The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview


1
Meharry Medical College School of Medicine, Department of Biochemistry, Cancer Biology, Neuroscience and Pharmacology, Nashville, TN 37208, USA
2
Vanderbilt University School of Medicine, Department of Surgery, Colon and Rectal Surgery, Nashville, TN 37232, USA
3
The American Society of Colon and Rectal Surgeons (ASCRS), Arlington Heights, IL 60005, USA
4
The American Gastroenterological Association (AGA), Bethesda, MD 20814, USA
5
Vanderbilt-Ingram Cancer Center (VICC), Vanderbilt University Medical Center, Nashville, TN 37232, USA
Gastrointest. Disord. 2019, 1(1), 75-105; https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1010007
Submission received: 18 September 2018
/
Revised: 9 October 2018
/
Accepted: 12 October 2018
/
Published: 18 October 2018
(This article belongs to the Special Issue The Interplay between Genetic Risk Factors, the Intestinal Microbiota, the Environment and Immune Responses in the Pathogenesis of IBD)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The gastrointestinal system where inflammatory bowel disease occurs is central to the immune system where the innate and the adaptive/acquired immune systems are balanced in interactions with gut microbes under homeostasis conditions. This article overviews the high-throughput research screening on multifactorial interplay between genetic risk factors, the intestinal microbiota, urbanization, modernization, Westernization, the environmental influences and immune responses in the etiopathogenesis of inflammatory bowel disease in humans. Inflammatory bowel disease is an expensive multifactorial debilitating disease that affects thousands new people annually worldwide with no known etiology or cure. The conservative therapeutics focus on the established pathology where the immune dysfunction and gut injury have already happened but do not preclude or delay the progression. Inflammatory bowel disease is evolving globally and has become a global emergence disease. It is largely known to be a disease in industrial-urbanized societies attributed to modernization and Westernized lifestyle associated with environmental factors to genetically susceptible individuals with determined failure to process certain commensal antigens. In the developing nations, increasing incidence and prevalence of inflammatory bowel disease (IBD) has been associated with rapid urbanization, modernization and Westernization of the population. In summary, there are identified multiple associations to host exposures potentiating the landscape risk hazards of inflammatory bowel disease trigger, that include: Western life-style and diet, host genetics, altered innate and/or acquired/adaptive host immune responses, early-life microbiota exposure, change in microbiome symbiotic relationship (dysbiosis/dysbacteriosis), pollution, changing hygiene status, socioeconomic status and several other environmental factors have long-standing effects/influence tolerance. The ongoing multipronged robotic studies on gut microbiota composition disparate patterns between the rural vs. urban locations may help elucidate and better understand the contribution of microbiome disciplines/ecology and evolutionary biology in potentially protecting against the development of inflammatory bowel disease.
Keywords:
gastrointestinal disorders; inflammatory bowel disease; ulcerative colitis; Crohn’s disease; indeterminate colitis; inflammation; rural lifestyle; urbanization; diet/nutrition; environment; intestinal microbiota; dysbiosis/dysbacteriosis); genetics; ethnicity; innate immune system; adaptive/acquired immune system1. Introduction
The “Colitides” also known as Inflammatory Bowel Disease (IBD), include ulcerative colitis (UC) and Crohn’s disease (CD), is intestinal disease that cause prolonged chronic relapsing and remitting inflammation of the digestive tract due to multifactorial interplay between genetic risk, the immune system, environmental exposures, and the intestinal microbiota in genetically susceptible individuals [1,2,3]. The aetiopathogenesis of UC and CD remains enigmatic [4,5]. The incidence of IBD is alarmingly evolving in pediatrics and young adults worldwide [4,5]. In the beginning of the 21st century, in some developed nations the incidence of IBD declined with a prevalence of as lower as up to 0.5% of the general population, while it has continued to rise in developing countries [6,7,8] as well as in some Western and developed countries [9,10,11,12,13].
IBD incidence and prevalence is evolving worldwide [14,15] and is now contemplated to be an emergence global disease [4]. The burden of IBD varies in different countries and locations, especially when compared between developing [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31] and developed nations [32,33]. Data suggest that younger populations are more affected [6,34] and that the peak incidence of IBD occurs in children and adolescents [34,35]. It is estimated that 25 to 30 percent of cases diagnosed with CD and 20 percent of patients with UC present early in life and in most cases before the age of 20 years [35,36,37].
Rapid urbanization represents a major demographic shift and has been associated with an escalated incidence of several autoimmune diseases, including IBD [36,38,39,40]. In this long extensive literature search overview article we discuss: (i) pathogenesis of IBD, seeking to better understand accurately the aetiopathogenesis of IBD. (ii) environmental factors, building on the knowledge of how factors like diet, microbiota and psychological stress are reflected to play a role in IBD (iii) preclinical human IBD mechanisms, paying close attention to how IBD manifests in patients and ensuring research in the laboratory reflects this understanding, (iv) novel technologies, applying the latest multipronged innovations like non-invasive imaging and biosensors to IBD and (v) pragmatic clinical research, working in collaboration between basic scientist, clinical teams and patients to ultimately answer questions relevant to daily clinical practice and evaluate the effectiveness of current practices in diagnostics and treatments.
2. Methods
Performed literature review using multipronged search engine predetermined protocol in accordance with the quality assurance of reporting meta-analyses of observational contemplations (MOOSE) [41,42]. Preferred reporting items for review and meta-analysis protocols (PRISMA-P) was followed [43]. A comprehensive multipronged search of the “inflammatory bowel disease (IBD)” etiopathogenesis was carried out through 30 of June 2018 using Medical Literature Analysis, PubMed, and Retrieval System Online (MEDLINE), Current Nursing, Excerpta Medica database (EMBASE), and Allied Health Literature (CINAHL), Web of Science, the Cochrane library, and Google® search engine. The following search terms were used: inflammatory bowel disease, indeterminate colitis, ulcerative colitis, Crohn’s disease, Crohn’s colitis, inflammation, etiology, pathogenesis, intestinal microbiota, genetic risk factors, environmental factors, diet, immune responses, Westernization culture, developed countries, urbanization, developing nations, diagnostics, and treatment. Subordinate and hand/manual searches of reference lists, other studies cross-indexed by reviews, authors, books, commentaries, and conference abstracts were also carry out. Published reports in language other than English, non-human studies and editorials were eliminated. Manuscript inclusions were based on the available supportive evidence for each particular detailed item of interest. Final, conclusive consensus was statistically evaluated with the k-statistic during the title and abstract reviews. As a result, titles were examined and divided into two sets when the value was ≥0.6; each was reviewed by the researcher. Assessable discrepancies were corrected, followed by other assessments of agreement when the value was <0.6.
3. Results
There were 51,671 publications identified in the review search of the possible etiopathogenesis of IBD (22,925 for UC, 27,536 for CD and 1210 for indeterminate colitis (IC). Of the 51,671 publications, 13,773 were duplicate publications and were excluded. Further, following a review of the abstract or title, additional 23,970 were removed because were found not to be relevant to the topic of etiopathogenesis, leaving 13,928 full-text articles. Of the 13,928 reviewed articles, 277 were further excluded because they were not English-language publications and/or non-human studies leaving 13,651 articles qualified for inclusion in this extensive summarized overview.
4. Etiopathogenesis of IBD
Inflammatory bowel disease has been understood to be idiopathic attributable to several possible effectuates that include genetic, gut microbiota, dysbiosis; pollution, hygiene, environmental, immune response, urbanization and dietary/nutritional factors [44,45]. These factors, and more, are discussed point by point below. The digestive system in which IBD takes place is central to both the innate and adaptive/acquired immune systems where are balanced in complex reciprocal influence with intestine luminal microbes under homeostasis premises [46]. In IBD however, etiopathogenesis has become better elucidated owing to scientific technology advances in biological genetic, environmental and immunology that normal homeostasis physiology is disrupted and uncontrolled intestinal inflammation is perpetuated [47,48]. Customary pattern of thought, Th1 cell have been observed to play a crucial role in etiopathogenesis related to the chronicity of intestinal inflammation, especially in CD, where Th2 cells have been thought to play an important influence in UC [46,49]. Recently, however, it has been reported that activation of Th17 cells and imbalance of Th17/regulatory T (Treg) cells are recognized to be a vital segmental component in the trigger and development of intestinal inflammation, such as IBD [50]. Since tumor necrosis factor (TNF)-α is a strong candidate and has been identified as a potential cytokine in IBD etiopathogenesis, the establishment of anti-TNF-α treatment has contributed towards the initiation of disease-remodeling drugs [50,51,52].
4.1. Genetic Risk Factors
Despite the fact that the exact cause of IBD still remains unclear, there are susceptibilities that has broadly been recognized to have a genetic component ground, a defective immune system [53,54,55], and environmental basis combined are thought to partly play role in the etiopathogenesis [56,57]. This is singled out by the development of IBD in immigrants to high-prevalence countries [58] and contention of IBD among monozygotic twins [59]. The significance of environmental components is also well recognized by a rising trend in the incidence and prevalence of IBD in countries undergoing rapid Westernization [4,16,60]. As mentioned, genetics is observed to play role as observed by the greater prevalence of IBD in Ashkenazim Jews with trace ancestry in northern-European Jewish groups than Sephardic Jewish population [61,62]. Analyzing data materials from 5685 Ashkenazi Jewish exomes, Rivas et al. [62] bring forth a systemic analysis of Ashkenazi Jewish enriched protein-coding alleles, which contribute to distinct in genetic risk to IBD of which are transmitted via autosomal recessive inheritance. Other similar such genome-wide scan studies are herewith in-depth discussed [63,64]. Genetic population isolates like the Ashkenazim, Jews who trace their ancestry to eleventh century central European Jewish groups [65], have hitherto made it possible the mapping of alleles to play a part in to human disorder predisposition [66,67,68,69]. The documented 2–4 fold enrichment of CD prevalence in the Ashkenazi Jewish population [70,71] prompted enthusiasm for the use of exome sequencing and genome-wide array studies to evaluate the degree to which bottle-neck-enriched protein-altering alleles and undeniably implicated common variants contribute an excess CD genetic risk to Ashkenazi Jewish [70]. Despite efforts in the advance in the mapping genes and alleles for physical injuries and/or disorders with increased prevalence in the Ashkenazi Jewish population, precise estimates of the risk-allele frequency and the carrier rate in the Ashkenazi Jewish population have unfortunately not yet been resolved to date [72]. In addition, the disproportion of immune responses to intestinal bacterial antigens is thought to play a critical role in the etiopathogenesis of IBD in genetically receptive host individuals [73].
The CARD family plays an important mechanistic role in innate immune response by the activation of nuclear factor-κB (NF-κB). Studies to determine the gene expression and enumeration of the protein-expressing cells of some members of the CARD family (CARD9, CARD10, CARD11, CARD14 and CARD15) in patients with IBD vs. normal controls have demonstrated that the CARD9 and CARD10 gene expression was significantly elevated in UC as compared to CD. CARD11 gene expression was significant decreased in UC than in CD patients while CARD14 gene expression was significantly heightened in the group with active UC compared to non-inflamed controls. The down expression of CARD14 gene was associated with a benign clinical course of UC, characterized by initial activity followed by long-term remission longer than 5 years. CARD15 gene expression was significantly reduced in UC patients vs. CD. CARD9 protein expression was detected in inflammatory infiltrates; CARD14 in parenchymal cells, while CARD15 in inflammatory and parenchymal cells. CARD9-, CARD14- and CARD15-expressing cells were observed significantly higher in patients with active UC vs. non-inflamed controls. Therefore the CARD family looks indisputably involved in the inflammatory process and might be elaborated in the IBD etiopathophysiology.
There are about 71 CD and 47 UC susceptibility loci/genes to date. Approximately one-third of loci described confer susceptibility to both CD and UC. Amongst these are multiple genes involved in IL23/Th17 signaling (IL23R, IL12B, JAK2, TYK2 and STAT3), IL10, IL1R2, REL, CARD9, NKX2.3, ICOSLG, PRDM1, SMAD3 and ORMDL3. The evolving genetic architecture of IBD has furthered the understanding of disease etiopathogenesis. For CD, defective processing of intracellular bacteria has become a central theme, following gene discoveries in autophagy and innate immunity (associations with NOD2, IRGM, ATG16L1 are specific to CD). Genetic evidence has also demonstrated the importance of barrier function to the development of ulcerative colitis (HNF4A, LAMB1, CDH1 and GNA12). According to Chua et al., 2012, there is a strong association between both inflammatory bowel disease gene 5 (IBD5) locus variants but not the IL23R gene variant with CD (in the Malaysian population) but the IBD5 locus variants were highest in Indians, which may explain the increased susceptibility of this particular ethnic group to the disease [74].
4.2. Intestinal Microbiota
The symbiotically benefit of the intestinal microbiota to the host’s physiology can be divided into three different group categories—(i) nutrition, (ii) immune development, and (iii) host defense [75]. An inauspicious alteration of the constitutional composition and variety of the gastrointestinal microbiota (dysbiosis) is observed and reported in IBD patients which affects the host immune system functionality and barrier integrity, resulting in chronic inflammation and aberrant immune responses [76]. Studies into host-microbe interactions, involving both innate and acquired/adaptive immune responses, have shown to be of particular interest in understanding the possible etiopathogenesis of IBD [76,77]. Evolutions in sequencing advance technology have triumphed to the groundbreaking findings and characterization of the gut microbiota and its role in health and disease. While an altered microbiome has been described in IBD, whether it is a causative source or an effect of the local intestinal response to cellular injury (inflammation) has yet to be illuminated. Moreover, the bidirectional relationship between the intestinal microbiota and the mucosal immune system (discussed on 4.3) adds to the multifaceted complexity of intestinal homeostasis at large. A better understanding of how host genetics, including NOD2, influence immune-microbe interactions and alter susceptibility to IBD is still a challenge endeavor and potentially essential in order to gradually manifest therapeutic and preventative precision measures [77].
When compared patients with IBD to healthy individuals, the decrease of bacteria with anti-inflammatory capacities and the increase of bacteria with inflammatory capacities have been reported [78,79]. The most consistent observations are a downsizing in the diversity of gut microbiota and pruning abundance of Formicates [78,80,81,82]. Expansion in abundance of Proteobacteria and Bacteroidetes have been outlined [78], but downsizing have also been outlined [82]. It has been communicated that F. prausnitzii, Blautia faecis, Roseburia inulinivorans, Ruminococcus torgues, and Clostridium lavalense are reduced in cases with CD when collated to healthy subjects [83,84] and that the number of F. prausnitzii is correlated with the risk of subsequent relapse of ileal CD following surgery. The defect of colonization of F. prausnitzii was noted in UC patients during remission and the recovery of the F. prausnitzii population after relapse is seen to be associated with the maintenance of clinical remission [85]. In addition, human peripheral blood mononuclear cells simulated with F. prausnitzii induced the production of IL-10 and inhibit the population of inflammatory cytokines, such as IL-12 and IFN-γ [86]. Further, a significant reduction of Roseburia spp. was noted in the gut microbiota of healthy individual with a high vulnerable genetic risk for IBD. In contrast, a relative increase in Proteobacteria, mainly E. coli, was reported in CD patients, in particular, on mucosa-associated microbiota compared to fecal samples [87,88,89,90,91,92,93,94]. CD-associated E. coli with pro-inflammatory properties is adhesion-invasive E. coli (AIEC), which was initially isolated from adult CD patients [79]. It has been communicated that the number of AIEC increased in about 38% of patients with active CD compared to only 6% in healthy subjects [95]. The increase of pathogenic bacteria with the ability to adhere to the gut mucosa affects the permeability of the intestine, revamps the diversity and composition of gut microbiota, and induces inflammatory reactions by regulating the expression of inflammatory genes, consequently leading to the causing of intestinal inflammation [96]. Further, fluorescence in situ hybridization analyses have shown an enhanced abundance of mucosa-associated bacteria in IBD [97,98,99]. This may be caused by the altered ecology and increased volume of mucolytic bacteria, such as Runinococcus gnavas and Ruminococcus torques in IBD patients [99].
The subsequent yield of metabolites affected by the disruption of gut microbiota is reported attributable to the etiopathogenesis of IBD [84]. For example, the concentration of SCFAs has been communicated to decrease in IBD patients, as a result of butyrate-producing bacteria, such as F. prausnizzi and Clostridium clusters IV, XIVa, XVIII [84]. The reduced production of SCFAs affects the differentiation and expansion of Treg cells and the growth of mucosal cells [100], which play an important part in conserving intestinal homeostasis. On the other hand, the number of sulfate-lessening bacteria, such as Desulfovibrio, is abundantly increased in IBD patients [101,102], stemming in the fabrication of hydrogen-sulfate that cause severe injure to the intestinal epithelial cells and induces mucosal damage and inflammation [101]. Collectively, these data forcefully demonstrate that dysbiosis partly is associated with the etiopathogenesis of IBD.
4.3. The Intestinal Epithelium and Microbiota
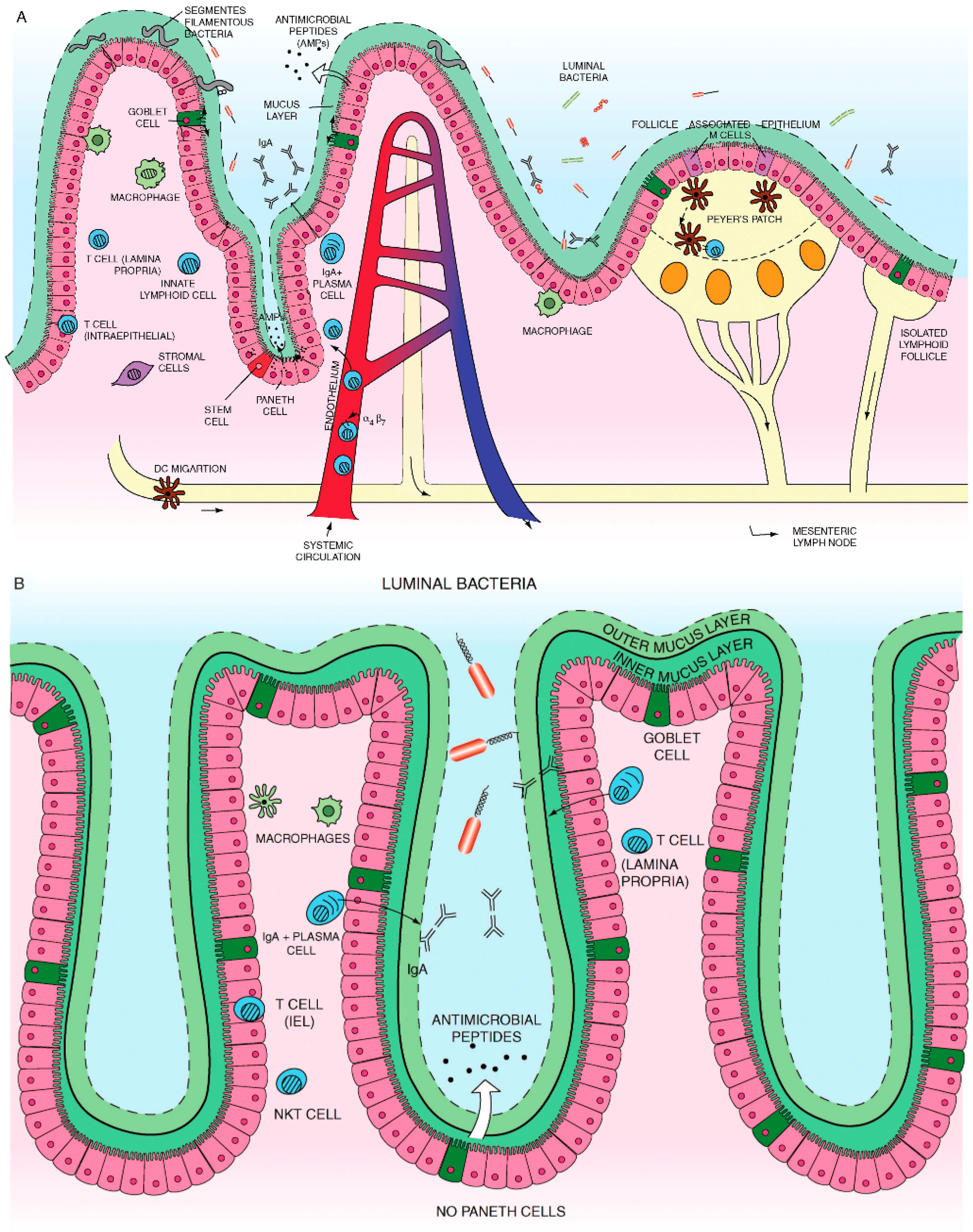
A number of different cells, including enterocytes, goblet cells, neuro-endocrine cells, Paneth cells, M cells, and epithelia resident intestinal stem cells together make the intestinal epithelial cell (IEC) compartment, Figure 1. These monolayer cells structurally self-possessed crypt and villi, with a single columnar cell inner surface with an impervious jointure secreting anti-microbial peptides accommodated mucus; these cells separate intra-luminal pathogens from the sub-epithelial lamina propria [48,103,104].
Normally, there are roughly 1011~1014 enteric commensal microorganisms from 300~500 different bacterial types [105,106]. These indigenous commensal bacteria play an important duty in defending intestinal homeostasis which has essential impact crucial to nutrient provision, development of the immune system, and regulation of energy metabolism [49,107]. Under certain acquired circumstances these microorganisms can become harmful and can cause intestinal inflammation [108]. Iatrogenically, when patients are treated with a systemic antibiotic drug(s) two or even three times the indigenous/commensal intestinal microbiota get lost and should rebuild to normalize and that could take months. In a compromised luminal innate immune system mechanisms there are some indications that commensal bacteria play crucial role in the developmental trigger of IBD. These include, (i) empiric antibiotic therapy experiences has been satisfying in certain IBD patients [109], (ii) IBD patients have enhanced concentrations against indigenous commensal bacteria [110], (iii) genetic deviants that are consociated with bacterial spotting, such as NOD2 [111], and T cell immunity, such as IL23R, are incriminated in IBD [112] and (iv) most animal model studies of colitis require commensal bacteria for the initiation or trigger of intestinal inflammation [113]. In addition, recent observations have concentrated on the benefaction of other enteric microorganism, such as viruses or fungi, for IBD elaboration [114,115].
Intriguing observation on stem cell regenerative enrichment report by Marlicz et al. [116]. They observed that developmentally early cells, including hematopoietic stem progenitor cells (HSPCs), mesenchymal stem cells (MSCs), endothelial progenitor cells (EPCs), and very small embryonic-like stem cells (VSELs), were observed mobilized into circulating peripheral blood (PB) in CD patients possibly in response to intestinal tissue injury [116,117]. The mobilized cells also expressed at the mRNA level genes playing a role in development and regeneration of gastrointestinal epithelium accompanied by increased serum concentrations of VEGF and HGF. Therefore it was concluded that CD triggers the mobilization of MSCs, EPCs, and VSELs, while the significance and precise role of these mobilized cells in repair of damaged intestine is still obscure and requires further studies.
4.3.1. Escherichia coli (AIEC)
A number of pathogens have been reported as possible causative microorganisms for IBD establishment trigger. Current studies reveal Proteobacteria, especially adherent-invasive Escherichia coli (AIEC), as one of the candidates. AIEC has been more frequently recognized in patients diagnosed with CD as compared to control subjects [88,90,118]. AIEC is known to be able to capture epithelium and clone within macrophage [119]. Some studies removed AIEC from the small intestine of patients with CD (Crohn’s ileitis) [82,120]. Interestingly, AIEC was infrequently seen in the colon tissue of CD (Crohn’s colitis) patients and was not recognized in UC patients [95], meaning that AIEC performs a vital role in the event of inflammation [121].
4.3.2. Clostridium
Clostridium cluster XIVa and IV are crucial part of gut homeostasis through Treg cell accumulation which is in contrast to AIEC, [122]. Foxp3+CD4+ Tregs are plenteous in the lamina propria of the large intestine and are crucial immune-regulating cells [123]. Studies revealed that Treg cells were significantly contrived by ileal microbiota [124]. In particular, Treg cells stimulated by Cbir1, a microbiota flagellin, induce IgA + B cells in the intestine. As a result, reduced pathogenic loading by IgA leads to down-regulation of systemic Tcell activation [125]. A development murine quintessential study with an escalated Clostridium XIVa/IV population was observed to be resistant to allergy and intestinal inflammation [122]. Contrastingly, patients with IBD demonstrated a decreased Clostridium XIVa/IV compared to that in non-IBD controls [78,86,126].
Observational studies of dysbiosis in IBD [127] and the disparities in host-microbe relationships which play part to the extent, severity, and chronicity of intestinal inflammation led to efforts to restore microbiota to a normal composition [128]. Further, a successful novel management approach in patients suffering from IBD using fecal microbial transplantation (FMT) has been introduced [129,130]. One randomized control trial involving 75 UC cases demonstrated a significantly escalated remission rate (24%) in individuals cases treated with FMT from unrelated/discrete donor enemas than that in the placebo array (5%) [131]. In another randomized control trial with 48 UC cases yielded antagonistic negative result [132]. To date, there are no randomized control trials contrasting FMT with placebo management in CD population. However, a meta-analysis using four patient series data in 38 CD patients unveiled a 60.5% pooled result rate [133]. It looks likely their outcome was not that of epithelial remission but of clinical response. Consequently, the effectiveness of FMT as a therapeutic use for IBD is still preliminary. Furthermore, optimal donor selection, delivery methods, and donor feces processing, which are both critically important, have not yet been formalized and remain unsettled to date. Probiotics are nutritional supplements that contain microorganisms that when consumed or administered in the proper amount restores beneficial bacteria to the digestive tract and benefit the host’s health. There has been trials made to manage IBD patients by improving intestinal microbial balance through probiotics. In a pilot colitis model study, probiotics demonstrated an anti-inflammatory outcome via TLR9 signaling [134]. In a recent meta-analysis study using 23 randomized controlled trials demonstrated that administering of probiotics was seen to be associated with benefits concerning induction and continuance of remission in patients suffering from UC but painstakingly not in cases suffering from CD [135]. Obviously, these studies are vindicated to draw a concrete conclusion in terms of the management sequels of probiotics in IBD. As IBD-related research advances, expounding of IBD pathologies is accentuating and some of such advances are illustrated in Figure 2. With the opening of the era of biologic and biosimilar agents, it has become realizable to anticipate deep sustainable remission in IBD patients, unalike in the erstwhile; however, about one-third of sufferers painstakingly still do not demonstrate clinical benefit to these modern agents. There are different new biologic and biosimilar agents specific to IBD etiopathogenesis that are now surfacing and are under different phases of clinical trials, Figure 3. With this advancement, more and more patients will likely benefit from these new unfamiliar agents. Moreover, future IBD clinical settings should be used in terms of patient-customized management, and it is expected greatly to shed light clinical practice to have a feasible drug repertoire targeting different mechanisms of the disorder.
4.4. Environmental Factors
An environmental factor is an identifiable element in the cultural, demographic, physical, economic, technological environment, or political, regulatory that impacts the operations, survival and growth of an organization and/or institution in health and in disease [136]. IBD is believed to result from several interactions between genetic susceptibility and environmental agents that affect the normal intestinal indigenous/commensal flora to activate an inappropriate mucosal immune response [137]. Despite the fact that IBD susceptibility genes have been elucidated [138,139], similar developments in outlining environmental risk factors have lagged [140,141]. Numerous environmental risk factors have been investigated, including smoking, appendicitis, nutrition, cultural influences on diet, breastfeeding, infections/vaccinations, oral contraceptives, antibiotics, helminths, psychological stress, urban life style, air pollution and childhood hygiene, all portray shared vulnerabilities that could constitute risk for IBD [142,143,144,145,146,147]. Most of these factors construe the evident relationship between Westernization-urbanization culture and the risk of IBD trigger, as has been described in China and offspring of South Asian immigrants to the United States and Canada [10] and/or United Kingdom [148,149]. These surveillances again raise the issue of how vivid culture influences, such as diet, regulating and/or adjusting the risk of IBD in certain ethnic communities and in certain geographic regions. It will be meaningful to identify the role of these environmental influences/factors in IBD etiopathogenesis triggers [150].
Smoking has been reported to be associated with IBD risk, specifically CD [151,152] and is also reported to be associated with increased intestinal permeability [153], but what remains vague is whether the influence is mediated through the gut microbiome [154]. It also not known whether secondary smoke exposure can increase risk of IBD onset. A meta-analysis study did not identify a relationship between childhood passive-smoke vulnerability and CD [155]; although, additional recent studies have opposite observational results, thus the influence of secondary smoke on IBD assault warrants further investigational studies [156,157,158].
In humans, psychological stress has been observed to play a significant role in the etiopathogenesis of IBD due to the chronic, relapsing, and remitting nature of this condition [146,147]. The commonly seen chronic and acute stress in these pathologies do alter immune function [142]. Results from experimental studies have been conflicting, with observations endorsing both positive and null connections [146,147]. However, due to the retrospective nature of these studies, recall bias may have influenced the results [142]. Evidence from animal model studies demonstrates that chronic psychological stress may exacerbate IBD by upgrading damage to the gut luminal epithelium, thereby disrupting barrier function [159,160].
Clearly, the environmental risk factors that have been recognized have not nailed down the etiopathogenesis of the “Colitides” to date [161]. These factors mentioned herewith, have been implicated in the increased global incidence of IBD [161,162]. However, even the most conflictingly substantiated environmental risk factor such as smoking is seen to contribute only partially to disease etiopathogenesis. We now know that most people with smoking habits do not have CD and most patients diagnosed with CD are not smokers [161,162]. Agreeably thus, more studies are warranted to better elucidate the environmental determinants of IBD [161,162].
4.5. Immune Response
In Colitides, the immune defense against intestinal microbes’ compromises in two different levels [163]: (i) the impairment of epithelial mucosal barrier and (ii) the altered innate and adaptive/acquired host immune responses. The immunopathogenesis of IBD may occur in three distinct stages [163]: (i) penetration of luminal inner contents into amenable tissues which may be facilitated by environmental components such as inherent defects in epithelial barrier or infection, (ii) defective secretion of pro-inflammatory cytokines by macrophages due to compromised clearance of foreign materials from the gut wall and (iii) a compensatory acquired/adaptive immune reaction which results to a chronic inflammatory reaction and gives rise to distinctive IBD abrasions. Briefly, chronic improper activation of the acquired immune system against indigenous commensal microorganism has been observed to be the main etiopathogenesis of IBD [46]. During the process there is increase secretion of IFN-γ from Th1 cells and cytokines associated with Th17 cell, such as IL-17A/F, IL-21, IL-22, and CXCL8, are seen in the intestine of CD cases, while T cells from the lamina propria of UC patients’ significantly produce Th2 cell-associated cytokines, such as IL-5 and IL-13 [48,164,165].
Recently, IL-9-secreting Th9 cells are known to be involved in the pathogenesis of IBD [166]. However, the role of Th9 cells and their secretory cytokine IL-9 in IBD is poorly elucidated and studies on its functional importance in IBD are underway. Clearly, studying the actual role and mechanisms of different T helper cell subsets including Th9 cells in IBD is critical to develop novel IBD therapies. An understanding of the mechanisms that employed by Th9 cells and IL-9 to cause IBD could help contemplate potential targets for the treatment of Th9 cell-mediated IBD.
Conservatively, immune-modulating management of IBD have aligned on acquired/adaptive immunity [167,168,169]. The NOD2 gene was the first susceptibility gene established within the IBD 1 locus for CD. Subsequently, over 230 genetic risk loci have been identified with IBD and yet NOD2 remains the most robust/powerful association to date [77].
4.6. Urbanization
Urbanization is a multidimensional undertaking that manifests as rapidly changing population characteristics and land cover [170]. The rapid urbanization has been observed to correlate with an increasing incidence and prevalence of IBD [4,14]. In the past six decades, IBD has recognized as a rapidly evolving challenge in previously low incidence countries, especially in recently urbanize-industrialized nations, including Africa, Asia, the Middle East and South America [4,16,171,172,173,174,175]. The emergence of IBD in these countries, which are undergoing rapid modernization and urbanization, resembles patterns that were seen in the Western world during the early 20th century, with the upsizing prevalence of UC preceding that of CD in urban areas [10,176]. A meta-analysis of 40 multipronged studies investigating the association between urban environment and IBD observed that the pooled incidence rate ratios (IRRs) for urban vs. rural environments were 1.17 (95% CI 1.03–1.32) and 1.42 (95% CI 1.26–1.60) for UC and CD, respectively [177]. This suggests a bond between urbanization and the incidence of IBD. The relative risk in the urban areas is reported to be 1.3 versus countryside [177]. The effects of swift urbanization are likely to be reflected in the human intestinal microbiome that we herewith discussed earlier (Section 4.2), and alterations in the gut microbiome have been reported to be associated with IBD incidences [178]. The role of the gut bacterial microbiome in human health and diseases has been herewith extensively presented [179,180]. Additional studies have outlined that inhabitants residing in non-Western and/or rural areas have a higher bacterial diversity when compared with populations in the Western nations such as United States, Canada and Europe [181,182,183,184]. The fecal microbiota of children from a rural African villages, e.g., of Burkina Faso, who mostly eat a diet high in fibre [185], is similar to that of the microbiome of early human settlement at the time of the birth [181]. Children from Burkina Faso demonstrate a significant (p < 0.001) enhancement of Bacteroidetes and a reduction of Formicates compared with children from the urban locations, e.g., of Florence, Italy, with a unique plenteous of bacteria from the genera Prevotella and Xylanibacter, which are recgnized to contain a set of bacterial genes for cellulose and xylan hydrolysis. These bacteria were totally lacking in the 15 European country children studied [181]. Corresponding observations have been seen in children and adults in Malawi, Amazonian American Indians [183] and adult Hadza hunter-gatherers in mainland Tanzania [182]. The Hadza of Tanzania, in Eastern Africa, despite human civilization, are one of the very few societies in the world who still live by hunting and gathering [182]. These studies have shown that urbanization is consociated with an upsizing proportion of Bacteroides, Alistipes (Bacteroidetes), Balautia, Faecalibacterium, Ruminococcus (Formicates) Bifidobacterium (Actinobacteria) and Bilophila (Proteobacteria), whereas Prevotella (Bacteroidetes) is significantly elevated in the gut microbiota of individuals residing in non-industrialized communities [181,182,183].
Studies comparing the rural and urban microbiome within a population of homogeneous ethnicity is a scarcity [185]. In one study comparing the fecal microbiota composition of African descendants residing in rural, semi-urban communities with those settling in urban locations, substantial dissimilarities were observed, with Prevotella predominating in semi-urban individuals and Bacteroides predominating in urban African Americans [186]. These observations indicate that the intestinal microbiota content contrasts between genetically similar populations living in diverse communities, such as rural vs. urban. Comparison of the fecal microbiota of elderly persons residing in rural and urban areas in Japan demonstarted that individuals living in Yuzurihara (a rural village) had a larger number of bifidobacteria, whereas larger proportions of bacilli and lecithinase-positive clostridia were found in residents of Tokyo [187]. An underway countrywide study of IBD incidence in Asia shows that Inner Mongolia has a low IBD incidence than other regions [185]. Microbial profiling of Inner Mongolia residents indicates that the high-level presence of Phascolarctobacterium, Lactobacillus and Bifidobacterium might be associated to a pasturing lifestyle and a diary diet. Lactobacillus helvetucus is oftentimes found in individuals from every rural pasturing area in Inner Mongolia but not in Mongolians living in Hohhot city (urban), indicating that diet affects the intestine luminal microbial composition of Mongolians [188]. A Russian study [189] demonstrated that microbial communities from inhabitants in rural areas had a 2.6-fold increase in the frequency of new microbial community structures distinct from the common three enterotypes [190] compared with the microbial communities of urban hosts. The predominant microbial populations in rural inhabitants were from the Formicates and Actinobacteria phyla. These bacterial communities are accommodated by the consumption of starch-rich bread and potatoes, typical staple foods utilized in rural Russia, and natural foods that are available to low-income socioeconomic communities from their household gardens [189,191]. Correspondent with the theory of vanishing microbiota and its relation with the emergence of autoimmune and chronic GI disorders [192], the constantly witnessed loss of microbiota affluent and diversity during urbanization might largely account for the upsizing in IBD incidence. These experimental and observational studies were mostly based on 16S ribosomal RNA (rRNA) gene sequencing [185]. All-in-all, an in-depth understanding of rural vs. urban intestinal bacterial species or strains and their functions in IBD etiopathogenesis is largely still lacking warranting more experimental studies.
Indisputably, an air contamination is taking place in parallel with urbanization [170,193], and it is thought to have significant detrimental effect on a wide range of public health issues, including IBD [143,194]. Persistently long-term exposure to a high concentration of nitrogen dioxide pollution and particulate matters has been reported to be related with an escalated risk of early-onset of CD, indicating a linear increase proportional to high concentrations of pollutants [143]. Other forms of inhaled environmental exposures observed to promote the susceptibility to IBD via alteration of the intestinal microbiota are herewith in-depth discussed [195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220].
4.7. Dietary/Nutrition
Intestinal mucosal microbiota composition is dependently influenced and modified by diet [221,222]. According to the IBD-EPIC (IBD European Prospective Cohort) Study, several dietary factors have been found to be related with IBD onset [223,224,225,226]. Milk and milk product consumption was observed to be potentially related with a low risk of developing CD (p = 0.23) but not for with UC (p = 0.60) [224], whereas a role of flavones (C15H10O2) and resveratrol (C14H12O3) in the risk of triggering CD was also shown [225]. The IBD-EPIC study results also indicate dietary role for linoleic acid in the etiopathogenesis of UC [225]. According to this calculated study cohort, overweight, as measured by BMI (body mass index), was observed not consociated with the etiopathogenesis of either UC or CD, respectively [226]. Altogether, total fiber consumption from vegetables, cereals or fruits, and the ensuing evolution of either CD or UC were found not consociated [223]. Nonetheless, it is acknowledged that most of these studies are grounded on food intake frequentness questionnaire data that may have significant boundaries/limitations [227]. There is an indisputable evidence indicating a role for diet, particularly among genetically susceptible individuals and the development of IBD [44]. In addition, data from animal model studies demonstrate that dietary change and/or modification impacts the risk of IBD [44]. More studies report to have seen affirmation suggesting that artificial sweeteners such as saccharin, sucralose, acesulfame potassium (ace K) and cyclamate may have played causative role in the pathogenesis of IBD [228,229,230]. These sweeteners are observed to be distinctive by high stability with little metabolism by the body and long-lasting in the environment but high inhibitory effect on bacteria [231]. In one EPIC (European Prospective Investigation into cancer and Nutrition) demonstrated that a dietary pattern obtaining of high intake of sugar and sweetened beverages and low consumption of vegetables is linked with elevated hazard of UC (incidence rate ratios for the fifth vs. first quintile, 1.68 [1.00–2.82]; Ptrend = 0.02) [228]. In contrast, a recent sizable prospective cohort study from Scandinavian demonstrated no corroborations between consumption of sweetened beverage and subsequent endangered CD or UC [232]. The study established 143 incident cases of CD (incidence rate = 11 cases/100,000 persons-years) and 349 incidence cases of UC (incidence rate = 28 cases/100,000 person-years) over 1,264,345 person-years of follow up. Consumption of sweetened beverages does not appear to increase endanger of CD (Ptrend = 0.34) or UC (Ptrend = 0.40) [232]. Compared to participants who reported no consumption of sweetened beverage, the calculated multivariable-adjusted HRs for 1 or more consumptions per day were 1.02 for CD (95% CI, 0.60–1.73) and 1.14 for UC (95% CI, 0.83–157), respectively. The association between consumption of sugar-sweetened beverages and risk of CD or UC were not modified by age, sex (cohort), body mass index, or smoking (all Pinteraction ≥ 0.12) [232].
Gene-environmental studies on the interrelationship between environmental factors and genetic variant in functionally annotated genes have largely been encouraged to assist infer causal affiliation [233] to shed light into potential biological mechanistic pathways through which an environmental component such as diet might shed light to the aetiopathogenesis of IBD. Genetic loci related with IBD risk can be widely designated into those involving the innate and/or acquired immune response anomalies and mucosal barrier functional event [234]. Experimental studies also show that a number of these pathways are impacted by nutritional/dietary factors [235]. Thus, it is biologically creditable that special and/or specific dietary ingredients have unique differential outcomes on the incidence of IBD, coinciding with the genetic background of every individual per sig.
Introductory analyses of gene-environmental interplay in studies involving dietary factors in IBD have been increasingly promising [236,237,238,239]. A study from the NHS and NHSII, which involved 169 patients with CD and 202 cases with UC matched to 740 participants as control, analyzed the reciprocal action between total dietary consumption of iron and heam iron and genetic variants related with risk factors of IBD [240]. This analysis illustrated a relationship between iron and haem iron consumption and the UC vulnerability locus rs1801274, a coding variant in the ECGR2A (which encodes the low affection immunoglobulin-γ Fc location receptor IIa) gene. Peculiarly, among females with the GG genotype, low haem iron consumption was linked with a consequentially reduced risk of UC (or 0.11, 95% CI 0.03–0.37 for each 1g increase in heam iron consumption. In contrast, increasing haem iron consumption was correlated with an almost threefold elevated risk of UC among females with the TT genotype (OR 2.26, 95% CI 1.02–7.48). Owing to the important role of FCGR2A in controlling humoral reaction to infection [241] and the known importance of the rs1801274 variant in changing the binding volume capacity of the encoded protein product of C-reactive protein (CRP) and immunoglobulin G2 (IgG2) [242,243,244], these results offer supportive attestation for an interesting interplay between dietary haem consumption and immune physiological function in the etiopathogenesis of UC.
In an identical population, in a NCC (nested case-control) study of 202 cases with UC and 169 patients with CD in the NHS and NHSII cohorts matched to 740 participants as control based on age, menopausal situation, period of blood withdrawal collection and fasting status, an interplay between dietary potassium consumption and genetic variants in the IL-23 pathway that have been previously related with risk of IBD in GWAS was identified [234,236]. Particularly, the rs7657746 variant of IL21 (which encodes IL-21) appeared to modify the relationship between potassium consumption and risk of IBD pathogenesis. Each additional 200 mg more in dietary potassium consumption was inversely consociated with risk of UC (OR 0.90, 95% CI 0.82–0.98) among participants with the AA genotype, but not among those with the AG or GG genotypes. Similar observations were reported in cases with CD in the study. As IL-21 play a key developmental role of TH17 cells through signal transducer and driver of transcription 3 (STAT3), a transcription factor needed for the differentiation of TH17 cells in vivo, the results from gene-environment interaction studies indicate an existence of a potential mechanism for the identified relationship [245]. IL-21 and IL-23 induced expression of the nuclear receptor RORγ (also known as RORC), which, in synergy with STAT3, upstairs IL-17 expression in CD4+ T cells, leading to the activation of TH17 cells [245]. In addition, IL-21 hinders the transforming growth factors-β (TGFβ)-dependent generation of FOXP3+ Treg cells and catalyzes TH17 cells activation [246]. Intriguingly, the gen-environment interplay finding was encouraged further supported by in vitro studies showing that potassium generates FOXP3 expression in naïve and memory T cells and in pro-inflammatory TH17 cells. This effect was noted even in the presence of pro-inflammatory cytokine, indicating that potassium curbs inflammation in a pro-inflammatory milieu.
Chassaing et al., reported in animal model that, relatively list content of emulsifiers, such as carboxymethylcellulose and polysorbate-80, caused low-grade inflammation and obesity/metabolic syndrome in WT hosts and incubated robust colitis in mice predisposed to this gastrointestinal disorder [247]. Emulsifier-induced metabolic syndrome was associated with microbiota encroachment, altered species composition, and increased pro-inflammatory potential. Use of germ-free mice and fecal transplants demonstrated that such alterations in microbiota were essential and enough for both low-grade inflammation and metabolic syndrome. These observations support the emerging idea that perturbed host-microbiota associations resulting in low-grade inflammation can increase adiposity and its associated metabolic effects. Moreover, they suggest that broad use of emulsifying agents might be contributing to increased societal incidence of obesity/metabolic syndrome and other chronic inflammatory diseases such as IBD.
Finally, studies from the NHS and NHSII have vividly indicated that two variants in CYP4F3, which encodes the cytochrome P40 4F3 enzyme (CYP4F3) elaborated in PUFA metabolism, might modify the relation between n-3 and n-6 PUFA intake and risk of UC [238]. Potentially, the relation between n-3:n-6 PUFA intake ratio and UC was modified by rs4646904 single nucleotide polymorphism (SNP) in CYP4F3 (Pinteraction = 0.049). A high (greater than or equal to the medium) n-3:n-6 PUFA intake ratio was related with a lower risk of UC among women with the GG or AG genotypes (OR 0.57, 95% CI 0.32–0.99), but not among those with the AA genotype (OR 0.95, 95% CI 0.47–193). Identical observations were also reported earlier in a pediatric case-control study with newly diagnosed CD [239], suggesting that the interaction is indeed robust.
4.8. Role of miRNA in IBD trigger
Advances in the field of miRNA (microRNA) research technology is speedily expanding [246] and are strongly associated in the etiopathogenesis of IBD, having an important role in the development, regulation and differentiation of both the innate and acquired/adaptive immune system [248]. A number of studies have demonstrated a differential expression of miRNA in tissue and blood samples from cases suffering from IBD compared with normal/healthy controls, indicating that miRNAs may be shortlisted not only in the development of immune system component but as new candidate biomarkers of these disorders [249]. Due to the fact that CD and UC differ in their clinical presentations, genetic consociations, gene expression patterns, and immune reactions, differing miRNA profiles are anticipated for these two IBD pathologies. It is now been realized that CD and UC patients have unique miRNA expression profiles in their target organs. Not surprising that while some uniquely expressed miRNA are commonly routine to other immune-related disorders, most are different. Further, studies have demonstrated peculiar miRNA expression profile bio-fingerprints in IBD and preliminary operational analyses relate these deregulated miRNA to canonical pathways related with IBD etiopathogenesis [250]. In order to elucidate precision roles of miRNAs is the human context more studies are required despite current promising to advance understanding of miRNAs in the pathogenesis and diagnosis of IBD which may be useful for the development of miRNA-based therapies [251].
In recent years, blood-derived microparticle biochemical peptides have become invaluable for IBD monitoring and more frequently used as surrogate markers of intestinal inflammation. Emerging concepts that revolve around measurement of cell-derived microvesicles (MVs) in the circulating blood vascular bed of IBD patients is another emerging advances advantageous in future disease understanding and management. Extracellular microvesicles (ExMVs) are part of the cell secretome baroque in chronic autoimmune diseases, such as IBD. ExMVs capture functional RNA species and proteins from one cell to another, an observation that paved up the new way to the new field of research of bioactive molecules in cell-to-cell communications [252,253,254]. This observation disclosed up the gates to novel idea, in which the presence of mRNA, noncoding RNA, and miRNA in ExMVs in blood and other biological body fluids gave the possibility of employing ExMVs as new fingerprint biomarkers for pathological disorders. Subsequently, ExMVs has become a target for “liquid biopsy” strategies. Tziatzios et al. [255] observed that circulating levels of platelet derived microparticles (PDMPs) were enhanced in CD patients but did not correlate with disease activity. 5-ASA treatment was associated with lower levels of PDMPs, while anti-TNF-α treatment did not influence expression of ExMVs in IBD patients. Similarly, circulating PDMPs were increased in IBD patients with active disease.
5. Clinical Diagnosis
Currently, “Diagnostic Gold Standard” test for IBD does not existent. Also, there is no accurate tools to predict whether a patient with newly diagnosed IBD will progress to complications of disease before and after colectomy for UC or indeterminate colitis such as cancer, pouchits, cufittis or fistulae, structuring and penetrating complications in de novo CD. In the IBD clinical setting, clinicians use state-of-the-art criteria, yet engage in invasive and inexact testing classification systems such as endoscopy (gastroscopy and colonoscopy), radiologic imaging, and histopathology to diagnose IBD patients, resulting to a substantial number of incorrect or delayed diagnosis [256,257,258,259,260]. It is possible that early accurate diagnosis and timely treatment could improve outcomes for those high-risk patients [13]. Even with a combination of recommended state-of-the-art diagnostic system modalities IBD patients cannot be accurately diagnosed. Up to 15% of colonic IBD cases are classified as “indeterminate colitis (IC)” because the established criteria for UC and CC are non-definitive [257,261,262]. In addition, another 15% of colonic IBD cases that are prescribed pouch surgery (restorative proctocolectomy (RPC) with ileal pouch-anal anastomosis (IPAA), which are standard surgical procedures for treating UC or IC predicted as UC, are in fact CC cases. Therefore, a total of 30% of colonic IBD patients are not diagnosed accurately [258,259,260]. For these reasons, efforts at identifying accurate, noninvasive biomarkers have been undertaken [263,264,265]. Another really challenge is a significant subgroup of IBD patients, especially UC patients undergoing proctocolectomy that convert to de novo Crohn’s, are thought to be “misdiagnosed” [265]. This may not be accurate because there is a possibility that these patients with UC were “transformed” due to an altered microbiome ecology in the pouch and immune environment and that the patient actually “convert”. Since there is significant overlap between disease-associated genes, it is possible that disease phenotype may change within a given individual. More elucidation studies are needed in this area.
A recent breakthrough finding that Paneth cell specific peptide “Human alpha-defensin 5 (DEFA5)” delineate colonic IBD (CC versus UC) may solve diagnostic dilemma in IBD clinical settings [265]. Detection of DEFA5 more accurately circumvented the IC cases into UC or CC phenotype and identified CC cases initially treated as UC cases [265]. Among patients with IC, DEFA5 is a reliable delineator with a positive predictive value of 96 percent [265]. The distinction between UC and CC is of utmost importance when prescribing a patient’s candidacy for pouch surgery, RPC and IPAA [40,266,267].
6. Management and Challenges
As discussed on Section 4.3, persons suffering from IBD are frequent users of the healthcare system, with an annual frequency of hospitalization exceeding 20 percent [32]. Studies from the United States economic implications report of IBD, showed in 2014, that CD and UC were related with annual direct and indirect costs ranging between US $14.6 and $31.6 billion [268]. Costs include invasive endoscopic and radiologic procedures for diagnostics and management decisions, as well as medications, hospitalizations, and surgical interventions [269]. Further costs accrue to community in loss of productivity and disability of impacted patients with deprived quality of life. While a noninvasive, easier, accurate and fast screening diagnostics tool is needed to downsize costs and burden of disease [265] the unmet need for noninvasive markers has outpaced the evidence. Thus, IBD is indeed expensive to treat and manage [270]. Due to an incomparable infrastructural gape in terms of access to care between developing vs. developed nations and the uneven representation of IBD across socioeconomic strata, a serious plan is required in the developing countries concerning how to tackle this emerging human health challenge [4,137].
There are significant advances in genetic and immunologic analytical technologies and as a result new therapeutic approached are now in place that accurately target the mechanistic pathways of IBD [46]. Apart from conventional immune-suppressive treatment, the evolution of biological and biosimilar agents that are target specific has lead in more frequent and deeper remission in the IBD patients, with mucosal healing as a therapeutic goal. In not too distant future, targeted novel biologic and biosimilar agents should defeat the obstacles of customary treatments and ensure that each patient can be managed with optimal medications that are nontoxic and precisely target IBD. These drugs are unfortunately the quickest-cultivating partition of the prescription drug market in the West as is IBD in the developing countries [4]. The healthcare system, and certainly the patients, in developing nations will struggle and will not be able to afford such costly managements which will lead into a series of events that are life threatening in terms of health, safety or well-being of large group of people.
Fecal microbiota transplantation (FMT) is emerging advances as a novel approach to therapy for UC. However, the interpretation of efficacy of FMT for UC is disputably complicated based on various study contentions, FMT administration procedures, intensity of therapy (dose) and donor stool processing methodologies. In a systemic review with meta-analysis including randomized controlled trials (RCTs), Costello et al. reported that despite variation in stool processes, FMT appears to be effective for induction of remission in UC patients, with no major short-term safety signals [271]. However, further elucidative observational studies are required to better define, establish, and verify dose frequency and preparation methods, and to explore its feasibility, efficacy and safety as a maintenance agent.
Author Contributions
A.E.M.—Invitation recipient for “Gastrointestinal Disorders—Special Issue”. Original idea of the paper, formulation of the protocol and contribution to data abstraction and analysis. Critical drafting, writing and revision of the manuscript, data analysis. Carried out the literature search, selection, and validity assessment.
Funding
This work was supported by: Research Foundation, American Society of Colon and Rectal Surgeons, LPG-086); National Institute of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)—R21DK095186; Vanderbilt University Institute for Clinical and Translational Research (VICTR-CTSA)—1UL1RR024975; NIH/National Center for Advancing Translational Science VICTR-2UL1TR000445-06; NIH/National Cancer Institute (NCI)-3U54CA091408-09S1; and NIH/NCI-3U54CA091408-09S2.
Conflicts of Interest
A.E.M., has received Honoraria fees for Educational Presentation from Lipscomb University. Further, he receives institutional grants from Meharry Medical College Schools of Medicine (SOM) and Graduate Studies and Research (SOGRS) and Vanderbilt Institute for Clinical and Translational Research (VICTR).
Abbreviations
| IBD | inflammatory bowel Disease |
| UC | ulcerative colitis |
| CD | Crohn’s disease |
| MOOSE | meta-analyses of observational studies |
| PRISMA-P | preferred reporting items for systematic review and meta-analysis protocols |
| MEDLINE | Medical Literature Analysis and retrieval system online |
| EMBASE | Excerpta Medica dataBASE |
| CINAHL | Cumulative index of Nursing and Allied Health Literature |
| SCFAs | short-chain fatty acids |
| NOD2 | Nucleotide-binding oligomerization domain-containing protein 2 |
| TNF-α | tumor necrosis factor-alpha |
References
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Love, J.R.; Irvine, E.J.; Fedorak, R.N. Quality of life in inflammatory bowel disease. J. Clin. Gastroenterol. 1992, 14, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Sands, B.E. Therapy of inflammatory bowel disease. Gastroenterology 2000, 118, S68–S82. [Google Scholar] [CrossRef]
- M’Koma, A.E. Inflammatory Bowel Disease: An Expanding Global Health Problem. Clin. Med. Insights Gastroenterol. 2013, 6, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Ventham, N.T.; Kennedy, N.A.; Nimmo, E.R.; Satsangi, J. Beyond gene discovery in inflammatory bowel disease: The emerging role of epigenetics. Gastroenterology 2013, 145, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; Benchimol, E.I.; Bernstein, C.N.; Bitton, A.; Feagan, B.; Panaccione, R.; Glasgow, K.W.; Fernandes, A.; Ghosh, S. Inflammatory bowel disease: A Canadian burden of illness review. Can. J. Gastroenterol. 2012, 26, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Hammer, T.; Nielsen, K.R.; Munkholm, P.; Burisch, J.; Lynge, E. The Faroese IBD Study: Incidence of Inflammatory Bowel Diseases across 54 Years of Population-based Data. J. Crohns Colitis 2016, 10, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.I.; Manuel, D.G.; Guttmann, A.; Nguyen, G.C.; Mojaverian, N.; Quach, P.; Mack, D.R. Changing age demographics of inflammatory bowel disease in Ontario, Canada: A population-based cohort study of epidemiology trends. Inflamm. Bowel Dis. 2014, 20, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.I.; Guttmann, A.; Griffiths, A.M.; Rabeneck, L.; Mack, D.R.; Brill, H.; Howard, J.; Guan, J.; To, T. Increasing incidence of paediatric inflammatory bowel disease in Ontario, Canada: Evidence from health administrative data. Gut 2009, 58, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- M’Koma, A.E. Diagnosis of inflammatory bowel disease: Potential role of molecular biometrics. World J. Gastrointest. Surg. 2014, 6, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Globalisation of inflammatory bowel disease: Perspectives from the evolution of inflammatory bowel disease in the UK and China. Lancet Gastroenterol. Hepatol. 2016, 1, 307–316. [Google Scholar] [CrossRef]
- Everhov, Å.H.; Halfvarson, J.; Myrelid, P.; Sachs, M.C.; Nordenvall, C.; Söderling, J.; Ekbom, A.; Neovius, M.; Ludvigsson, J.F.; Askling, J.; Olén, O. Incidence and Treatment of Patients Diagnosed With Inflammatory Bowel Diseases at 60 Years or Older in Sweden. Gastroenterology 2018, 154, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Archampong, T.N.; Nkrumah, K.N. Inflammatory bowel disease in Accra: What new trends. West Afr. J. Med. 2013, 32, 40–44. [Google Scholar] [PubMed]
- Ukwenya, A.Y.; Ahmed, A.; Odigie, V.I.; Mohammed, A. Inflammatory bowel disease in Nigerians: Still a rare diagnosis? Ann. Afr. Med. 2011, 10, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Agoda-Koussema, L.K.; Anoukoum, T.; Djibril, A.M.; Balaka, A.; Folligan, K.; Adjenou, V.; Amouzou, K.D.; N’dakéna, K.; Redah, R. Ulcerative colitis: A case in Togo. Med. Sante Trop. 2012, 22, 79–81. [Google Scholar] [PubMed]
- Mebazaa, A.; Aounallah, A.; Naija, N.; Cheikh Rouhou, R.; Kallel, L.; El Euch, D.; Boubaker, J.; Mokni, M.; Filali, A.; Ben Osman, A. Dermatologic manifestations in inflammatory bowel disease in Tunisia. Tunis Med. 2012, 90, 252–257. [Google Scholar] [PubMed]
- Senbanjo, I.O.; Oshikoya, K.A.; Onyekwere, C.A.; Abdulkareem, F.B.; Njokanma, O.F. Ulcerative colitis in a Nigerian girl: A case report. BMC Res. Notes 2012, 5, 564. [Google Scholar] [CrossRef] [PubMed]
- Bouzid, D.; Fourati, H.; Amouri, A.; Marques, I.; Abida, O.; Haddouk, S.; Ben Ayed, M.; Tahri, N.; Penha-Gonçalves, C.; Masmoudi, H. The CREM gene is involved in genetic predisposition to inflammatory bowel disease in the Tunisian population. Hum. Immunol. 2011, 72, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, E.A.; Wright, J.P.; Froggatt, J.; Cuming, L.; Elliot, M. Medium-term follow-up of ulcerative colitis in Cape Town. S. Afr. Med. J. 1989, 76, 142–145. [Google Scholar] [PubMed]
- O’Keefe, E.A.; Wright, J.P.; Froggatt, J.; Zabow, D. Medium-term follow-up of Crohn’s disease in Cape Town. S. Afr. Med. J. 1989, 76, 139–141. [Google Scholar] [PubMed]
- Segal, I. Ulcerative colitis in a developing country of Africa: The Baragwanath experience of the first 46 patients. Int. J. Colorectal Dis. 1988, 3, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Segal, I.; Tim, L.O.; Hamilton, D.G.; Walker, A.R. The rarity of ulcerative colitis in South African blacks. Am. J. Gastroenterol. 1980, 74, 332–336. [Google Scholar] [PubMed]
- Wright, J.P.; Marks, I.N.; Jameson, C.; Garisch, J.A.; Burns, D.G.; Kottler, R.E. Inflammatory bowel disease in Cape Town, 1975–1980. Part II. Crohn’s disease. S. Afr. Med. J. 1983, 63, 226–229. [Google Scholar] [PubMed]
- Wright, J.P.; Marks, I.N.; Jameson, C.; Garisch, J.A.; Burns, D.G.; Kottler, R.E. Inflammatory bowel disease in Cape Town, 1975–1980. Part I. Ulcerative colitis. S. Afr. Med. J. 1983, 63, 223–226. [Google Scholar] [PubMed]
- Brom, B.; Bank, S.; Marks, I.N.; Barbezat, G.O.; Raynham, B. Crohn’s disease in the Cape: A follow-up study of 24 cases and a review of the diagnosis and management. S. Afr. Med. J. 1968, 42, 1099–1107. [Google Scholar] [PubMed]
- Novis, B.H.; Marks, I.N.; Bank, S.; Louw, J.H. Incidence of Crohn’s disease at Groote Schuur Hospital during 1970–1974. S. Afr. Med. J. 1975, 49, 693–697. [Google Scholar] [PubMed]
- Sobel, J.D.; Schamroth, L. Ulcerative colitis in the South African Bantu. Gut 1970, 11, 760–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraud, R.M.; Luke, I.; Schmaman, A. Crohn’s disease in the Transvaal Bantu: A report of 5 cases. S. Afr. Med. J. 1969, 43, 610–613. [Google Scholar] [PubMed]
- Ananthakrishnan, A.N.; Kwon, J.; Raffals, L.; Sands, B.; Stenson, W.F.; McGovern, D.; Kwon, J.H.; Rheaume, R.L.; Sandler, R.S. Variation in treatment of patients with inflammatory bowel diseases at major referral centers in the United States. Clin. Gastroenterol. Hepatol. 2015, 13, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Kappelman, M.D.; Rifas-Shiman, S.L.; Porter, C.Q.; Ollendorf, D.A.; Sandler, R.S.; Galanko, J.A.; Finkelstein, J.A. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology 2008, 135, 1907–1913. [Google Scholar] [CrossRef] [PubMed]
- Chouraki, V.; Savoye, G.; Dauchet, L.; Vernier-Massouille, G.; Dupas, J.L.; Merle, V.; Laberenne, J.E.; Salomez, J.L.; Lerebours, E.; Turck, D.; et al. The changing pattern of Crohn’s disease incidence in northern France: A continuing increase in the 10- to 19-year-old age bracket (1988–2007). Aliment. Pharmacol. Ther. 2011, 33, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, C.; Paerregaard, A.; Munkholm, P.; Faerk, J.; Lange, A.; Andersen, J.; Jakobsen, M.; Kramer, I.; Czernia-Mazurkiewicz, J.; Wewer, V. Pediatric inflammatory bowel disease: Increasing incidence, decreasing surgery rate, and compromised nutritional status: A prospective population-based cohort study 2007–2009. Inflamm. Bowel Dis. 2011, 17, 2541–2550. [Google Scholar] [CrossRef] [PubMed]
- North American Society for Pediatric Gastroenterology Hepatology, and Nutrition and the Crohn’s Colitis Foundation of America. Differentiating ulcerative colitis from Crohn disease in children and young adults. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 653–674. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.M. Specificities of inflammatory bowel disease in childhood. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N.; Shanahan, F. Disorders of a modern lifestyle: Reconciling the epidemiology of inflammatory bowel diseases. Gut 2008, 57, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Bernstein, C.N.; Sood, A.; Goh, K.L.; Yamamoto-Furusho, J.K.; Abbas, Z.; Fried, M. Role of biological therapy for inflammatory bowel disease in developing countries. Gut 2012, 61, 706–712. [Google Scholar] [CrossRef] [PubMed]
- M’Koma, A.E.; Wise, P.E.; Muldoon, R.L.; Schwartz, D.A.; Washington, M.K.; Herline, A.J. Evolution of the restorative proctocolectomy and its effects on gastrointestinal hormones. Int. J. Colorectal Dis. 2007, 22, 1143–1163. [Google Scholar] [CrossRef] [PubMed]
- Stroup, D.F.; Berlin, J.A.; Morton, S.C.; Olkin, I.; Williamson, G.D.; Rennie, D.; Moher, D.; Becker, B.J.; Sipe, T.A.; Thacker, S.B. Meta-analysis of observational studies in epidemiology: A proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA 2000, 283, 2008–2012. [Google Scholar] [CrossRef] [PubMed]
- Greenland, S. Quantitative methods in the review of epidemiologic literature. Epidemiol. Rev. 1987, 9, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Shamseer, L.; Moher, D.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; PRISMA-P Group. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: Elaboration and explanation. BMJ 2015, 350, g7647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, H.; Chan, S.S.M.; Lochhead, P.; Ananthakrishnan, A.N.; Hart, A.R.; Chan, A.T. The role of diet in the aetiopathogenesis of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Turpin, W.; Goethel, A.; Bedrani, L.; Croitoru Mdcm, K. Determinants of IBD Heritability: Genes, Bugs, and More. Inflamm. Bowel Dis. 2018, 24, 1133–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Cheon, J.H. Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies. Immune Netw. 2017, 17, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cader, M.Z.; Kaser, A. Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation. Gut 2013, 62, 1653–1664. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.H. Genetics of inflammatory bowel diseases: A comparison between Western and Eastern perspectives. J. Gastroenterol. Hepatol. 2013, 8, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, K.A.; Targan, S.R. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu. Rev. Med. 2000, 51, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Song, I.D.; Kim, Y.H.; Koo, J.S.; Kim, Y.S.; Kim, J.S.; Kim, N.; Kim, E.S.; Kim, J.H.; Kim, J.W. Efficacy and Safety of Infliximab Therapy and Predictors of Response in Korean Patients with Crohn’s Disease: A Nationwide, Multicenter Study. Yonsei Med. J. 2016, 57, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Anderson, C.A. Genetic studies of Crohn’s disease: Past, present and future. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Moller, F.T.; Andersen, V.; Wohlfahrt, J.; Jess, T. Familial risk of inflammatory bowel disease: A population-based cohort study 1977–2011. Am. J. Gastroenterol. 2015, 110, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Brant, S.R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Desai, H.G.; Gupte, P.A. Increasing incidence of Crohn’s disease in India: Is it related to improved sanitation? Indian J. Gastroenterol. 2005, 24, 23–24. [Google Scholar] [PubMed]
- Zheng, J.J.; Zhu, X.S.; Huangfu, Z.; Gao, Z.X.; Guo, Z.R.; Wang, Z. Crohn’s disease in mainland China: A systematic analysis of 50 years of research. Chin. J. Dig. Dis. 2005, 6, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Tsironi, E.; Feakins, R.M.; Probert, C.S.; Rampton, D.S.; Phil, D. Incidence of inflammatory bowel disease is rising and abdominal tuberculosis is falling in Bangladeshis in East London, United Kingdom. Am. J. Gastroenterol. 2004, 99, 1749–1755. [Google Scholar] [CrossRef] [PubMed]
- Halme, L.; Paavola-Sakki, P.; Turunen, U.; Lappalainen, M.; Farkkila, M.; Kontula, K. Family and twin studies in inflammatory bowel disease. World J. Gastroenterol. 2006, 12, 3668–3672. [Google Scholar] [CrossRef] [PubMed]
- Thia, K.T.; Loftus, E.V., Jr.; Sandborn, W.J.; Yang, S.K. An update on the epidemiology of inflammatory bowel disease in Asia. Am. J. Gastroenterol. 2008, 103, 3167–3182. [Google Scholar] [CrossRef] [PubMed]
- Levi, Z.; Shamiss, A.; Fraser, G.M.; Furman, M.; Derazne, E.; Tzur, D.; Gordon, B.; Welinsky, S.; Gingold Belfer, R.; Afek, A. The Increasing Prevalence of Inflammatory Bowel Diseases Among Jewish Adolescents and the Sociodemographic Factors Associated with Diagnosis. Inflamm. Bowel. Dis. 2013, 19, 1867–1871. [Google Scholar] [CrossRef] [PubMed]
- Rivas, M.A.; Avila, B.E.; Koskela, J.; Huang, H.; Stevens, C.; Pirinen, M.; Haritunians, T.; Neale, B.M.; Kurki, M.; Ganna, A. Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet. 2018, 14, e1007329. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, S.; Mano, S.; Kozlowski, L.; Bujnicki, J.M.; Shibata, H.; Fukumaki, Y.; Kidd, J.R.; Kidd, K.K.; Kawamura, S.; Oota, H. Crohn’s disease risk alleles on the NOD2 locus have been maintained by natural selection on standing variation. Mol. Biol. Evol. 2012, 29, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Schurr, E.; Gros, P. A common genetic fingerprint in leprosy and Crohn’s disease? N. Engl. J. Med. 2009, 361, 2666–2668. [Google Scholar] [CrossRef] [PubMed]
- Ostrer, H.; Skorecki, K. The population genetics of the Jewish people. Hum. Genet. 2013, 132, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Moltke, I.; Grarup, N.; Jørgensen, M.E.; Bjerregaard, P.; Treebak, J.T.; Fumagalli, M.; Korneliussen, T.S.; Andersen, M.A.; Nielsen, T.S.; Krarup, N.T. A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 2014, 512, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.T.; Würtz, P.; Havulinna, A.S.; Palta, P.; Tukiainen, T.; Rehnström, K.; Esko, T.; Mägi, R.; Inouye, M.; Lappalainen, T. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014, 10, e1004494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuk, O.; Schaffner, S.F.; Samocha, K.; Do, R.; Hechter, E.; Kathiresan, S.; Daly, M.J.; Neale, B.M.; Sunyaev, S.; Lander, E.S. Searching for missing heritability: Designing rare variant association studies. Proc. Natl. Acad. Sci. USA 2014, 111, E455–E464. [Google Scholar] [CrossRef] [PubMed]
- Bahcall, O.; Orli, B. Rare variant association studies. Nat. Genet. 2014, 46, 219. [Google Scholar] [CrossRef]
- Auer, P.L.; Nalls, M.; Meschia, J.F.; Worrall, B.B.; Longstreth, W.T., Jr.; Seshadri, S.; Kooperberg, C.; Burger, K.M.; Carlson, C.S.; Carty, C.L. A genome-wide scan of Ashkenazi Jewish Crohn’s disease suggests novel susceptibility loci. PLoS Genet. 2012, 8, e1002559. [Google Scholar]
- Karban, A.; Eliakim, R.; Brant, S.R. Genetics of inflammatory bowel disease. Isr. Med. Assoc. J. 2002, 4, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Baskovich, B.; Hiraki, S.; Upadhyay, K.; Meyer, P.; Carmi, S.; Barzilai, N.; Darvasi, A.; Ozelius, L.; Peter, I.; Cho, J.H. Expanded genetic screening panel for the Ashkenazi Jewish population. Genet. Med. 2016, 18, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 8, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.H.; Hilmi, I.; Lian, L.; Patmanathan, S.N.; Hoe, S.Z.; Lee, W.S.; Goh, K.L. Association between inflammatory bowel disease gene 5 (IBD5) and interleukin-23 receptor (IL23R) genetic polymorphisms in Malaysian patients with Crohn’s disease. J. Dig. Dis. 2012, 13, 459–465. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, A.M.; Shanahan, F. The gut flora as a forgotten organ. EMBO Rep. 2006, 7, 688–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Goethel, A.; Croitoru, K.; Philpott, D.J. The interplay between microbes and the immune response in inflammatory bowel disease. J. Physiol. 2018, 596, 3869–3882. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, D.A.; Frank, D.N.; Pace, N.R.; Gordon, J.I. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 2008, 3, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Moran, C.; Shanahan, F. The microbiota in inflammatory bowel disease. J. Gastroenterol. 2015, 50, 495–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, A.W.; Sanderson, J.D.; Churcher, C.; Parkes, G.C.; Hudspith, B.N.; Rayment, N.; Brostoff, J.; Parkhill, J.; Dougan, G.; Petrovska, L. High-throughput clone library analysis of the mucosa-associated microbiota reveals dysbiosis and differences between inflamed and non-inflamed regions of the intestine in inflammatory bowel disease. BMC Microbiol. 2011, 11, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–565. [Google Scholar] [CrossRef] [PubMed]
- Varela, E.; Manichanh, C.; Gallart, M.; Torrejón, A.; Borruel, N.; Casellas, F.; Guarner, F.; Antolin, M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment. Pharmacol. Ther. 2013, 38, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.J.; Blugeon, S.; Bridonneau, C.; Furet, J.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA. 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishino, K.; Nishida, A.; Inoue, R.; Kawada, Y.; Ohno, M.; Sakai, S.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Kawahara, M.; et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 2018, 53, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Dogan, B.; Rishniw, M.; Weitzman, G.; Bosworth, B.; Yantiss, R.; Orsi, R.H.; Wiedmann, M.; McDonough, P.; Kim, S.G.; et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007, 1, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Kotlowski, R.; Bernstein, C.N.; Sepehri, S.; Krause, D.O. High prevalence of Escherichia coli belonging to the B2+D phylogenetic group in inflammatory bowel disease. Gut 2007, 56, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.; Antolin, M.; Santos, J.; Torrejon, A.; Casellas, F.; Borruel, N.; Guarner, F.; Malagelada, J.R. Unstable composition of the fecal microbiota in ulcerative colitis during clinical remission. Am. J. Gastroenterol. 2008, 103, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Mylonaki, M.; Rayment, N.B.; Rampton, D.S.; Hudspith, B.N.; Brostoff, J. Molecular characterization of rectal mucosa-associated bacterial flora in inflammatory bowel disease. Inflamm. Bowel. Dis. 2005, 11, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Neut, C.; Bulois, P.; Desreumaux, P.; Membré, J.M.; Lederman, E.; Gambiez, L.; Cortot, A.; Quandalle, P.; van Kruiningen, H.; Colombel, J.F. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Ladhoff, A.; Pernthaler, A.; Swidsinski, S.; Loening-Baucke, V.; Ortner, M.; Weber, J.; Hoffmann, U.; Schreiber, S.; Dietel, M.; et al. Mucosal flora in inflammatory bowel disease. Gastroenterology 2002, 122, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.F. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- van der Waaij, L.A.; Harmsen, H.J.; Madjipour, M.; Kroese, F.G.; Zwiers, M.; van Dullemen, H.M.; de Boer, N.K.; Welling, G.W.; Jansen, P.L. Bacterial population analysis of human colon and terminal ileum biopsies with 16S rRNA-based fluorescent probes: Commensal bacteria live in suspension and have no direct contact with epithelial cells. Inflamm. Bowel. Dis. 2005, 11, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Schultsz, C.; Van Den Berg, F.M.; Ten Kate, F.W.; Tytgat, G.N.; Dankert, J. The intestinal mucus layer from patients with inflammatory bowel disease harbors high numbers of bacteria compared with controls. Gastroenterology 1999, 117, 1089–1097. [Google Scholar] [CrossRef]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Loubinoux, J.; Bronowicki, J.P.; Pereira, I.A.; Mougenel, J.L.; Faou, A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002, 40, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinkevich, V.V.; Beech, I.B. Screening of sulfate-reducing bacteria in colonoscopy samples from healthy and colitic human gut mucosa. FEMS Microbiol. Ecol. 2000, 34, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Ann. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun. Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.; Quera, R. Small intestinal bacterial overgrowth: Roles of antibiotics, prebiotics, and probiotics. Gastroenterology 2006, 130, S78–S90. [Google Scholar] [CrossRef] [PubMed]
- Pickard, K.M.; Bremner, A.R.; Gordon, J.N.; MacDonald, T.T. Microbial-gut interactions in health and disease. Immune responses. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Nagao-Kitamoto, H.; Kitamoto, S.; Kuffa, P.; Kamada, N. Pathogenic role of the gut microbiota in gastrointestinal diseases. Intest. Res. 2016, 14, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.J.; Ullman, T.A.; Ford, A.C.; Abreu, M.T.; Abadir, A.; Marshall, J.K.; Talley, N.J.; Moayyedi, P. Antibiotic therapy in inflammatory bowel disease: A systematic review and meta-analysis. Am. J. Gastroenterol. 2011, 106, 661–673. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyama, K.; Niwa, M.; Takedatsu, H.; Yamasaki, H.; Kuwaki, K.; Yoshioka, S.; Yamauchi, R.; Fukunaga, S.; Torimura, T. Antibody markers in the diagnosis of inflammatory bowel disease. World J. Gastroenterol. 2016, 22, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef] [PubMed]
- Elson, C.O.; Cong, Y.; McCracken, V.J.; Dimmitt, R.A.; Lorenz, R.G.; Weaver, C.T. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol. Rev. 2005, 206, 260–276. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Kim, M.S.; Kim, E.; Cheon, J.H.; Lee, Y.S.; Kim, Y.; Lee, S.H.; Seo, S.U.; Shin, S.H.; Choi, S.S.; et al. Enteric Viruses Ameliorate Gut Inflammation via Toll-like Receptor 3 and Toll-like Receptor 7-Mediated Interferon-beta Production. Immunity 2016, 44, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Marlicz, W.; Zuba-Surma, E.; Kucia, M.; Blogowski, W.; Starzynska, T.; Ratajczak, M.Z. Various types of stem cells, including a population of very small embryonic-like stem cells, are mobilized into peripheral blood in patients with Crohn’s disease. Inflamm. Bowel Dis. 2012, 18, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Marlicz, W.; Skonieczna-Zydecka, K.; Dabos, K.J.; Loniewski, I.; Koulaouzidis, A. Emerging concepts in non-invasive monitoring of Crohn’s disease. Therap. Adv. Gastroenterol. 2018, 11, 1756284818769076. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sitaraman, S.V.; Babbin, B.A.; Gerner-Smidt, P.; Ribot, E.M.; Garrett, N.; Alpern, J.A.; Akyildiz, A.; Theiss, A.L.; Nusrat, A.; et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab. Investig. 2007, 87, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A. Adherent-invasive Escherichia coli: A putative new E. coli pathotype associated with Crohn’s disease. Int. J. Med. Microbiol. 2002, 292, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Medina, M.; Aldeguer, X.; Lopez-Siles, M.; González-Huix, F.; López-Oliu, C.; Dahbi, G.; Blanco, J.E.; Blanco, J.; Garcia-Gil, L.J.; Darfeuille-Michaud, A. Molecular diversity of Escherichia coli in the human gut: New ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.L.; Zheng, L.B.; Kanazawa, Y.; Shih, D.Q. Immunopathology of inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Littman, D.R. The microbiome in infectious disease and inflammation. Annu. Rev. Immunol. 2012, 30, 759–795. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Hill, J.A.; Kretschmer, K.; von Boehmer, H.; Mathis, D.; Benoist, C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. USA 2010, 107, 5919–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, Y.; Feng, T.; Fujihashi, K.; Schoeb, T.R.; Elson, C.O. A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2009, 106, 19256–19261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willing, B.; Halfvarson, J.; Dicksved, J.; Rosenquist, M.; Järnerot, G.; Engstrand, L.; Tysk, C.; Jansson, J.K. Twin studies reveal specific imbalances in the mucosa-associated microbiota of patients with ileal Crohn’s disease. Inflamm. Bowel Dis. 2009, 15, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Gollwitzer, E.S.; Marsland, B.J. Microbiota abnormalities in inflammatory airway diseases—Potential for therapy. Pharmacol. Ther. 2014, 141, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bellaguarda, E.; Chang, E.B. IBD and the gut microbiota—From bench to personalized medicine. Curr. Gastroenterol. Rep. 2015, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Pigneur, B.; Sokol, H. Fecal microbiota transplantation in inflammatory bowel disease: The quest for the holy grail. Mucosal Immunol. 2016, 9, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, K.A.; Boesmans, L.; Boets, E. Modulating the microbiota in inflammatory bowel diseases: Prebiotics, probiotics or faecal transplantation? Proc. Nutr. Soc. 2014, 73, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients With Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.; Duflou, A.; Löwenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.; de Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients With Ulcerative Colitis. Gastroenterology 2015, 149, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Rubin, D.T. Fecal microbiota transplantation as therapy for inflammatory bowel disease: A systematic review and meta-analysis. J. Crohns Colitis 2014, 8, 1569–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, D.; Katakura, K.; Karmeli, F.; Hayashi, T.; Reinus, C.; Rudensky, B.; Akira, S.; Takeda, K.; Lee, J.; Takabayashi, K.; et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology 2004, 126, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zuo, Z.X.; Mao, A.P. Effect of probiotics on inducing remission and maintaining therapy in ulcerative colitis, Crohn’s disease, and pouchitis: Meta-analysis of randomized controlled trials. Inflamm. Bowel Dis. 2014, 20, 21–35. [Google Scholar] [CrossRef] [PubMed]
- van der Sloot, K.W.J.; Weersma, R.K.; Dijkstra, G.; Alizadeh, B.Z. Development and validation of a web-based questionnaire to identify environmental risk factors for inflammatory bowel disease: The Groningen IBD Environmental Questionnaire (GIEQ). J. Gastroenterol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Molodecky, N.A.; Kaplan, G.G. Environmental risk factors for inflammatory bowel disease. Gastroenterol. Hepatol. 2010, 6, 339–346. [Google Scholar]
- Jess, T.; Riis, L.; Jespersgaard, C.; Hougs, L.; Andersen, P.S.; Orholm, M.K.; Binder, V.; Munkholm, P. Disease concordance, zygosity, and NOD2/CARD15 status: Follow-up of a population-based cohort of Danish twins with inflammatory bowel disease. Am. J. Gastroenterol. 2005, 100, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Gaya, D.R.; Russell, R.K.; Nimmo, E.R.; Satsangi, J. New genes in inflammatory bowel disease: Lessons for complex diseases? Lancet 2006, 367, 1271–1284. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Jess, T.; Magnuson, A.; Montgomery, S.M.; Orholm, M.; Tysk, C.; Binder, V.; Järnerot, G. Environmental factors in inflammatory bowel disease: A co-twin control study of a Swedish-Danish twin population. Inflamm. Bowel Dis. 2006, 12, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Benchimol, E.I.; Kaplan, G.G.; Otley, A.R.; Nguyen, G.C.; Underwood, F.E.; Guttmann, A.; Jones, J.L.; Potter, B.K.; Catley, C.A.; Nugent, Z.J.; et al. Rural and Urban Residence During Early Life is Associated with Risk of Inflammatory Bowel Disease: A Population-Based Inception and Birth Cohort Study. Am. J. Gastroenterol. 2017, 112, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Hubbard, J.; Korzenik, J.; Sands, B.E.; Panaccione, R.; Ghosh, S.; Wheeler, A.J.; Villeneuve, P.J. The inflammatory bowel diseases and ambient air pollution: A novel association. Am. J. Gastroenterol. 2010, 105, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Albenberg, L.; Compher, C.; Baldassano, R.; Piccoli, D.; Lewis, J.D.; Wu, G.D. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology 2015, 148, 1087–1106. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Mawdsley, J.E.; Rampton, D.S. Psychological stress in IBD: New insights into pathogenic and therapeutic implications. Gut 2005, 54, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Lerebours, E.; Gower-Rousseau, C.; Merle, V.; Brazier, F.; Debeugny, S.; Marti, R.; Salomez, J.L.; Hellot, M.F.; Dupas, J.L.; Colombel, J.F.; et al. Stressful life events as a risk factor for inflammatory bowel disease onset: A population-based case-control study. Am. J. Gastroenterol. 2007, 102, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Probert, C.S.; Jayanthi, V.; Pinder, D.; Wicks, A.C.; Mayberry, J.F. Epidemiological study of ulcerative proctocolitis in Indian migrants and the indigenous population of Leicestershire. Gut 1992, 33, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Sewell, J.L.; Yee, H.F., Jr.; Inadomi, J.M. Hospitalizations are increasing among minority patients with Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2010, 16, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Vavricka, S. Exposome in IBD: Recent insights in environmental factors that influence the onset and course of IBD. Inflamm. Bowel Dis. 2015, 21, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Lakatos, P.L.; Vegh, Z.; Lovasz, B.D.; David, G.; Pandur, T.; Erdelyi, Z.; Szita, I.; Mester, G.; Balogh, M.; Szipocs, I.; et al. Is current smoking still an important environmental factor in inflammatory bowel diseases? Results from a population-based incident cohort. Inflamm. Bowel Dis. 2013, 19, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, L.M.; Khalili, H.; Chan, A.T.; Richter, J.M.; Bousvaros, A.; Fuchs, C.S. A prospective study of cigarette smoking and the risk of inflammatory bowel disease in women. Am. J. Gastroenterol. 2012, 107, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Kevans, D.; Turpin, W.; Madsen, K.; Meddings, J.; Shestopaloff, K.; Xu, W.; Moreno-Hagelsieb, G.; Griffiths, A.; Silverberg, M.S.; Paterson, A.; et al. Determinants of intestinal permeability in healthy first-degree relatives of individuals with Crohn’s disease. Inflamm. Bowel Dis. 2015, 21, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, C.N. Review article: Changes in the epidemiology of inflammatory bowel disease-clues for aetiology. Aliment. Pharmacol. Ther. 2017, 46, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Osterman, M.T.; Bewtra, M.; Lewis, J.D. Passive smoking and inflammatory bowel disease: A meta-analysis. Am. J. Gastroenterol. 2008, 103, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Chivese, T.; Esterhuizen, T.M.; Basson, A.R.; Watermeyer, G. The Influence of Second-Hand Cigarette Smoke Exposure during Childhood and Active Cigarette Smoking on Crohn’s Disease Phenotype Defined by the Montreal Classification Scheme in a Western Cape Population, South Africa. PLoS ONE 2015, 10, e0139597. [Google Scholar] [CrossRef] [PubMed]
- Mahid, S.S.; Minor, K.S.; Stromberg, A.J.; Galandiuk, S. Active and passive smoking in childhood is related to the development of inflammatory bowel disease. Inflamm. Bowel. Dis. 2007, 13, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.Y.; Stevens, B.W.; Wilson, R.G.; Russell, C.N.; Cohen, M.A.; Sturgeon, H.C.; Thornton, A.; Giallourakis, C.; Khalili, H.; Nguyen, D.D.; et al. Early life environment and natural history of inflammatory bowel diseases. BMC Gastroenterol. 2014, 14, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söderholm, J.D.; Yang, P.C.; Ceponis, P.; Vohra, A.; Riddell, R.; Sherman, P.M.; Perdue, M.H. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology 2002, 123, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Mazzon, E.; Sturniolo, G.C.; Puzzolo, D.; Frisina, N.; Fries, W. Effect of stress on the paracellular barrier in the rat ileum. Gut 2002, 51, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Hu, J.; Ghadermarzi, S.; Raza, A.; O’Connell, D.; Xiao, A.; Ayyaz, F.; Zhi, M.; Zhang, Y.; Parekh, N.K.; et al. Smoking and Inflammatory Bowel Disease: A Comparison of China, India, and the USA. Dig. Dis. Sci. 2018, 63, 2703–2713. [Google Scholar] [CrossRef] [PubMed]
- Salih, A.; Widbom, L.; Hultdin, J.; Karling, P. Smoking is associated with risk for developing inflammatory bowel disease including late onset ulcerative colitis: A prospective study. Scand. J. Gastroenterol. 2018, 53, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Matricon, J.; Barnich, N.; Ardid, D. Immunopathogenesis of inflammatory bowel disease. Self Nonself 2010, 1, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, C.T.; Elson, C.O.; Fouser, L.A.; Kolls, J.K. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu. Rev. Pathol. 2013, 8, 477–512. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Goswami, R. A Decade of Th9 Cells: Role of Th9 Cells in Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 1139. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Raj, J.P. Role of biologics and biosimilars in inflammatory bowel disease: Current trends and future perspectives. J. Inflamm. Res. 2018, 11, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Morrow, T.; Felcone, L.H. Defining the difference: What Makes Biologics Unique. Biotechnol. Healthc. 2004, 1, 24–29. [Google Scholar] [PubMed]
- Chan, H.C.; Ng, S.C. Emerging biologics in inflammatory bowel disease. J. Gastroenterol. 2017, 52, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Seto, K.C.P.S.; Elmqvist, T. Urbanization, Biodiversity and Ecosystem Service: Challenges and Opportunities: A Global Assessment; Springer: Dordrecht, The Netherlands, 2013. [Google Scholar]
- Ng, S.C.; Tang, W.; Ching, J.Y.; Wong, M.; Chow, C.M.; Hui, A.J.; Wong, T.C.; Leung, V.K.; Tsang, S.W.; Yu, H.H.; et al. Incidence and phenotype of inflammatory bowel disease based on results from the Asia-pacific Crohn’s and colitis epidemiology study. Gastroenterology 2013, 145, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Bernstein, C.N.; Vatn, M.H.; Lakatos, P.L.; Loftus, E.V., Jr.; Tysk, C.; O’Morain, C.; Moum, B.; Colombel, J.F.; Epidemiology and Natural History Task Force of the International Organization of Inflammatory Bowel Disease (IOIBD). Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013, 62, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C. Emerging leadership lecture: Inflammatory bowel disease in Asia: Emergence of a “Western” disease. J. Gastroenterol. Hepatol. 2015, 30, 440–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Kim, W.H.; Cheon, J.H. Clinical characteristics and treatment of inflammatory bowel disease: A comparison of Eastern and Western perspectives. World J. Gastroenterol. 2014, 20, 11525–115237. [Google Scholar] [CrossRef] [PubMed]
- Tozun, N.; Atug, O.; Imeryuz, N.; Hamzaoglu, H.O.; Tiftikci, A.; Parlak, E.; Dagli, U.; Ulker, A.; Hulagu, S.; Akpinar, H.; et al. Clinical characteristics of inflammatory bowel disease in Turkey: A multicenter epidemiologic survey. J. Clin. Gastroenterol. 2009, 43, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Kirsner, J.B. Historical origins of current IBD concepts. World J. Gastroenterol. 2001, 7, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Soon, I.S.; Molodecky, N.A.; Rabi, D.M.; Ghali, W.A.; Barkema, H.W.; Kaplan, G.G. The relationship between urban environment and the inflammatory bowel diseases: A systematic review and meta-analysis. BMC Gastroenterol. 2012, 12, 51. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, I.; Blaser, M.J. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012, 13, 260–270. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The gut microbiota of rural papua new guineans: Composition, diversity patterns, and ecological processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Kamm, M.A.; Colombel, J.F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Zoetendal, E.G.; Puylaert, P.G.P.; Ou, J.; Vipperla, K.; Brouard, F.M.; Ruder, E.H.; Newton, K.; Carbonero, F.; Gaskins, H.R.; de Vos, W.M.; et al. Distinct Microbiotas Are Present in Urban and Rural Native South Africans, and in African Americans. Gastroenterology 2013, 144. [Google Scholar] [CrossRef]
- Benno, Y.; Endo, K.; Mizutani, T.; Namba, Y.; Komori, T.; Mitsuoka, T. Comparison of fecal microflora of elderly persons in rural and urban areas of Japan. Appl. Environ. Microbiol. 1989, 55, 1100–1105. [Google Scholar] [PubMed]
- Zhang, J.; Zheng, Y.; Guo, Z.; Qiao, J.; Gesudu, Q.; Sun, Z.; Huo, D.; Huang, W.; Huo, Q.; Kwok, L.; et al. The diversity of intestinal microbiota of Mongolians living in Inner Mongolia, China. Benef. Microbes 2013, 4, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Tyakht, A.V.; Kostryukova, E.S.; Popenko, A.S.; Belenikin, M.S.; Pavlenko, A.V.; Larin, A.K.; Karpova, I.Y.; Selezneva, O.V.; Semashko, T.A.; Ospanova, E.A.; et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat. Commun. 2013, 4, 2469. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyakht, A.V.; Alexeev, D.G.; Popenko, A.S.; Kostryukova, E.S.; Govorun, V.M. Rural and urban microbiota: To be or not to be? Gut Microbes 2014, 5, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J. The theory of disappearing microbiota and the epidemics of chronic diseases. Nat. Rev. Immunol. 2017, 17, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Liang, S.; Carlton, E.J.; Jiang, Q.; Wu, J.; Wang, L.; Remais, J.V. Urbanisation and health in China. Lancet 2012, 379, 843–852. [Google Scholar] [CrossRef] [Green Version]
- Ananthakrishnan, A.N.; McGinley, E.L.; Binion, D.G.; Saeian, K. Ambient air pollution correlates with hospitalizations for inflammatory bowel disease: An ecologic analysis. Inflamm. Bowel Dis. 2011, 17, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- To, N.; Gracie, D.J.; Ford, A.C. The Importance of Smoking Cessation in Improving Disease Course in Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1198. [Google Scholar] [CrossRef] [PubMed]
- Nunes, T.; Etchevers, M.J.; García-Sánchez, V.; Ginard, D.; Martí, E.; Barreiro-de Acosta, M.; Gomollón, F.; Arroyo, M.; Bastida, G.; Gonzalez, B.; et al. Impact of Smoking Cessation on the Clinical Course of Crohn’s Disease Under Current Therapeutic Algorithms: A Multicenter Prospective Study. Am. J. Gastroenterol. 2016, 111, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.J.; Cosnes, J.; Mansfield, J.C. Review article: Smoking cessation as primary therapy to modify the course of Crohn’s disease. Aliment. Pharmacol. Ther. 2005, 21, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.L.; Hedin, C.R.; Koutsoumpas, A.; Ng, S.C.; McCarthy, N.E.; Prescott, N.J.; Pessoa-Lopes, P.; Mathew, C.G.; Sanderson, J.; Hart, A.L.; et al. Smokers with active Crohn’s disease have a clinically relevant dysbiosis of the gastrointestinal microbiota. Inflamm. Bowel Dis. 2012, 18, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Van de Wiele, T.; Gallawa, C.M.; Kubachka, K.M.; Creed, J.T.; Basta, N.; Dayton, E.A.; Whitacre, S.; Du Laing, G.; Bradham, K. Arsenic metabolism by human gut microbiota upon in vitro digestion of contaminated soils. Environ. Health Perspect. 2010, 118, 1004–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Wiele, T.; Vanhaecke, L.; Boeckaert, C.; Peru, K.; Headley, J.; Verstraete, W.; Siciliano, S. Human colon microbiota transform polycyclic aromatic hydrocarbons to estrogenic metabolites. Environ. Health Perspect. 2005, 113, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, K.; Mundandhara, S.; Ghio, A.J.; Madden, M.C. The effects of ambient particulate matter on human alveolar macrophage oxidative and inflammatory responses. J. Toxicol. Environ. Health A 2010, 73, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Van Eeden, S.F.; Tan, W.C.; Suwa, T.; Mukae, H.; Terashima, T.; Fujii, T.; Qui, D.; Vincent, R.; Hogg, J.C. Cytokines involved in the systemic inflammatory response induced by exposure to particulate matter air pollutants (PM(10)). Am. J. Respir. Crit. Care. Med. 2001, 164, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Rickert, D.E.; Butterworth, B.E.; Popp, J.A. Dinitrotoluene: Acute toxicity, oncogenicity, genotoxicity, and metabolism. Crit. Rev. Toxicol. 1984, 13, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Rickert, D.E.; Long, R.M.; Krakowka, S.; Dent, J.G. Metabolism and excretion of 2,4-[14C]Dinitrotoluene in conventional and axenic Fischer-344 rats. Toxicol. Appl. Pharmacol. 1981, 59, 574–579. [Google Scholar] [CrossRef]
- Yim, Y.J.; Seo, J.; Kang, S.I.; Ahn, J.H.; Hur, H.G. Reductive dechlorination of methoxychlor and DDT by human intestinal bacterium Eubacterium limosum under anaerobic conditions. Arch. Environ. Contam. Toxicol. 2008, 54, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Joly, C.; Gay-Quéheillard, J.; Léké, A.; Chardon, K.; Delanaud, S.; Bach, V.; Khorsi-Cauet, H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ. Sci. Pollut. Res. Int. 2013, 20, 2726–2734. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, M.; Hassanzadeh, P.; Alaei, S. Cadmium chloride exhibits a profound toxic effect on bacterial microflora of the mice gastrointestinal tract. Hum. Exp. Toxicol. 2011, 30, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Breton, J.; Massart, S.; Vandamme, P.; De Brandt, E.; Pot, B.; Foligne, B. Ecotoxicology inside the gut: Impact of heavy metals on the mouse microbiome. BMC Pharmacol. Toxicol. 2013, 14, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; Abo, R.P.; Schlieper, K.A.; Graffam, M.E.; Levine, S.; Wishnok, J.S.; Swenberg, J.A.; Tannenbaum, S.R.; Fox, J.G. Arsenic exposure perturbs the gut microbiome and its metabolic profile in mice: An integrated metagenomics and metabolomics analysis. Environ. Health Perspect. 2014, 122, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Heinze, T.M.; Chen, S.; Cerniglia, C.E.; Chen, H. Anaerobic metabolism of 1-amino-2-naphthol-based azo dyes (Sudan dyes) by human intestinal microflora. Appl. Environ. Microbiol. 2007, 73, 7759–7762. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhao, A.; Xie, G.; Chi, Y.; Zhao, L.; Li, H.; Wang, C.; Bao, Y.; Jia, W.; Luther, M.; et al. Melamine-induced renal toxicity is mediated by the gut microbiota. Sci. Transl. Med. 2013, 5, 172ra22. [Google Scholar] [CrossRef] [PubMed]
- Kish, L.; Hotte, N.; Kaplan, G.G.; Vincent, R.; Tso, R.; Gänzle, M.; Rioux, K.P.; Thiesen, A.; Barkema, H.W.; Wine, E.; et al. Environmental particulate matter induces murine intestinal inflammatory responses and alters the gut microbiome. PLoS ONE 2013, 8, e62220. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.J.; Eum, S.Y.; Rampersaud, E.; Daunert, S.; Abreu, M.T.; Toborek, M. Exercise attenuates PCB-induced changes in the mouse gut microbiome. Environ. Health Perspect. 2013, 121, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Lepage, P.; Häsler, R.; Spehlmann, M.E.; Rehman, A.; Zvirbliene, A.; Begun, A.; Ott, S.; Kupcinskas, L.; Doré, J.; Raedler, A.; et al. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011, 141, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.L.; Finlay, B.B. The impact of gut microbes in allergic diseases. Curr. Opin. Gastroenterol. 2012, 28, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Gascon, M.; Morales, E.; Sunyer, J.; Vrijheid, M. Effects of persistent organic pollutants on the developing respiratory and immune systems: A systematic review. Environ. Int. 2013, 52, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Menard, S.; Guzylack-Piriou, L.; Leveque, M.; Braniste, V.; Lencina, C.; Naturel, M.; Moussa, L.; Sekkal, S.; Harkat, C.; Gaultier, E.; et al. Food intolerance at adulthood after perinatal exposure to the endocrine disruptor bisphenol A. FASEB J. 2014, 28, 4893–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belles-Isles, M.; Ayotte, P.; Dewailly, E.; Weber, J.P.; Roy, R. Cord blood lymphocyte functions in newborns from a remote maritime population exposed to organochlorines and methylmercury. J. Toxicol. Environ. Health A 2002, 65, 165–182. [Google Scholar] [CrossRef] [PubMed]
- MacGillivray, D.M.; Kollmann, T.R. The role of environmental factors in modulating immune responses in early life. Front. Immunol. 2014, 5, 434. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Andersen, V.; Chan, S.; Luben, R.; Khaw, K.T.; Olsen, A.; Tjonneland, A.; Kaaks, R.; Grip, O.; Bergmann, M.M.; Boeing, H.; et al. Fibre intake and the development of inflammatory bowel disease: A European prospective multi-centre cohort study (EPIC-IBD). J. Crohns Colitis 2018, 12, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Opstelten, J.L.; Leenders, M.; Dik, V.K.; Chan, S.S.; van Schaik, F.D.; Khaw, K.T.; Luben, R.; Hallmans, G.; Karling, P.; Lindgren, S.; et al. Dairy Products, Dietary Calcium, and Risk of Inflammatory Bowel Disease: Results From a European Prospective Cohort Investigation. Inflamm. Bowel Dis. 2016, 22, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zamora-Ros, R.; Chan, S.; Cross, A.J.; Ward, H.; Jakszyn, P.; Luben, R.; Opstelten, J.L.; Oldenburg, B.; Hallmans, G.; et al. Dietary Polyphenols in the Aetiology of Crohn’s Disease and Ulcerative Colitis-A Multicenter European Prospective Cohort Study (EPIC). Inflamm. Bowel Dis. 2017, 23, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Tjonneland, A.; Overvad, K.; Bergmann, M.M.; Nagel, G.; Linseisen, J.; Hallmans, G.; Palmqvist, R.; Sjodin, H.; Hagglund, G.; Berglund, G.; et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: A nested case-control study within a European prospective cohort study. Gut 2009, 58, 1606–1611. [Google Scholar] [PubMed]
- Shim, J.S.; Oh, K.; Kim, H.C. Dietary assessment methods in epidemiologic studies. Epidemiol. Health 2014, 36, e2014009. [Google Scholar] [CrossRef] [PubMed]
- Racine, A.; Carbonnel, F.; Chan, S.S.; Hart, A.R.; Bueno-de-Mesquita, H.B.; Oldenburg, B.; van Schaik, F.D.; Tjønneland, A.; Olsen, A.; Dahm, C.C.; et al. Dietary Patterns and Risk of Inflammatory Bowel Disease in Europe: Results from the EPIC Study. Inflamm. Bowel Dis. 2016, 22, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.F. Impaired inactivation of digestive proteases by deconjugated bilirubin: The possible mechanism for inflammatory bowel disease. Med. Hypotheses 2002, 59, 159–163. [Google Scholar] [CrossRef]
- Qin, X. Etiology of inflammatory bowel disease: A unified hypothesis. World J. Gastroenterol. 2012, 18, 1708–1722. [Google Scholar] [CrossRef] [PubMed]
- Lange, F.T.; Scheurer, M.; Brauch, H.J. Artificial sweeteners—A recently recognized class of emerging environmental contaminants: A review. Anal. Bioanal. Chem. 2012, 403, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Hakansson, N.; Chan, S.S.; Ludvigsson, J.F.; Olen, O.; Chan, A.T.; Hart, A.R.; Wolk, A. No Association Between Consumption of Sweetened Beverages and Later Risk of Crohn’s Disease or Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J. Gene-environment interactions in human diseases. Nat. Rev. Genet. 2005, 6, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, V.A.; Cham, C.M.; Chang, E.B. Diet, gut microbes, and genetics in immune function: Can we leverage our current knowledge to achieve better outcomes in inflammatory bowel diseases? Curr. Opin. Immunol. 2014, 31, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; Malik, S.; Ananthakrishnan, A.N.; Garber, J.J.; Higuchi, L.M.; Joshi, A.; Peloquin, J.; Richter, J.M.; Stewart, K.O.; Curhan, G.C.; et al. Identification and Characterization of a Novel Association between Dietary Potassium and Risk of Crohn’s Disease and Ulcerative Colitis. Front. Immunol. 2016, 7, 554. [Google Scholar] [CrossRef] [PubMed]
- Van Der Sloot, K.W.; Joshi, A.D.; Bellavance, D.R.; Gilpin, K.K.; Stewart, K.O.; Lochhead, P.; Garber, J.J.; Giallourakis, C.; Yajnik, V.; Ananthakrishnan, A.N.; et al. Visceral Adiposity, Genetic Susceptibility, and Risk of Complications Among Individuals with Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Khalili, H.; Song, M.; Higuchi, L.M.; Lochhead, P.; Richter, J.M.; Chan, A.T. Genetic Polymorphisms in Fatty Acid Metabolism Modify the Association Between Dietary n3: N6 Intake and Risk of Ulcerative Colitis: A Prospective Cohort Study. Inflamm. Bowel Dis. 2017, 23, 1898–1904. [Google Scholar] [CrossRef] [PubMed]
- Costea, I.; Mack, D.R.; Lemaitre, R.N.; Israel, D.; Marcil, V.; Ahmad, A.; Amre, D.K. Interactions between the dietary polyunsaturated fatty acid ratio and genetic factors determine susceptibility to pediatric Crohn’s disease. Gastroenterology 2014, 146, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Khalili, H.; de Silva, P.S.; Ananthakrishnan, A.N.; Lochhead, P.; Joshi, A.; Garber, J.J.; Richter, J.R.; Sauk, J.; Chan, A.T. Dietary Iron and Heme Iron Consumption, Genetic Susceptibility, and Risk of Crohn’s Disease and Ulcerative Colitis. Inflamm. Bowel Dis. 2017, 23, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, C.E.; Rose-Zerilli, M.J.; Machado, L.R.; Iriyama, C.; Hollox, E.J.; Cragg, M.S.; Strefford, J.C. Fcγ receptors: Genetic variation, function, and disease. Immunol. Rev. 2015, 268, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.P.; Edberg, J.C.; Kimberly, R.P.; Mangan, E.K.; Bharadwaj, D.; Mold, C.; Du Clos, T.W. C-reactive protein binding to FcgammaRIIa on human monocytes and neutrophils is allele-specific. J. Clin. Investig. 2000, 105, 369–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, M.P.; Mold, C.; Du Clos, T.W. C-reactive protein binding to murine leukocytes requires Fc gamma receptors. J. Immunol. 2000, 164, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, D.; Stein, M.P.; Volzer, M.; Mold, C.; Du Clos, T.W. The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J. Exp. Med. 1999, 190, 585–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Rizzo, A.; Fina, D.; Caruso, R.; Becker, C.; Neurath, M.F.; Macdonald, T.T.; Pallone, F.; Monteleone, G. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur. J. Immunol. 2007, 37, 3155–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Andersen, R.F.; Christensen, H.; Nathan, T.; Kjeldsen, J.; Madsen, J.S. Circulating microRNAs as biomarkers of adult Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1038–1044. [Google Scholar] [CrossRef] [PubMed]
- Chapman, C.G.; Pekow, J. The emerging role of miRNAs in inflammatory bowel disease: A review. Therap. Adv. Gastroenterol. 2015, 8, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Dalal, S.R.; Kwon, J.H. The Role of MicroRNA in Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2010, 6, 714–722. [Google Scholar]
- Ratajczak, M.Z.; Ratajczak, J. Horizontal transfer of RNA and proteins between cells by extracellular microvesicles: 14 years later. Clin. Transl. Med. 2016, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Ratajczak, D.; Pedziwiatr, D. Extracellular Microvesicles (ExMVs) in Cell to Cell Communication: A Role of Telocytes. Adv. Exp. Med. Biol. 2016, 913, 41–49. [Google Scholar] [PubMed]
- Ratajczak, M.Z.; Ratajczak, J. Extracellular Microvesicles as Game Changers in Better Understanding the Complexity of Cellular Interactions-From Bench to Clinical Applications. Am. J. Med. Sci. 2017, 354, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Tziatzios, G.; Polymeros, D.; Spathis, A.; Triantafyllou, M.; Gkolfakis, P.; Karakitsos, P.; Dimitriadis, G.; Triantafyllou, K. Increased levels of circulating platelet derived microparticles in Crohn’s disease patients. Scand. J. Gastroenterol. 2016, 51, 1184–1192. [Google Scholar] [CrossRef] [PubMed]
- Swanson, G.; Behara, R.; Braun, R.; Keshavarzian, A. Diagnostic medical radiation in inflammatory bowel disease: How to limit risk and maximize benefit. Inflamm. Bowel Dis. 2013, 19, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Cantoro, L.; Di Sabatino, A.; Papi, C.; Margagnoni, G.; Ardizzone, S.; Giuffrida, P.; Giannarelli, D.; Massari, A.; Monterubbianesi, R.; Lenti, M.V.; et al. The Time Course of Diagnostic Delay in Inflammatory Bowel Disease Over the Last Sixty Years: An Italian Multicentre Study. J. Crohns Colitis 2017, 11, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.L.; Stocchi, L.; Duraes, L.; Rencuzogullari, A.; Bennett, A.E.; Remzi, F.H. Long-Term Outcomes in Indeterminate Colitis Patients Undergoing Ileal Pouch-Anal Anastomosis: Function, Quality of Life, and Complications. J. Gastrointest. Surg. 2017, 21, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Tremaine, W.J. Is indeterminate colitis determinable? Curr. Gastroenterol. Rep. 2012, 14, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Tremaine, W.J. Review article: Indeterminate colitis—Definition, diagnosis and management. Aliment. Pharmacol. Ther. 2007, 25, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Conrad, K.; Roggenbuck, D.; Laass, M.W. Diagnosis and classification of ulcerative colitis. Autoimmun. Rev. 2014, 13, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Laass, M.W.; Roggenbuck, D.; Conrad, K. Diagnosis and classification of Crohn’s disease. Autoimmun. Rev. 2014, 13, 467–471. [Google Scholar] [CrossRef] [PubMed]
- M’Koma, A.E.; Seeley, E.H.; Washington, M.K.; Schwartz, D.A.; Muldoon, R.L.; Herline, A.J.; Wise, P.E.; Caprioli, R.M. Proteomic profiling of mucosal and submucosal colonic tissues yields protein signatures that differentiate the inflammatory colitides. Inflamm. Bowel Dis. 2011, 17, 875–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeley, E.H.; Washington, M.K.; Caprioli, R.M.; M’Koma, A.E. Proteomic patterns of colonic mucosal tissues delineate Crohn’s colitis and ulcerative colitis. Proteom. Clin. Appl. 2013, 7, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.D.; Korolkova, O.Y.; Sakwe, A.M.; Geiger, T.M.; James, S.D.; Muldoon, R.L.; Herline, A.J.; Goodwin, J.S.; Izban, M.G.; Washington, M.K.; et al. Human alpha defensin 5 is a candidate biomarker to delineate inflammatory bowel disease. PLoS ONE 2017, 12, e0179710. [Google Scholar]
- Shen, B.; Remzi, F.H.; Brzezinski, A.; Lopez, R.; Bennett, A.E.; Lavery, I.C.; Queener, E.; Fazio, V.W. Risk factors for pouch failure in patients with different phenotypes of Crohn’s disease of the pouch. Inflamm. Bowel Dis. 2008, 14, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Remzi, F.H.; Lavery, I.C.; Lashner, B.A.; Fazio, V.W. A proposed classification of ileal pouch disorders and associated complications after restorative proctocolectomy. Clin. Gastroenterol. Hepatol. 2008, 6, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Mehta, F. Report: Economic implications of inflammatory bowel disease and its management. Am. J. Manag. Care 2016, 22, s51–s60. [Google Scholar] [PubMed]
- Long, M.D.; Sands, B.E. What Is the Role of the Inflammatory Bowel Disease Panel in Diagnosis and Treatment? Clin. Gastroenterol. Hepatol. 2018, 16, 618–620. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Cagan, A.; Cai, T.; Gainer, V.S.; Shaw, S.Y.; Savova, G.; Churchill, S.; Karlson, E.W.; Kohane, I.; Liao, K.P.; et al. Comparative Effectiveness of Infliximab and Adalimumab in Crohn’s Disease and Ulcerative Colitis. Inflamm. Bowel Dis. 2016, 22, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.P.; Soo, W.; Bryant, R.V.; Jairath, V.; Hart, A.L.; Andrews, J.M. Systematic review with meta-analysis: Faecal microbiota transplantation for the induction of remission for active ulcerative colitis. Aliment. Pharmacol. Ther. 2017, 46, 213–224. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) Small intestine mucosal immune system landscape. The intestinal epithelial cell (IEC) layers form villi and crypt structures and are composed of different cell lineages. Goblet cells secrete mucus. Paneth cells, found only in the small intestine, can be found at the base of the crypts and are the main secretors of antimicrobial peptides. The base of the crypts also contains the IEC stem cell populations. Immune cells can be found in organized tissue such as Peyer’s patches and throughout the lamina propria. They include macrophages, dendritic cells, intra-epithelial lymphocytes, lamina propria effector T cells, IgA secreting plasma cells, innate lymphoid cells and stromal cells such as fibroblasts. Antigen presenting cells in Peyer’s patches or mesenteric lymph nodes interact with and activate local lymphocytes, which consequently upregulate expression of the integrin α4β7. Such cells then enter the systemic circulation but home towards the gut, in response to chemokine ligands such as CCL25. (B) Colon (large intestine) mucosal immune system. The colon has a much higher bacterial load and a markedly different immune cell composition. The colon contains only crypts, no villi. Also there are no Paneth cells, which mean that enterocytes have a much more important contribution to antimicrobial peptide production. However, there is a high prevalence of goblet cells. The mucus forms dual layers, with a thick largely sterile inner layer and a thinner outer layer. There are no Peyer’s patches. While the immune cell types present are similar to those found in the small intestine it is likely that there may be at least subtle differences. In particular natural killer T cells are found more frequently and have a more significant role in the colon. Adapted with permission from “Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation” by Cader and Kaser, Gut 2013, 62, 1653–1664, BMJ Publishing Group Limited [48].
Figure 1.
(A) Small intestine mucosal immune system landscape. The intestinal epithelial cell (IEC) layers form villi and crypt structures and are composed of different cell lineages. Goblet cells secrete mucus. Paneth cells, found only in the small intestine, can be found at the base of the crypts and are the main secretors of antimicrobial peptides. The base of the crypts also contains the IEC stem cell populations. Immune cells can be found in organized tissue such as Peyer’s patches and throughout the lamina propria. They include macrophages, dendritic cells, intra-epithelial lymphocytes, lamina propria effector T cells, IgA secreting plasma cells, innate lymphoid cells and stromal cells such as fibroblasts. Antigen presenting cells in Peyer’s patches or mesenteric lymph nodes interact with and activate local lymphocytes, which consequently upregulate expression of the integrin α4β7. Such cells then enter the systemic circulation but home towards the gut, in response to chemokine ligands such as CCL25. (B) Colon (large intestine) mucosal immune system. The colon has a much higher bacterial load and a markedly different immune cell composition. The colon contains only crypts, no villi. Also there are no Paneth cells, which mean that enterocytes have a much more important contribution to antimicrobial peptide production. However, there is a high prevalence of goblet cells. The mucus forms dual layers, with a thick largely sterile inner layer and a thinner outer layer. There are no Peyer’s patches. While the immune cell types present are similar to those found in the small intestine it is likely that there may be at least subtle differences. In particular natural killer T cells are found more frequently and have a more significant role in the colon. Adapted with permission from “Recent advances in inflammatory bowel disease: Mucosal immune cells in intestinal inflammation” by Cader and Kaser, Gut 2013, 62, 1653–1664, BMJ Publishing Group Limited [48].

Figure 2.
Intestinal immune system. IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; TGF, transforming growth factor; Th, helper T cell; Treg, regulatory T cell; TCR, T cell receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cell; TLR, toll-like receptor; NOD, nucleotide oligomerization domain. Adapted with permission from “Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies” by Kim and Cheon, Immune Netw. 2017, 17, 25–40 [46].
Figure 2.
Intestinal immune system. IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; TGF, transforming growth factor; Th, helper T cell; Treg, regulatory T cell; TCR, T cell receptor; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cell; TLR, toll-like receptor; NOD, nucleotide oligomerization domain. Adapted with permission from “Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies” by Kim and Cheon, Immune Netw. 2017, 17, 25–40 [46].
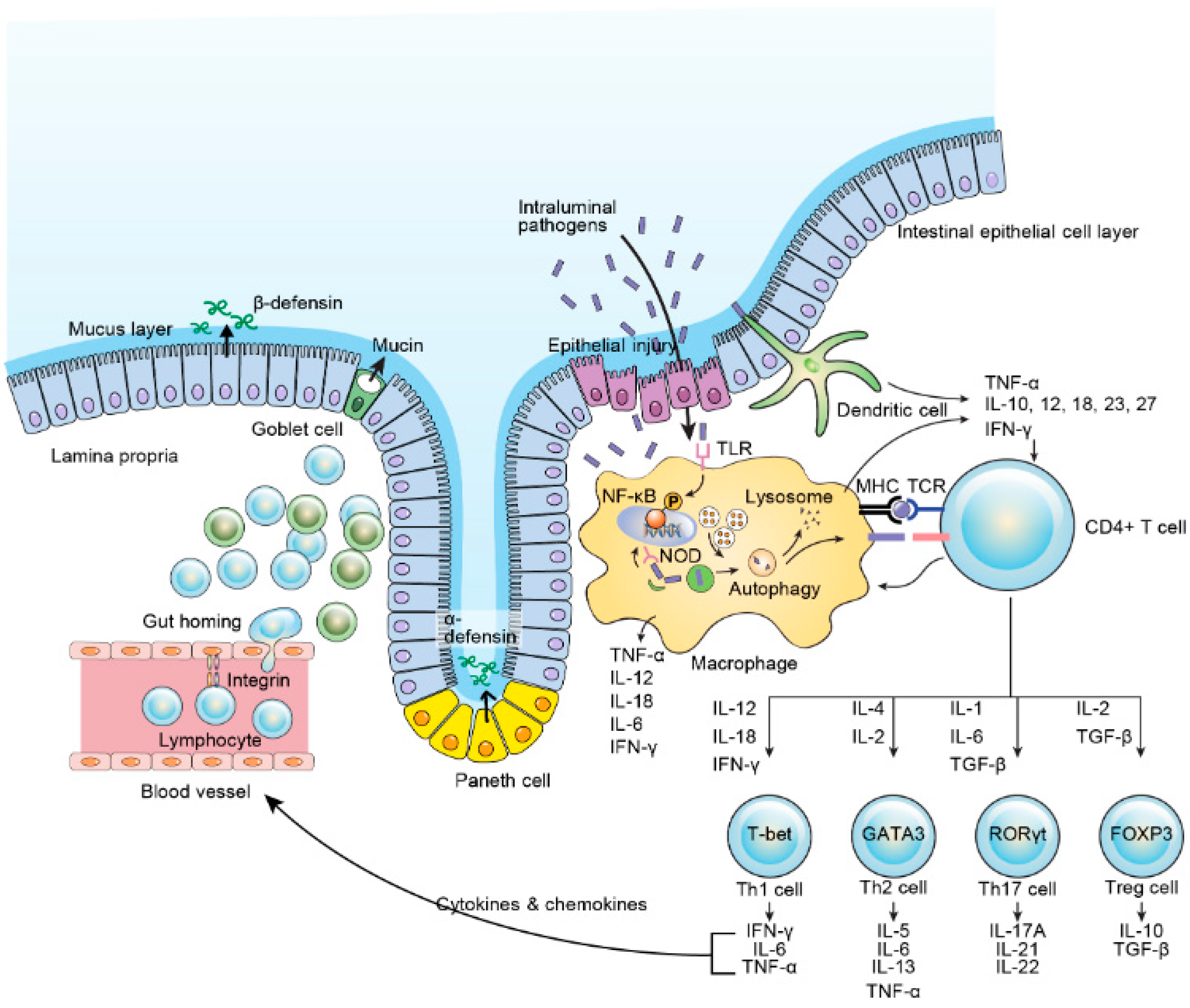
Figure 3.
Biologics regarding therapeutic targets (black: showed benefits; violet: no benefits). APC, antigen presenting cell; IEC, intestinal epithelial cell; TNF, tumor necrosis factor; MHC, major histocompatibility complex; TCR, T cell receptor; JAK, Janus kinase; TGF, transforming growth factor; IL, interleukin; MAdCAM, mucosal vascular addressing cell adhesion molecule. Adapted with permission from “Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies” by Kim and Cheon, Immune Netw. 2017, 17, 25–40 [46].
Figure 3.
Biologics regarding therapeutic targets (black: showed benefits; violet: no benefits). APC, antigen presenting cell; IEC, intestinal epithelial cell; TNF, tumor necrosis factor; MHC, major histocompatibility complex; TCR, T cell receptor; JAK, Janus kinase; TGF, transforming growth factor; IL, interleukin; MAdCAM, mucosal vascular addressing cell adhesion molecule. Adapted with permission from “Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies” by Kim and Cheon, Immune Netw. 2017, 17, 25–40 [46].

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
M’Koma, A.E. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointest. Disord. 2019, 1, 75-105. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1010007
AMA Style
M’Koma AE. The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview. Gastrointestinal Disorders. 2019; 1(1):75-105. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1010007
Chicago/Turabian StyleM’Koma, Amosy E. 2019. "The Multifactorial Etiopathogeneses Interplay of Inflammatory Bowel Disease: An Overview" Gastrointestinal Disorders 1, no. 1: 75-105. https://0-doi-org.brum.beds.ac.uk/10.3390/gidisord1010007