even aerial oxygen [46]. It is believed that the role of these species is to promote the dissolution of the bulk silver. In a similar way, the intentional sulfidation of silver immersed in a Na2S solution was prevented when the dissolved oxygen was purged from the solution [41]. Silver oxides can be produced in conditions with high ozone concentrations significantly exceeding typical atmospheric concentrations [43].
One of the major challenges of studying the atmospheric corrosion of silver in any systematic or quantitative fashion is that natural concentrations of the corroding gases are very low and vary significantly depending on the local environment. This difficulty was identified in the earliest studies of silver corrosion, where considerable variation in the corrosion rate between samples placed in different locations was observed [44]. Natural H2S concentrations in unpolluted areas may be as low as 0.02 ppb but can reach as high as 30 ppb in particularly adverse conditions [37,45]. Indoor concentrations of H2S and other reduced sulfur gases have been reported to be around 0.14–0.71 ppb [47]. In museum environments, concentrations of between 0.086–0.6 ppb for H2S and 0.40–0.85 ppb for OCS have been measured [48]. The H2S concentration in intestinal gas is 1–4 ppm, and in exhaled breath, it can range from 1–100 ppb [45], meaning that the presence of the experimenter themselves and other nearby individuals have the potential to influence the outcome of the experiment. Similarly, whether any sulfur producing materials are stored in the laboratory, as well as the nature of nearby industries and ecological or geological systems, will also influence the outcome. Measuring the atmospheric composition for such low concentrations of the corroding gases is not straightforward.
Many of the studies in the literature are performed in ambient conditions in a natural or laboratory environment where the corroding gas concentrations are neither measured, controlled, nor reported. RH, light, and NO2 concentrations can also influence the corrosion rate, and these are often not controlled or reported in ambient environments. The disadvantage of studying corrosion in these ambient environments is the lack of systematic control, which may explain the discrepancies observed by different researchers. This difficulty suggests the need for controlled environments. However, producing controlled concentrations at such low values (sub ppb) is exceedingly difficult. When the corroding gas concentrations have been controlled and reported in the literature, they are at concentrations significantly higher than those found naturally, such that accelerated corrosion occurs. The conditions of accelerated corrosion may not be representative of what occurs in ambient environments. These challenges of studying atmospheric corrosion processes are not unique to the study of silver corrosion [49] but are exacerbated by the very low concentrations of H2S that are usually responsible for silver corrosion.
One of the few quantitative investigations of the growth rate of the corrosion layer on a silver surface exposed to laboratory air (of <0.2 ppb H2S) showed an initial growth rate of about 0.1 nm/h for the first couple of hours, slowing to an average growth of about 0.4 nm/day during the first week of exposure and slowing even further to about 0.15 nm/day after longer periods (15–60 days). Highlighting the sensitivity to the details of the local environment, it was observed that the growth rate was found to vary depending on the number of people present in the laboratory (e.g., during the day versus the night) and in which room the samples were located [42]. These values contrast to a much higher rate of corrosion of approximately 2.5 nm per day, previously observed in a kitchen environment where, presumably, higher H2S concentrations might be expected [44].
The surface reaction rate is very fast, and corrosion rates are expected to be limited by the mass transfer of H2S to the surface at typical H2S concentrations and RH levels [50]. The corrosion layer forms rapidly and then shows a saturation-like behaviour, and it was suggested that the rate-limiting step in the early stage is the diffusion of the gases to the surface and in the later stage it is the diffusion of silver to the surface of the corroded layer [35]. The role of RH is to provide an adsorbed layer of water on the surface, which acts as a medium for the disassociation of atmospheric gases and for the dissolution of solid silver into Ag+ ions. The dissolution rate of silver, and thus the corrosion rate, will also be influenced by the precise chemical make-up of the adsorbed water layer, which itself is influenced by the presence of species other than H2S in the environment. This complex interplay between H2S concentrations, RH, and the presence of oxidising species in the environment may account for some of the discrepancies between observations in the literature.
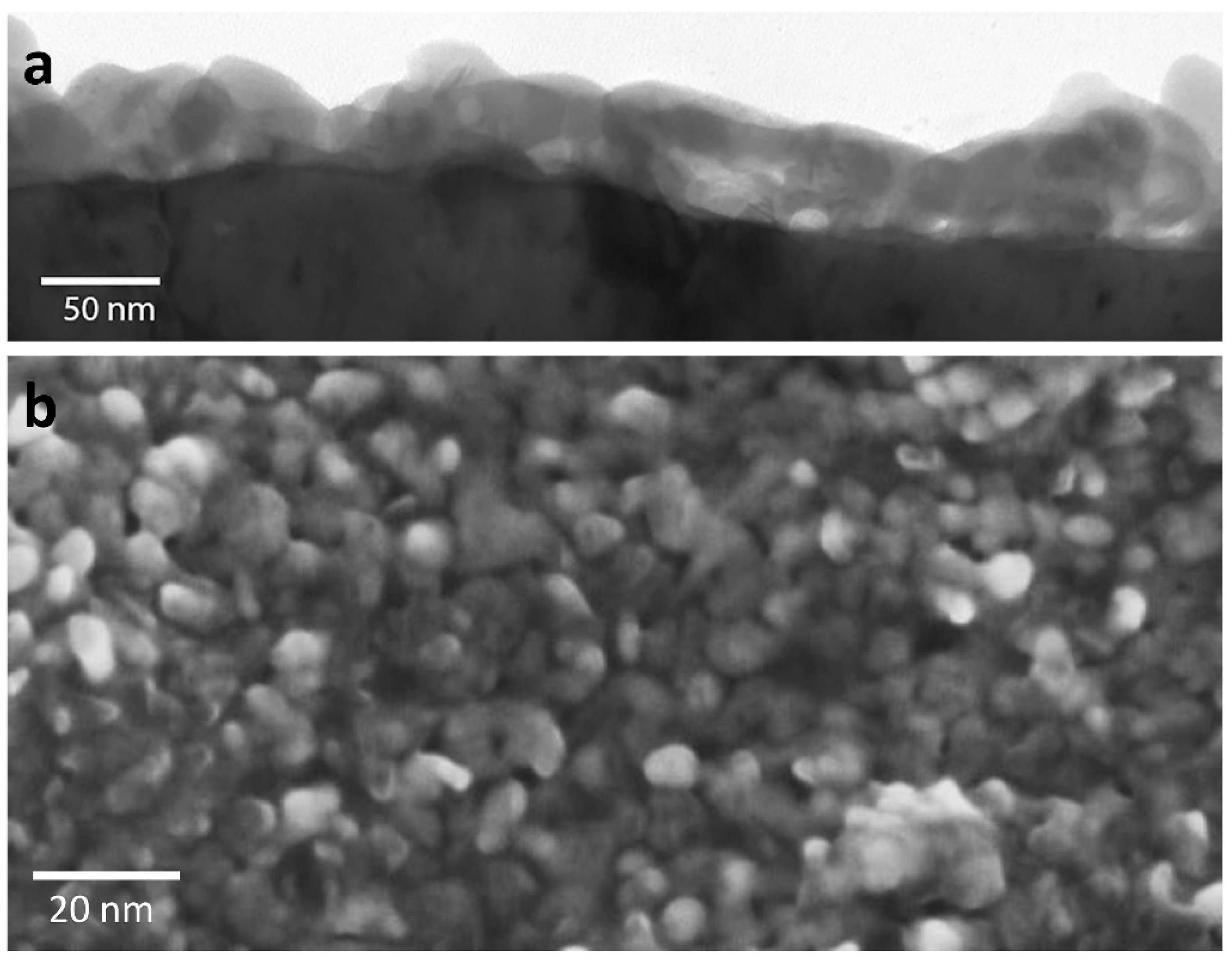
The corrosion product forms as a discontinuous layer of particles, and the particles increase in size but not in density over time [42]. The particulate nature of the corrosion product can be seen in Figure 3, which shows electron microscopy images of the corrosion layer formed after many years of exposure to ambient air. In accelerated corrosion environments, needle-like structures of Ag2S have been observed to form instead [40,51]. Corrosion occurs more rapidly on surface steps and edges and at the intersection of defects with the surface, such as stacking faults and dislocations [22,52,53]. It has also been observed that corrosion occurs more easily on (100) surfaces than (111) surfaces [54]. These results can be understood by recognising the higher reactivity and greater likelihood for Ag+ ion release for sites with a lower coordination number.
In addition to the formation of Ag2S as a corrosion product on bulk silver, there have been reports of the formation of secondary metallic silver particles. This has been observed to occur when bulk silver objects are in contact with another surface in humid conditions and was extremely rapid when a water droplet was present and was allowed to evaporate [55]. The formation of metallic silver particles as a corrosion product on a silver surface has also been observed in environments where ozone and UV light are present [29]. These observations can be understood as resulting from the release of Ag+ ions into the adsorbed water layer, followed by reduction back to metallic silver before the reaction to form Ag2S can occur. As will be discussed later, this process is also important for understanding the corrosion of nanoparticle silver.
3. Corrosion of Nanoparticle Silver
Although bulk silver has antibacterial properties, the release of Ag+ ions is relatively slow, so whilst it might have historically found application for its medical and sterilizing properties [10], in modern times, attention has switched to other forms. At the other extreme, ionic silver compounds used in medicine, such as silver nitrate and silver sulfadiazine, release a high load of Ag+ ions on application, but the rapid reactions with other species in the environment mean that this antibacterial load of Ag+ ions is not sustained. The advantage of silver in the form of nanoparticles is that it acts as a reservoir for the steady and controlled release of Ag+ ions in sufficient quantities to be an effective antibacterial agent [56]. Their relatively high surface area, where lowered coordination for the silver atoms is found, accounts for the enhanced Ag+ release and antibacterial activity of silver nanoparticles when compared to bulk silver. Correspondingly, we would expect a strongly enhanced corrosion rate for silver nanoparticles when compared to bulk silver, which is indeed the case.
Silver nanoparticles are most commonly prepared by solution phase synthesis, meaning the resultant product is a nanoparticle suspension in water or other solvent [52]. The solution may include surfactants or capping agents. Such suspensions can usually be stored for many months or even years [53], but as soon as the nanoparticles are exposed to atmosphere, by depositing on a substrate for example, they will rapidly corrode. Silver nanoparticles can also be prepared by electron or ion beam lithography techniques, and these will corrode very rapidly after synthesis unless stored in an inert environment [57].
The first published study of the corrosion of silver nanoparticles showed that degradation was observed within hours of exposure to laboratory air [57]. The corrosion was detected and monitored by the shift in the surface plasmon response. The presence of sulfur in the corrosion product was confirmed, and the corrosion rate increased with increasing RH. A later study on a similar nanoparticle array confirmed the degradation to a sulfur-containing product in laboratory air and that corrosion did not occur for nanoparticles stored in a vacuum [58]. The images presented of the corrosion product indicated they have a particulate appearance, similar to that seen in bulk corrosion.
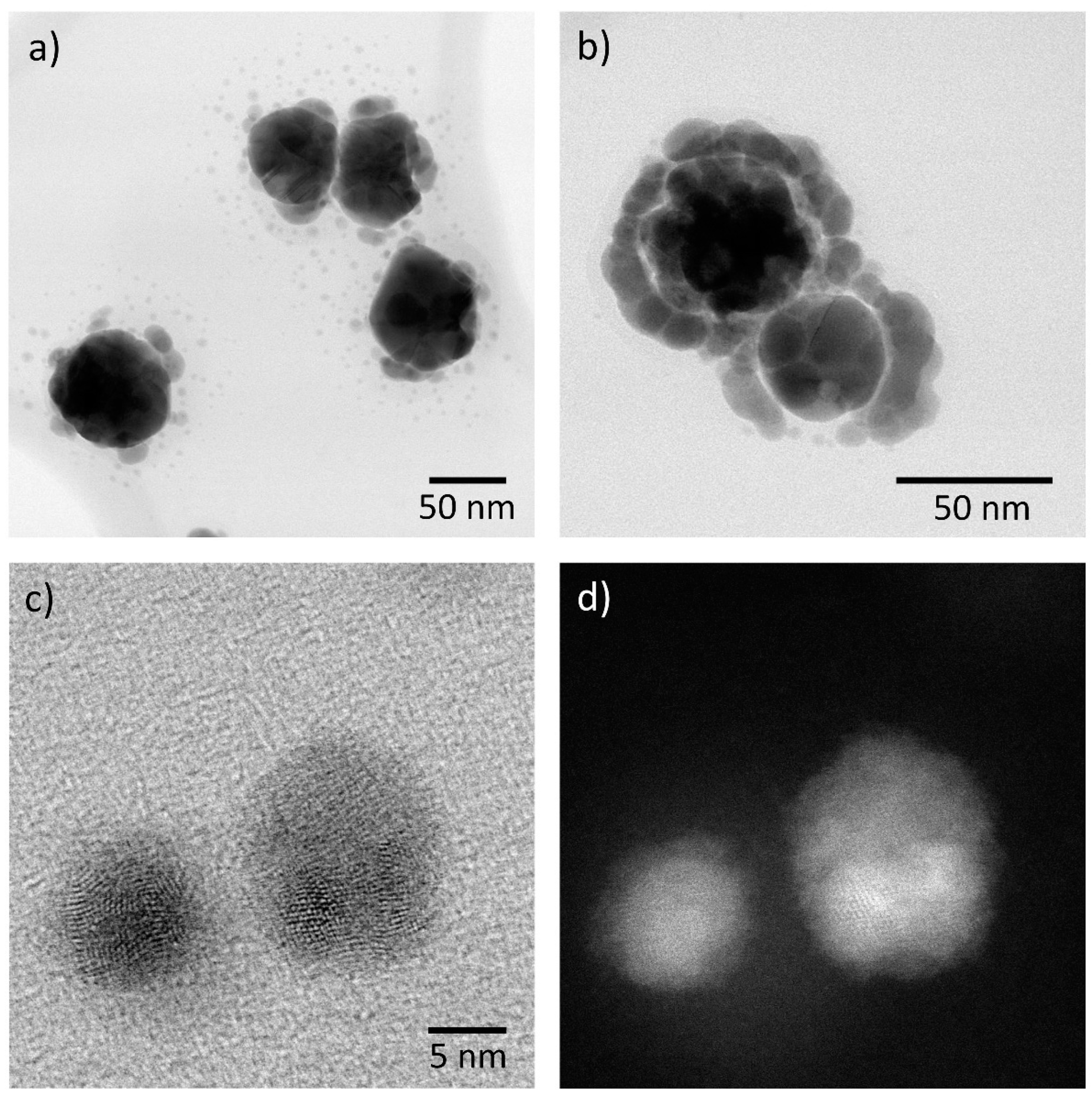
Around the same time, detailed imaging and analysis of silver particles fabricated using solution phase synthesis and then exposed to laboratory air was able to unambiguously identify the final corrosion product as Ag2S [59]. The corrosion product initially formed as new particles located around the fabricated particles, evolving to an inhomogeneous shell at longer times. Silver nanoparticles corroding to a sulfur-containing product with a particulate nature, where particles are found attached to and in the vicinity of the original nanoparticles, is a consistent result observed in the literature [60,61,62,63,64,65]. How closely the corrosion products are associated with the parent nanoparticle is dependent on the RH; a high RH allows for released Ag+ ions to migrate further over the substrate [62]. Typical morphologies for the corrosion products are shown in Figure 4. More rapid corrosion of long nanowires when compared to more regular nanoparticles was assumed to be connected to the greater number of crystalline defects in the former [59].
There are some studies of the corrosion of silver nanoparticles where an a priori assumption was made that the corrosion product was an oxide and the possibility of Ag2S as an explanation for the observed results do not appear to have been considered [66,67,68,69]. The oxidation of nanoparticles can be promoted using ozone concentrations greater than normal atmospheric levels [70].
An important insight into the role of the adsorbed water layer on the mechanism for corrosion of silver nanoparticles was provided by the work of Glover et al. [55], where the formation of a large number of secondary particles appeared when the samples were exposed to laboratory air at high RH but not at low RH. They identified the newly formed particles as secondary silver nanoparticles and did not detect the presence of sulfur, in contrast to earlier studies. It is not certain whether this is because the sulfur was below the detection limit of their methods or because their laboratory atmosphere was particularly low in H2S and OCS. There are some indications in their images of the distinctive bimodal morphology (e.g., see Figure 4c,d) which are indicative of secondary silver nanoparticles corroding to Ag2S [62,63]. They also found that exposure to light rapidly increased the corrosion rate. The work of Glover et al. [55] suggests that the formation of secondary silver nanoparticles, as an intermediary for the transformation to Ag2S, is an important aspect of the mechanism of both bulk and nanoparticle corrosion.
Many studies have investigated the behaviour of silver nanoparticles in solution, and because corrosion in atmosphere is mediated by the adsorbed water layer, these studies can assist with understanding atmospheric corrosion as well. It was shown that the dissolution of silver nanoparticles in solution to Ag+ required both H+ and dissolved O2 [71]. Hence, even though silver corrodes to a sulfide, not an oxide, storing samples in an oxygen-free environment can slow or prevent corrosion (for both bulk and nanoparticle silver). It was proposed that there are three forms of Ag in a colloidal solution: Ag0 in the form of nanoparticles; Ag+ in solution; and Ag+ adsorbed on the surface of the nanoparticles.
Other authors have investigated the intentional sulfidation of nanoparticles using a solution containing Na2S to convert all or a part of the particles to Ag2S [72,73,74,75,76]. With the aid of sulfidation experiments in Na2S solutions, Liu et al. proposed that two processes are possible for the sulfidation of silver nanoparticles: oxidative dissolution of Ag to Ag+ followed by precipitation as Ag2S; or direct conversion to Ag2S, where the former would dominate at low sulfur concentrations and the latter at high concentrations [41]. As shown by Yu et al., in a complex aqueous environment, there is a dynamic process involving the simultaneous release of Ag+ ions, precipitation of new smaller metal nanoparticles, and reactions with other species present [77]. These observations can be expected to have parallels in atmospheric corrosion at high RH. There is evidence that the precipitation of secondary silver nanoparticles occurs for silver nanoparticles in normal atmospheres and that corrosion of these secondary particles to Ag2S is an important aspect of the corrosion process [62,63]. Thus, a third possible mechanism can be added: the oxidative dissolution of Ag to Ag+ followed by precipitation to nanoparticulate Ag, then followed by conversion to Ag2S.
The high reactivity of silver nanoparticles to H2S and their strong plasmonic response have led to them being proposed as atmospheric gas sensors by monitoring the change in the surface plasmon peak intensity with exposure [78]. In contrast to other studies [61,66,79], these authors didn’t see an effect of RH and oxygen on the reaction rate. However, they were operating at accelerated corrosion rates (ppm concentrations of H2S), giving credence to the idea that direct conversion to Ag2S is the corrosion mechanism that dominates at high exposures, in which case RH and the associated Ag+ ion release might not be expected to play a significant role.
In a related experiment, a decrease and shift of the surface plasmon peak was monitored, but with exposure to ambient air rather than at elevated H2S concentrations [69]. Exposure to UV light was necessary for the corrosion to continue, and in this case, oxygen flow did enhance the observed corrosion. The RH was not reported. The changes in the surface plasmon energy were associated with the appearance of secondary particles attached to and in the vicinity of the exposed nanoparticles. Unfortunately, this is an example of a silver corrosion study where it was assumed that the corrosion product was an oxide, and the possibility of a sulfide to explain the observed results does not appear to have been considered.
On the other hand, oxygen, and not sulfur, was detected in an X-ray photoelectron spectroscopy (XPS) study of a nanoparticulate thin film of silver after exposure to laboratory air, except for one sample intentionally exposed to a high S environment (Oates et al., 2013). However, the composition and RH of the laboratory environment was not reported, and a low RH can suppress silver corrosion. Furthermore, XPS reports the composition and valence of the top few atomic layers, and the observations may represent either the adsorbed oxygen-containing species on the surface or the monolayer of AgO that has been proposed to be associated with the transformation of silver in aqueous environments (Johnston et al. 2019). Another XPS study on a silver film also observed the presence of oxygen, rather than sulfur [80], but then assumed the observed corrosion product of silver nanoparticles was an oxide without performing a direct verification. As the corrosion of nanoparticles to Ag2S is strongly enhanced when compared to bulk, this assumption may not be warranted, and the appearance of the corroded nanoparticles resembled those seen for an Ag2S corrosion product. However, it cannot be excluded that in these two studies, an unusually low concentration of H2S and OCS in the ambient air inhibited the formation of Ag2S and a thin oxide layer was formed instead. Another study of silver nanowire electrodes observed no degradation in ambient air after two and a half years [81]. These examples highlight some of the contradictions that exist in the literature when studying corrosion in uncontrolled and unknown ambient atmospheres.
In keeping with the observations from bulk silver that crystal defects are likely to be associated with more rapid corrosion, a number of studies have noted that the antibacterial effectiveness of nanoparticles is enhanced for nanoparticles with more defects [64,82,83]. In some of these cases, enhanced Ag+ ion release with defected structures was also measured. Although it has been noted that inhomogeneous corrosion and the higher corrosion of rods may be associated with defect-enhanced corrosion [59], there has not been any systematic investigation of the role of defects in nanoparticle corrosion. It should be noted that the secondary silver nanoparticles produced by the reduction of Ag+ are often polycrystalline and highly defected when compared to the parent nanoparticle (see Figure 4c,d) and also appear to be corrode very rapidly.
4. Corrosion in Other Environments
The transformation of silver nanoparticles in biological and ecological systems is of considerable interest, and there have been many studies of the fate of silver nanoparticles in relevant environments. As observations in any aqueous environment may have relevance to atmospheric corrosion at elevated humidity, it is worth describing some of the research in this area.
In most environments, transformation to Ag2S is observed, as is the case for atmospheric corrosion. For example, antimicrobial silver nanoparticles in a titanium implant were transformed to Ag2S and located in the newly regenerated bones of rats [84]. The corrosion of silver nanowires to small particles, identified as containing sulfur, occurred in human alveolar epithelial cells (in vitro) [85]. These particles were observed within one hour of exposure, and further decay of the morphology of nanowires occurred up to seven days later. The particle formation appeared to occur more at the tip of the wire, and shell-like structures appeared after longer times. It was proposed that the early stages involved the release and migration of Ag+ ions and re-precipitation as Ag2S and the later stages involved direct transformation at the surface of the particles. It was suggested the rapid transformation to Ag2S would limit the toxicity, alleviating concerns regarding the effect of silver nanoparticle ingestion on human health.
In contrast, X-ray absorption spectroscopy (XAS) of the fate of silver in rat lungs after an inhalation exposure to silver nanoparticles showed that it partially remained in the form of metallic silver and also suggested the formation of small, secondary silver nanoparticles [79]. An examination of silver nanowires incubated in lung lining fluid (LLF) showed the formation of small secondary nanoparticles and the Ag+ dissolution rates varied with pH and LLF components [86].
The potential of introducing artefacts due to the sulfidising effects of some cell culture media have been noted [87]. A comparison of the sulfidation of silver nanowires under incubation in a variety of cell-culture media showed the formation of Ag2S particles in a shell-like structure on surface of the wires in short time frames [82]. Similarly, some sample preparation protocols can enhance the reduction of Ag+ ions back to silver [87].
With the introduction of a variety of consumer products containing antibacterial silver, there have been a number of studies of silver nanoparticles in sewer and wastewater treatment plants. The high availability of sulfur-containing materials in these environments mean that corrosion to Ag2S occurs very readily [83,88,89,90,91,92]. These results may help to alleviate concerns regarding the potential ecotoxic effects of silver nanoparticles upon environmental release. With the high sulfur concentrations, the direct transformation mechanism has been implicated in these environments [90].
A study of commercially available silver containing textiles showed that different textiles produce different nanoparticle products after washing, and these included AgCl, Ag2S, and silver nanoparticles [93,94,95]. Some of these will have been released directly from the textile in the process of washing, and others produced from released Ag+ have been precipitated out from solution during the washing process.
The observations in natural water and soil environments are complex. In a simulated large-scale freshwater wetland it was found that silver nanoparticles were transformed to Ag2S and Ag-sulfhydryl compounds, but silver still remained in significant bio-available quantities after eighteen months [96]. Similarly, it was observed that bioavailable silver remained for an extended period after spiking soil with uncoated silver nanoparticles [97]. The deployment of silver nanoparticles in freshwater and marine environments showed a complex behaviour [98] with the fate of the nanoparticles, depending on the location. The transformation of silver nanoparticles in soil was found to be predominantly to Ag2S under anaerobic conditions, but they persisted under aerobic conditions [99].
These few examples illustrate that many of the themes for the atmospheric corrosion of silver also appear in other environments: the high reactivity of silver to sulfur species; the important role of Ag+ ion release in silver transformation; the formation of secondary silver nanoparticles under certain conditions; and the challenges in understanding the corrosion processes in such complex and variable environments.
5. Summary and Areas for Future Research
Silver corrosion in indoor environments, for both bulk and nanoparticle forms, is dominated by transformations to Ag2S. Although rare, there are a sufficient number of reports of oxide formation for further investigation into whether an oxide layer can appear after extended periods in ambient environments with low H2S, OCS, and/or high ozone concentrations is warranted.
There are three proposed mechanisms for silver corrosion:
- direct conversion to Ag2S;
- oxidative dissolution of Ag to Ag+ followed by precipitation as Ag2S; and
- oxidative dissolution of Ag to Ag+ followed by precipitation as nanoparticulate Ag, then followed by conversion to Ag2S.
The literature is suggestive that direct conversion predominates at high H2S concentrations, but this has not been systematically studied. The other two mechanisms rely on RH and the presence of oxidising species to facilitate the release of Ag+ ions. In general, our understanding what conditions determine which of the three processes occur is not well developed. Studies of silver corrosion have generally relied on unknown and uncontrolled ambient environments or have been performed under accelerated corrosion conditions. In my experience, which has involved studying nanoparticle corrosion in three different laboratories and two different office environments, it is not possible to obtain consistency in the corrosion behaviour from uncontrolled ambient environments.
Given that the mechanism may be different at elevated concentrations of corroding gases, there is a clear need to perform systematic studies of the corrosion behaviour in controlled atmospheric environments, where the H2S (and/or OCS) concentrations are similar to those found in ambient environments (between 0 to ~2 ppb). This presents a considerable technological challenge. The control of H2S concentrations needs to be coupled with the control of RH, which is thankfully much more straightforward. Ideally, controlled concentrations of ozone and NO2 would also be investigated to explore the role of oxidising species and the extent to which aerial oxygen alone can play this role.
A consistent picture in the literature as to whether exposure to light enhances silver corrosion has not emerged. Some studies indicate enhanced corrosion, and others do not. The role of UV light is complicated by the different roles that it might play in separate aspects of the corrosion process. UV light can assist in the generation of oxidising species by decomposing the associated molecules and therefore increase corrosion rates by enhancing the release of Ag+ ions. On the other hand, UV light can promote the photoreduction of Ag+ ions to metallic silver, potentially slowing corrosion rates. A third complicating aspect is that the secondary nanoparticles produced by photoreduction will be small and possibly highly defective and therefore will corrode more quickly than bulk silver or the original nanoparticle. The role of light illumination and its interplay with oxidising species, H2S and OCS concentrations and RH is an area that warrants further systematic investigation.
As nanoparticles corrode much more quickly than bulk silver and are readily imaged and analysed using advanced electron microscopy methods, the corrosion processes in nanoparticles are more amenable to investigation than is the case for bulk silver. There is also significant technological interest in nanoparticle corrosion. Furthermore, many of the themes in silver corrosion are common to both bulk and nanoparticle forms. However, the appearance of small, secondary silver nanoparticles is a key factor in nanoparticle corrosion, and there are some indications that it may also occur for bulk silver. Given the advances that have occurred in imaging and analysis since bulk silver corrosion was first investigated in earlier decades, there is an argument that the atomistic mechanisms of bulk silver corrosion should be revisited.
Further analysis of the role of defects such as stacking faults, dislocation, edges, steps, and grain boundaries using high resolution imaging and analysis would also be valuable. Finally, the outdoor corrosion of silver has received only limited attention to date and is another area worthy of more detailed study.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
The author acknowledges the use of facilities within the University of Wollongong Electron Microscopy Centre.
Conflicts of Interest
The author declares no conflict of interest.
References
- Emsley, J. Nature’s Building Blocks: An A-Z Guide to the Elements, 2nd ed.; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Rycenga, M.; Cobley, C.M.; Zeng, J.; Li, W.; Moran, C.H.; Zhang, Q.; Qin, D.; Xia, Y. Controlling the synthesis and assembly of silver nanostructures for plasmonic applications. Chem. Rev. 2011, 111, 3699–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Peng, Y.; Yang, Y.; Li, Z.Y. Plasmon-enhanced light–matter interactions and applications. NPJ Comput. Mater. 2019, 5, 45. [Google Scholar] [CrossRef]
- Ardakani, L.S.; Surendar, A.; Thangavelu, L.; Mandal, T. Silver nanoparticles (Ag NPs) as catalyst in chemical reactions. Synth. Commun. 2021, 51, 1516–1536. [Google Scholar]
- Cortie, M.B.; Arnold, M.D.; Keast, V.J. The quest for zero loss: Unconventional materials for plasmonics. Adv. Mater. 2019, 32, 1904532. [Google Scholar] [CrossRef]
- Naik, G.V.; Shalaev, V.M.; Boltasseva, A. Alternative plasmonic materials: Beyond gold and silver. Adv. Mater. 2013, 25, 3264–3294. [Google Scholar] [CrossRef]
- Burdușel, A.C.; Gherasim, O.; Grumezescu, A.M.; Mogoantă, L.; Ficai, A.; Andronescu, E. Biomedical applications of silver nanoparticles: An up-to-date review. Nanomaterials 2018, 8, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Sonshine, D.A.; Shervani, S.; Hurt, R.H. Controlled release of biologically active silver form nanosilver surfaces. ACS Nano 2010, 4, 6903–6913. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Heras, M.; Theodorou, I.G.; Leo, B.F.; Ryan, M.P.; Porter, A.E. Towards understanding the antibacterial activity of Ag nanoparticles: Electron microscopy in the analysis of the materials-biology interface in the lung. Environ. Sci. Nano 2015, 2, 312–326. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.W. History of the medical use of silver. Surg. Infect. 2009, 10, 289–292. [Google Scholar] [CrossRef] [Green Version]
- Barillo, D.J.; Marx, D.E. Silver in medicine: A brief history BC 335 to present. Burns 2014, 40S, S3–S8. [Google Scholar] [CrossRef]
- Reidy, B.; Haase, A.; Luch, A.; Dawson, K.A.; Lynch, I. Mechanisms of silver nanoparticle release, transformation and toxicity: A critical review of current knowledge and recommendations for future studies and applications. Materials 2013, 6, 2295–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, D.E.; Barillo, D.J. Silver in medicine: The basic science. Burns 2014, 405, S9–S18. [Google Scholar] [CrossRef] [PubMed]
- McQueen, R.H.; Keelan, M.; Xu, Y.; Mah, T. In vivo assessment of odour retention in an antimicrobial silver chloride-treated polyester textile. J. Text. Inst. 2013, 104, 108–117. [Google Scholar] [CrossRef]
- Toy, L.W.; Macera, L. Evidence-based review of silver dressing use on chronic wounds. J. Am. Acad. Nurse Pract. 2011, 23, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Walter, N.; McQueen, R.H.; Keelan, M. In vivo assessment of antimicrobial treated textiles on skin microflora. Int. J. Cloth. Sci. Technol. 2014, 26, 330–342. [Google Scholar] [CrossRef]
- Tortella, G.R.; Rubilar, O.; Durán, N.; Diez, M.C.; Martinez, M.; Parada, J.; Seabra, A.B. Silver nanoparticles: Toxicity in model organisms as an overview of is hazard for human health and the environment. J. Hazard. Mater. 2020, 390, 121974. [Google Scholar] [CrossRef]
- Storme, P.; Schalm, O.; Wiesinger, R. The sulfidation process of sterling silver in different corrosive environments: Impact of the process on the surface films formed and consequences for the conservation-restoration community. Herit. Sci. 2015, 3, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Tissot, I.; Monteiro, O.C.; Barreiros, M.A.; Correia, J.; Guerra, M.F. The influence of the constituent elements on the corrosion mechanisms of silver alloys in sulphide environments: The case of sterling silver. RSC Adv. 2017, 7, 28564–28572. [Google Scholar] [CrossRef] [Green Version]
- Phillips, V.A. Role of defects in evaporated silver films on the nucleation of sulfide “patches”. J. App. Phys. 1962, 33, 712–717. [Google Scholar] [CrossRef]
- Guan, R.; Yu, Y.D. A TEM study of Ag8S formed in the early stage of sulfidization of silver. Scripta Met. 1990, 24, 869–872. [Google Scholar] [CrossRef]
- Salvado, N.; Buti, S.; Labrador, A.; Cinque, G.; Emerich, H.; Pradell, T. SR-XRD and SR-FTIR study of the alteration of silver foils in medieval paintings. Anal. Bioanal. Chem. 2011, 399, 3041–3052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.D.; Guan, R. Electron-microscope study of the structure of Ag8S formed in the initial stage of silver sulfidation. Acta Cryst. 1995, B51, 149–155. [Google Scholar] [CrossRef]
- Sadanaga, R.; Sueno, S. X-ray study on the α-β transition of Ag2S. Mineral. J. 1967, 5, 124–148. [Google Scholar] [CrossRef] [Green Version]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Sanders, C.E.; Verreault, D.; Frankel, G.S.; Allen, H.C. The role of sulfur in the atmospheric corrosion of silver. J. Electrochem. Soc. 2015, 162, C630–C637. [Google Scholar] [CrossRef]
- Wan, Y.; Wang, X.; Wang, X.; Li, Y.; Sun, H.; Zhang, K. Determination and generation of the corrosion compounds in silver exposed to the atmospheres. Int. J. Electrochem. Sci. 2015, 10, 2336–2354. [Google Scholar]
- Lin, H.; Frankel, G.S.; Abbott, W.H. Analysis of Ag corrosion products. J. Electrochem. Soc. 2013, 160, C345–C355. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Macha, E.N.; Kelly, R.G. Modification of ASTM B117 salt spray corrosion test and its correlation to field measurements of silver corrosion. Corrosion 2012, 68, 036001. [Google Scholar] [CrossRef]
- Watanabe, M.; Hokazono, A.; Handa, T.; Ichino, T.; Kuwaki, N. Corrosion of copper and silver plates by volcanic gases. Corros. Sci. 2006, 48, 3759–3766. [Google Scholar] [CrossRef]
- Watanabe, M.; Shinozaki, S.; Toyoda, E.; Asakura, K.; Ichino, T.; Kuwaki, N.; Higashi, Y.; Tanaka, T. Corrosion products formed on silver after a one-month exposure to urban atmospheres. Corrosion 2006, 62, 243–250. [Google Scholar] [CrossRef]
- Lin, H.; Frankel, G.S. Accelerated atmospheric corrosion testing of Ag. Corrosion 2013, 69, 1060–1072. [Google Scholar] [CrossRef]
- Liang, D.; Allen, H.C.; Frankel, G.S.; Chen, Z.Y.; Kelly, R.G.; Wu, Y.; Wyslouzil, B.E. Effects of sodium chloride particles, ozone, UV and relative humidity on atmospheric corrosion of silver. J. Electrochem. Soc. 2010, 157, C146–C156. [Google Scholar] [CrossRef] [Green Version]
- Franey, J.P.; Kammlott, G.W.; Graedel, T.E. The corrosion of silver by atmospheric sulfur gases. Corros. Sci. 1985, 25, 133–143. [Google Scholar] [CrossRef]
- Graedel, T.E.; Franey, J.P.; Gualtieri, G.J.; Kammlott, G.W.; Malm, D.L. On the mechanism of silver and copper sulfidation by atmospheric H2S and OCS. Corros. Sci. 1985, 25, 1163–1180. [Google Scholar] [CrossRef]
- Graedel, T.E. Corrosion mechanisms for silver exposed to the atmosphere. J. Electrochem. Soc. 1992, 139, 1963–1970. [Google Scholar] [CrossRef]
- Pope, D.; Gibbens, H.R.; Moss, R.L. The tarnishing of Ag at naturally-occurring H2S and SO2 levels. Corros. Sci. 1968, 8, 883–887. [Google Scholar] [CrossRef]
- Rice, D.W.; Peterson, P.; Rigby, E.B.; Phipps, P.B.P.; Coppell, R.J.; Tremoureux, R. Atmospheric corrosion of copper and silver. J. Electrochem. Soc. 1981, 128, 275–284. [Google Scholar] [CrossRef]
- Martina, I.; Wiesinger, R.; Schreiner, M. Micro-Raman investigations of early stage silver corrosion products occurring in sulfur containing atmospheres. J. Raman Spectrosc. 2013, 44, 770–775. [Google Scholar] [CrossRef]
- Sinclair, J.D. Tarnishing of solver by organic sulfur vapours: Rates and film characteristics. Electrochem. Sci. Technol. 1982, 239, 33–40. [Google Scholar] [CrossRef]
- Liu, J.; Pennell, K.G.; Hurt, R.H. Kinetics and mechanisms of nanosilver oxysulfidation. Environ. Sci. Technol. 2011, 45, 7345–7353. [Google Scholar] [CrossRef] [Green Version]
- Bennett, H.E.; Peck, R.L.; Burge, D.K.; Bennett, J.M. Formation and growth of tarnish on evaporated silver films. J. Appl. Phys. 1969, 40, 3351–3360. [Google Scholar] [CrossRef]
- Wiesinger, R.; Grayburn, R.; Dowsett, M.; Sabbe, P.J.; Thompson, P.; Adriaens, A.; Schreiner, M. In situ time-lapse synchrotron radiation X-ray diffraction of silver corrosion. J. Anal. At. Spectrom. 2015, 30, 694–701. [Google Scholar] [CrossRef]
- Price, L.E.; Thomas, G.J. The tarnishing of silver and silver alloys and its prevention. J. Inst. Metals 1938, 63, 29–65. [Google Scholar]
- Chou, C.H. Hydrogen Sulfide: Human Health Aspects. In Concise International Chemical Assessment Document; World Health Organization: Geneva, Switzerland, 2003; Volume 53. [Google Scholar]
- Kleber, C.; Wiesinger, R.; Schnöller, J.; Hilfrich, U.; Hutter, H.; Schreiner, M. Initial oxidation of silver surfaces by S2− and S4+ species. Corros. Sci. 2008, 50, 1112–1121. [Google Scholar] [CrossRef]
- Rice, D.W.; Cappell, R.J.; Kinsolving, W.; Laskowski, J.J. Indoor corrosion of metals. J. Electrochem. Soc. 1980, 127, 891–901. [Google Scholar] [CrossRef]
- Ankersmit, H.A.; Tennant, N.H.; Watts, S.F. Hydrogen sulfide and carbonyl sulfide in the museum environment—Part I. Atmos. Environ. 2005, 39, 695–707. [Google Scholar] [CrossRef]
- Leygraf, C.; Wallinde, I.O.; Tidblad, J.; Graedel, T. Atmospheric Corrosion, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Volpe, L.; Peterson, P.J. The atmospheric sulfidation of silver in a tubular corrosion reactor. Corros. Sci. 1989, 29, 1179–1196. [Google Scholar] [CrossRef]
- Fischmeister, H.; Drott, J. Reaction rate and growth forms of reaction product in the system Ag-H2S. Acta Metall. 1959, 7, 777–781. [Google Scholar] [CrossRef]
- Lee, S.H.; Jun, B.H. Silver nanoparticles: Synthesis and application for nanomedicine. Int. J. Mol. Sci. 2019, 20, 865. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Li, Z.Y.; Zhong, Z.; Gates, B.; Xia, Y.; Venkateswaran, S. Synthesis and characterization of stable aqueous dispersions of silver nanoparticles through the Tollens process. J. Mater. Chem. 2002, 12, 522–527. [Google Scholar] [CrossRef]
- Allpress, J.G.; Sanders, J.V. The influence of surface structure on a tarnishing reaction. Philos. Mag. 1964, 10, 829–836. [Google Scholar] [CrossRef]
- Glover, R.D.; Miller, J.M.; Hutchinson, J.E. Generation of metal nanoparticles from silver and copper objects: Nanoparticle dynamics on surfaces and potential sources of nanoparticles in the environment. ACS Nano 2011, 5, 8950–8957. [Google Scholar] [CrossRef] [PubMed]
- Le Ouay, B.; Stellacci, F. Antibacterial activity of silver nanoparticles: A surface science insight. Nano Today 2015, 10, 339–354. [Google Scholar] [CrossRef] [Green Version]
- McMahon, M.D.; Lopez, R.; Meyer, H.M.; Feldmen, L.C.; Haglund, R.F., Jr. Rapid tarnishing of silver nanoparticles in ambient laboratory air. Appl. Phys. B 2005, 80, 915–921. [Google Scholar] [CrossRef]
- Cao, W.; Elsayed-Ali, H.E. Stability of Ag nanoparticles fabricated by electron beam lithography. Mater. Lett. 2009, 63, 2263–2266. [Google Scholar] [CrossRef]
- Elechiguerra, J.L.; Larios-Lopez, L.; Lui, C.; Garcia-Gutierrez, D.; Camacho-Bragado, A.; Yacaman, M.J. Corrosion at the nanoscale: The case of silver nanowires and nanoparticles. Chem. Mater. 2005, 17, 6042–6052. [Google Scholar] [CrossRef]
- Lu, W.; Yao, K.; Wang, J.; Yuan, J. Ionic liquids-water interfacial preparation of triangular Ag nanoplates and their shape-dependent antibacterial activity. J. Colloid Interface Sci. 2015, 437, 35–41. [Google Scholar] [CrossRef]
- Wang, L.; Xiong, W.; Nishijima, Y.; Yokota, Y.; Ueno, K.; Misawa, H.; Bi, G.; Qiu, J.R. Spectral properties and mechanism of instability of nanoengineered silver blocks. Opt. Express 2011, 19, 10640–10646. [Google Scholar] [CrossRef]
- Keast, V.J. Corrosion processes of silver nanoparticles. Appl. Nanosci. 2022, 12, 1859–1868. [Google Scholar] [CrossRef]
- Keast, V.J.; Myles, T.A.; Shahcheraghi, N.; Cortie, M.B. Corrosion processes of triangular silver nanoparticles compared to bulk silver. J. Nanopart. Res. 2016, 18, 45. [Google Scholar] [CrossRef]
- Scuderi, M.; Esposito, M.; Todisco, F.; Simeone, D.; Tatamtini, I.; De Marco, L.; De Giorgi, M.; Nicotra, G.; Carbone, L.; Sanvitto, D.; et al. Nanoscale study of the tarnishing process in electron beam lithography-fabricated silver nanoparticles for plasmonic applications. J. Phys. Chem. C 2016, 120, 24314–24323. [Google Scholar] [CrossRef]
- Trautmann, S.; Dathe, A.; Csáki, A.; Thiele, M.; Müller, R.; Fritzche, W.; Stranik, O. Time-resolved study of site-specific corrosion in a single crystalline silver nanoparticle. Nanoscale Res. Lett. 2019, 14, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Zhong, H.; Zhang, L. Optical measurements of oxidation behavior of silver nanometer particle within pores of silica host. J. Appl. Phys. 1998, 83, 1705–1710. [Google Scholar] [CrossRef]
- Qi, H.; Alexson, D.; Glembocki, O.; Prokes, S.M. The effect of size and size distribution on the oxidation kinetics and plasmonics of nanoscale Ag particles. Nanotechnology 2010, 21, 215706. [Google Scholar] [CrossRef]
- Kuzma, A.; Weis, M.; Flickyngerova, S.; Jakabovic, J.; Satka, A.; Dobrocka, E.; Chlpik, J.; Cirak, J.; Donoval, M.; Telek, P.; et al. Influence of surface oxidation on plasmon resonance in monolayer of gold and silver nanoparticles. J. Appl. Phys. 2012, 112, 130531. [Google Scholar] [CrossRef]
- Grillet, N.; Manchon, D.; Cottancin, E.; Bertorelle, F.; Bonnet, C.; Broyer, M.; Lerme, J.; Pellarin, M. Photo-oxidation of individual silver nanoparticles: A real-time tracking of optical and morphological changes. J. Phys. Chem. C 2013, 117, 2274–2282. [Google Scholar] [CrossRef]
- Han, Y.; Lupitsky, R.; Chou, T.M.; Stafford, C.M.; Du, H.; Sukhishvila, S. Effect of oxidation on surface-enhanced Raman scattering activity of silver nanoparticles: A quantitative correlation. Anal. Chem. 2011, 83, 5873–5880. [Google Scholar] [CrossRef]
- Liu, J.; Hurt, R.H. Ion release kinetics and particle persistence in aqueous nano-silver colloids. Environ. Sci. Technol. 2010, 44, 2169–2175. [Google Scholar] [CrossRef]
- Levard, C.; Reinsch, B.C.; Michel, F.M.; Oumahi, C.; Lowry, G.V.; Brown, G.E., Jr. Sulfidation processes of PVP-coated silver nanoparticles in aqueous solution: Impact on dissolution rate. Environ. Sci. Technol. 2011, 45, 5260–5266. [Google Scholar] [CrossRef]
- Liu, B.; Ma, Z. Synthesis of Ag2S-Ag nanoprisms and their use as DNA hybridization probes. Small 2011, 7, 1587–1592. [Google Scholar] [CrossRef]
- Zeng, J.; Tao, J.; Su, D.; Zhu, Y.; Qin, D.; Xia, Y. Selective sulfuration at the corner sites of silver nanocrystal and it’s use in stabilization of the shape. Nano Lett. 2011, 11, 3010–3015. [Google Scholar] [CrossRef] [PubMed]
- Park, G.; Lee, C.; Seo, D.; Song, H. Full-color tuning of surface plasmon resonance by compositional variation of Ay@Ag core-shell nanocubes with sulfides. Langmuir 2012, 28, 9003–9009. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, Q.; Wang, J.; Li, Z.; Yu, X.F.; Chu, P.K. Sensitive and robust colorimetric sensing of sulfide anion by plasmonic nanosensors based on quick crystal growth. Plasmonics 2014, 9, 11–16. [Google Scholar] [CrossRef]
- Yu, S.; Yin, Y.; Chao, J.; Shen, M.; Liu, J. Highly dynamic PVP-coated silver nanoparticles in aquatic environments: Chemical and morphology change induced by oxidation of Ag0 and reduction of Ag+. Environ. Sci. Technol. 2014, 48, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Morris, H.R.; Whitmore, P.M. Fast detection of hydrogen sulfide gas in the ppm range with silver nanoparticle films at ambient conditions. Sens. Actuators B Chem. 2013, 186, 431–438. [Google Scholar] [CrossRef]
- Davidson, R.A.; Anderson, D.S.; Van Winkle, L.S.; Pinkerton, K.E.; Guo, T. Evolution of silver nanoparticles in the rat lung investigated by X-ray absorption spectroscopy. J. Phys. Chem. A 2014, 119, 281–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Santschi, C.; Martin, O.J.F. Strong improvement of long-Term chemical and thermal stability of plasmonic silver nanoantennas and films. Small 2017, 13, 1700044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayousse, C.; Celle, C.; Fraczkiewicz, A.; Simonato, J.P. Stability of silver nanowire based electrodes under environmental and electrical stresses. Nanoscale 2015, 7, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Theodorou, I.G.; Goode, A.E.; Gow, A.; Schwander, S.; Zhang, J.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.; Ryan, M.P.; et al. High-resolution analytical electron microscopy reveals cell culture media-induced changes to the chemistry of silver nanowires. Environ. Sci. Technol. 2013, 47, 13813–13821. [Google Scholar] [CrossRef] [Green Version]
- Impellitteri, C.A.; Harmon, S.; Silva, R.G.; Miller, B.W.; Scheckel, K.G.; Luxton, T.P.; Schupp, D.; Panguluri, S. Transformation of silver nanoparticles in fresh, ages and incinerated biosolids. Water Res. 2013, 47, 3678–3886. [Google Scholar] [CrossRef]
- Geng, H.; Poologasundarampillai, G.; Todd, N.; Devlin-Mullin, A.; Moore, K.L.; Golrokhi, Z.; Gilchrist, J.B.; Jones, E.; Potter, R.J.; Sutcliffe, C.; et al. Biotransformation of silver released from nanoparticle coated titanium implants revealed in regenerating bone. Appl. Mater. Interfaces 2017, 9, 21169–21180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Goode, A.E.; Sweeney, S.; Theodorou, I.G.; Thorley, A.J.; Ruenraroengsak, P.; Chang, Y.; Gow, A.; Schwander, S.; Skepper, J.; et al. Sulfidation of silver nanowires inside human alveolar epithelial cells: A potential detoxification mechanism. Nanoscale 2013, 5, 9839–9847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodorou, I.G.; Botelho, D.; Schwander, S.; Zhang, J.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.P.; Gow, A.; Ryan, M.P.; Porter, A.E. Static and dynamic microscopy of the chemical stability and aggregation state of silver nanowires in components of Murine pulmonary surfactant. Environ. Sci. Technol. 2015, 49, 8048–8056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Goode, A.E.; Skepper, J.N.; Thorley, A.J.; Seiffert, J.M.; Chung, K.F.; Tetley, T.D.; Shaffer, M.S.P.; Ryan, M.P.; Porter, A.E. Avoiding artefacts during electron microscopy of silver nanomaterials exposed to biological environments. J. Microsc. 2015, 261, 157–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaegi, R.; Voegelin, A.; Ort, C.; Sinnet, B.; Thalmann, B.; Krismer, J.; Hagendorfer, H.; Elumela, M.; Mueller, E. Fate and transformation of silver nanoparticles in urban wastewater systems. Water Res. 2013, 47, 3866–3877. [Google Scholar] [CrossRef]
- Kaegi, R.; Voegelin, A.; Sinnet, B.; Zuleeg, S.; Hagendorfer, H.; Burkhardt, M.; Siegrist, H. Behavior of metallic silver nanoparticles in a pilot wastewater treatment plant. Environ. Sci. Technol. 2011, 45, 3902–3908. [Google Scholar] [CrossRef]
- Kent, R.D.; Oser, J.G.; Vikesland, P.J. Controlled evaluation of silver nanoparticle sulfidation in a full-scale wastewater treatment plant. Environ. Sci. Technol. 2014, 48, 8564–8572. [Google Scholar] [CrossRef]
- Lombi, E.; Donner, E.; Taheri, S.; Tavakkoli, E.; Jamting, A.K.; McClure, S.; Naidu, R.; Miller, B.W.; Scheckel, K.G.; Vasilev, K. Transformation of four silver/silver chloride nanoparticles during anaerobic treatment of wastewater and post-processing of sewage sludge. Environ. Pollut. 2013, 176, 193–197. [Google Scholar] [CrossRef]
- Ma, R.; Levard, C.; Judy, J.D.; Unrine, J.M.; Durenkamp, M.; Martin, B.; Jefferson, B.; Lowry, G.V. Fate of zinc oxide and silver nanoparticles in a pilot wastewater treatment plant and in processed biosolids. Environ. Sci. Technol. 2013, 48, 104–112. [Google Scholar] [CrossRef]
- Lorenz, C.; Windler, L.; von Goetz, N.; Lehman, R.P.; Schuppler, M.; Hungerbuhler, K.; Heuberger, M.; Nowack, B. Characterization of silver release from commercially available functional (nano)textiles. Chemosphere 2012, 89, 817–824. [Google Scholar] [CrossRef]
- Mitrano, D.M.; Rimmele, E.; Wischer, A.; Erni, R.; Height, M.; Nowack, B. Presence of nanoparticles in wash water from conventional silver and nano-silver textiles. ACS Nano 2014, 8, 7208–7219. [Google Scholar] [CrossRef] [PubMed]
- Impellitteri, C.A.; Tolaymat, T.M.; Scheckel, K.G. The speciation of silver nanoparticles in antimicrobial fabric before and after exposure to a hypochlorite/detergent solution. J. Environ. Qual. 2009, 38, 1528–1530. [Google Scholar] [CrossRef] [PubMed]
- Lowry, G.V.; Epinasse, B.P.; Badireddy, A.R.; Richardson, C.J.; Reinsch, B.C.; Bryant, L.D.; Bone, A.J.; Deonarine, A.; Chae, S.; Therezin, M.; et al. Long-term transformation and fate of manufactured Ag nanoparticles in a simulated large scale freshwater emergent wetland. Environ. Sci. Technol. 2012, 46, 7027–7036. [Google Scholar] [CrossRef] [PubMed]
- Coutris, C.; Joner, E.J.; Oughton, D.H. Aging and soil organic matter content affect the fate of silver nanoparticles in soil. Sci. Total Environ. 2012, 420, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Khaksar, M.; Jolley, D.E.; Sekine, R.; Vasilev, K.; Johannessen, B.; Donner, E.; Lombi, E. In situ chemical transformations of silver nanoparticles along the water-sediment continuum. Environ. Sci. Technol. 2015, 49, 318–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, Y.; Takeuchi, S.; Mitsunobu, S.; Ok, Y.S. Chemical speciation of silver (Ag) in soils under aerobic and anaerobic conditions: Ag nanoparticles vs. ionic Ag. J. Hazard. Mater. 2015, 322, 318–324. [Google Scholar] [CrossRef]
Figure 1.
Pure silver sheet after exposure to ambient office and laboratory environments for a period of a couple of years (left) as compared to a couple of decades (right).
Figure 1.
Pure silver sheet after exposure to ambient office and laboratory environments for a period of a couple of years (left) as compared to a couple of decades (right).

Figure 2.
Crystal structure of acanthite Ag2S. Image generated using VESTA 3 [25].
Figure 2.
Crystal structure of acanthite Ag2S. Image generated using VESTA 3 [25].

Figure 3.
Corrosion layer formed on bulk silver imaged with (a) transmission electron microscopy (TEM) in cross-section and (b) scanning electron microscopy of the surface. This corrosion layer was one that had formed after many years of exposure to ambient air and can be seen to be particulate in nature.
Figure 3.
Corrosion layer formed on bulk silver imaged with (a) transmission electron microscopy (TEM) in cross-section and (b) scanning electron microscopy of the surface. This corrosion layer was one that had formed after many years of exposure to ambient air and can be seen to be particulate in nature.

Figure 4.
TEM images of silver nanoparticles exposed to ambient air at (a) medium and (b) low levels of humidity. Characteristic bimodal appearance of the corrosion product in (c) bright field (BF) and (d) annular dark field images (ADF).
Figure 4.
TEM images of silver nanoparticles exposed to ambient air at (a) medium and (b) low levels of humidity. Characteristic bimodal appearance of the corrosion product in (c) bright field (BF) and (d) annular dark field images (ADF).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Keast, V.J. Atmospheric Corrosion of Silver and Silver Nanoparticles. Corros. Mater. Degrad. 2022, 3, 221-234. https://0-doi-org.brum.beds.ac.uk/10.3390/cmd3020013
AMA Style
Keast VJ. Atmospheric Corrosion of Silver and Silver Nanoparticles. Corrosion and Materials Degradation. 2022; 3(2):221-234. https://0-doi-org.brum.beds.ac.uk/10.3390/cmd3020013
Chicago/Turabian StyleKeast, Vicki J. 2022. "Atmospheric Corrosion of Silver and Silver Nanoparticles" Corrosion and Materials Degradation 3, no. 2: 221-234. https://0-doi-org.brum.beds.ac.uk/10.3390/cmd3020013