Hemiacetal Ester Exchanges, Study of Reaction Conditions and Mechanistic Pathway

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. NMR Analyses
2.3. Flash Chromatography
2.4. Hemiacetal Ester Syntheses
2.5. Dihemiacetal Ester Syntheses
2.6. Hemiacetal Ester Exchange Demonstration
2.7. Kinetics of the Hemiacetal Ester Exchange Reaction
3. Results
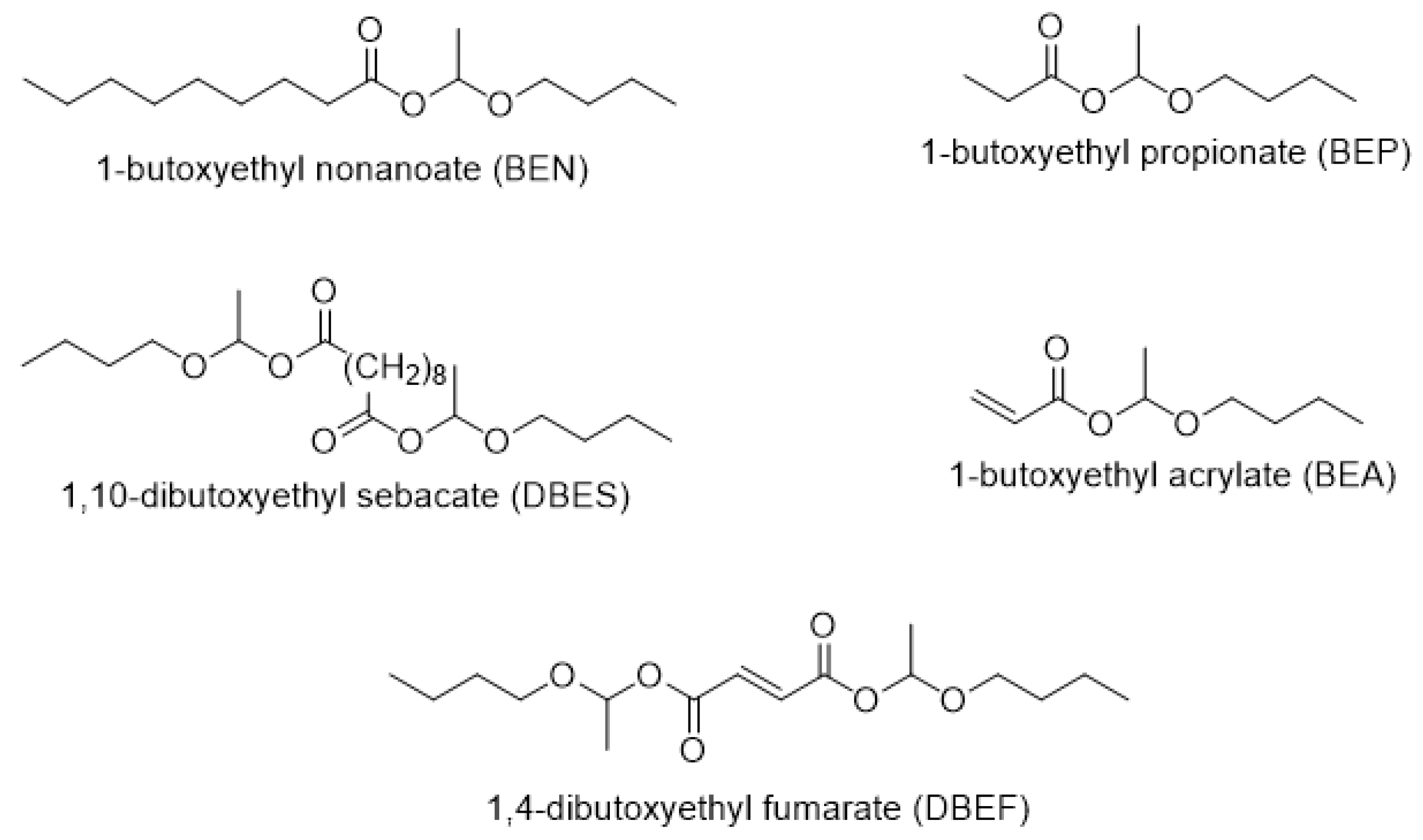
3.1. Synthesis of Hemiacetal Esters
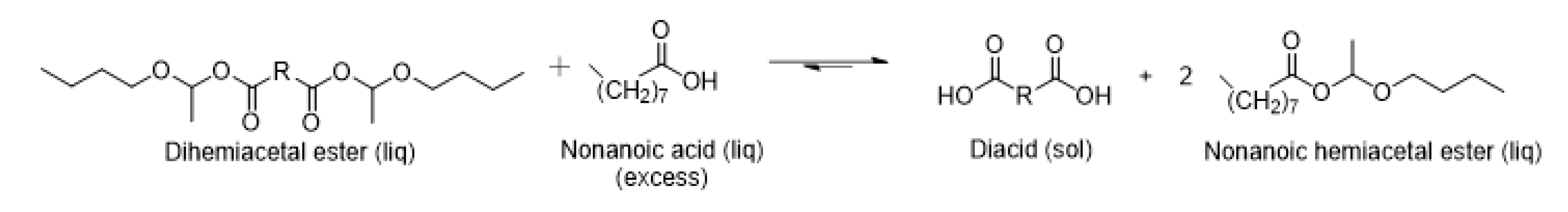
3.2. Exchange Reaction with Carboxylic Acids of Comparable Acid Strength
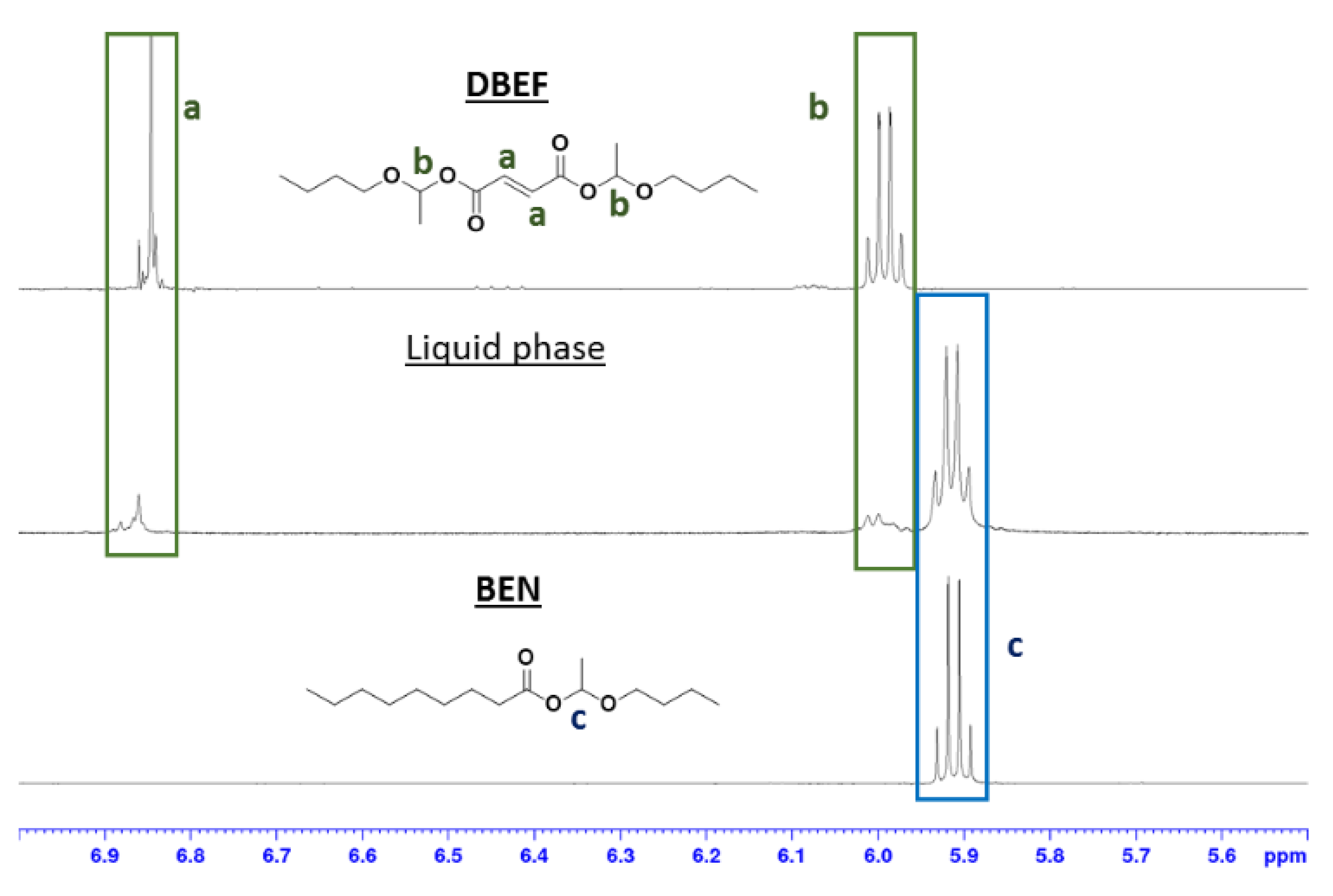
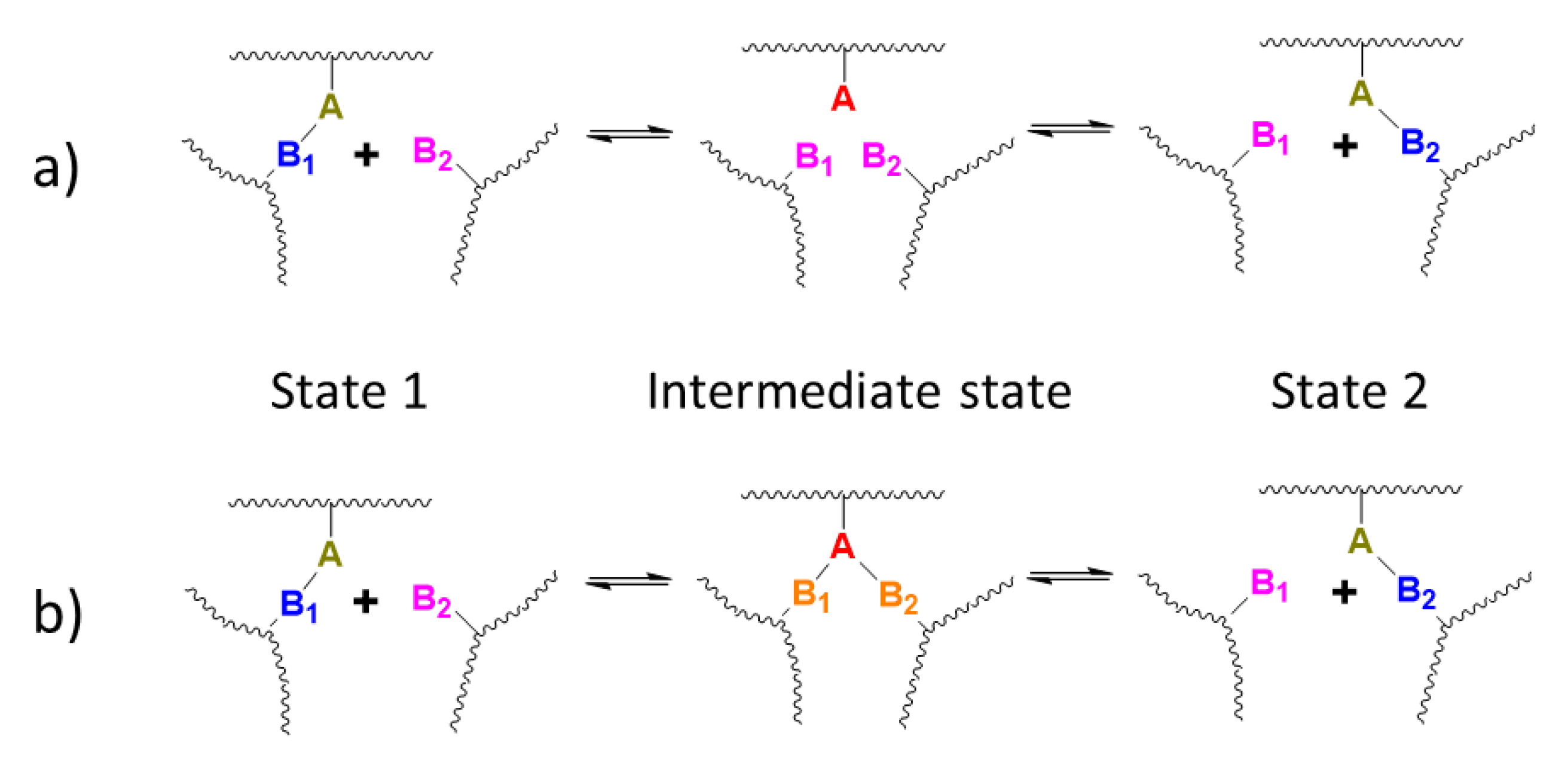
3.3. Exchange Mechanism
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gallucci, R.R.; Going, R.C. Reaction of hemiacetal esters, acetals, and acylals with alcohols or acetic acid. J. Org. Chem. 1982, 47, 3517–3521. [Google Scholar] [CrossRef]
- Kammiyada, H.; Konishi, A.; Ouchi, M.; Sawamoto, M. Ring-Expansion Living Cationic Polymerization via Reversible Activation of a Hemiacetal Ester Bond. ACS Macro Lett. 2013, 2, 531–534. [Google Scholar] [CrossRef]
- Okada, M.; Sumitomo, H.; Atsumi, M. Specific formation of a polymer containing five-membered oxalactone rings in the main chain in the cationic ring-opening polymerization of 6,8-dioxabicyclo[3.2.1]octan-7-one. Macromolecules 1984, 17, 1840–1843. [Google Scholar] [CrossRef]
- Otsuka, H.; Endo, T. Poly(hemiacetal ester)s: New Class of Polymers with Thermally Dissociative Units in the Main Chain. Macromolecules 1999, 32, 9059–9061. [Google Scholar] [CrossRef]
- Otsuka, H.; Fujiwara, H.; Endo, T. Thermal dissociation behavior of polymers with hemiacetal ester moieties in the side chain: The effect of structure on dissociation temperature. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 4478–4482. [Google Scholar] [CrossRef]
- Otsuka, H.; Fujiwara, H.; Endo, T. Fine-tuning of thermal dissociation temperature using copolymers with hemiacetal ester moieties in the side chain: Effect of comonomer on dissociation temperature. React. Funct. Polym. 2001, 46, 293–298. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Zhang, H. Living Anionic Copolymerization of 1-(Alkoxy)ethyl Methacrylates with Polar and/or Nonpolar Monomers and the Preparation of Amphiphilic Block Copolymers Containing Poly(methacrylic acid) Hydrophilic Segments at Higher Temperatures Than Usually Employed. Macromolecules 1998, 31, 9127–9133. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ishidoya, M. New thermosetting coatings using blocked carboxyl groups. Prog. Org. Coat. 2000, 40, 267–273. [Google Scholar] [CrossRef]
- Kovash, C.S.; Pavlacky, E.; Selvakumar, S.; Sibi, M.P.; Webster, D.C. Thermoset Coatings from Epoxidized Sucrose Soyate and Blocked, Bio-Based Dicarboxylic Acids. ChemSusChem 2014, 7, 2289–2294. [Google Scholar] [CrossRef]
- Komatsu, H.; Hino, T.; Endo, T. Novel thermally latent self-crosslinkable copolymers bearing oxetane and hemiacetal ester moieties: The synthesis, self-crosslinking behavior, and thermal properties. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 4260–4270. [Google Scholar] [CrossRef]
- Komatsu, H.; Ochiai, B.; Endo, T. Thermally latent synthesis of networked polymers from multifunctional hemiacetal ester and diepoxide catalyzed by Schiff-base-zinc chloride complex. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3682–3689. [Google Scholar] [CrossRef]
- Kohsaka, Y.; Matsumoto, Y.; Zhang, T.; Matsuhashi, Y.; Kitayama, T. α-exomethylene lactone possessing acetal-ester linkage: Polymerization and postpolymerization modification for water-soluble polymer. J. Polym. Sci. Part A Polym. Chem. 2015, 54, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, M.; Nakano, M.; Nakanishi, T.; Sawamoto, M. Alternating Sequence Control for Carboxylic Acid and Hydroxy Pendant Groups by Controlled Radical Cyclopolymerization of a Divinyl Monomer Carrying a Cleavable Spacer. Angew. Chem. Int. Ed. 2016, 55, 14584–14589. [Google Scholar] [CrossRef]
- Matsukawa, D.; Mukai, T.; Okamura, H.; Shirai, M. Photocurable oligo(hemiacetal ester)s having methacrylate side chains. Eur. Polym. J. 2009, 45, 2087–2095. [Google Scholar] [CrossRef]
- Matsukawa, D.; Okamura, H.; Shirai, M. Reworkable dimethacrylates with low shrinkage and their application to UV nanoimprint lithography. J. Mater. Chem. 2011, 21, 10407–10414. [Google Scholar] [CrossRef]
- Kazama, A.; Kohsaka, Y. Radical polymerization of ‘dehydroaspirin’ with the formation of a hemiacetal ester skeleton: A hint for recyclable vinyl polymers. Polym. Chem. 2019, 10, 2764–2768. [Google Scholar] [CrossRef]
- Neitzel, A.E.; Barreda, L.; Trotta, J.T.; Fahnhorst, G.W.; Haversang, T.J.; Hoye, T.R.; Fors, B.P.; Hillmyer, M.A. Hydrolytically-degradable homo- and copolymers of a strained exocyclic hemiacetal ester. Polym. Chem. 2019, 10, 4573–4583. [Google Scholar] [CrossRef]
- Ouchi, M.; Konishi, A.; Takenaka, M.; Sawamoto, M. Consecutive living polymerization from cationic to radical: A straightforward yet versatile methodology for the precision synthesis of “cleavable” block copolymers with a hemiacetal ester junction. Polym. Chem. 2012, 3, 2193–2199. [Google Scholar] [CrossRef]
- Kammiyada, H.; Ouchi, M.; Sawamoto, M. A Study on Physical Properties of Cyclic Poly(vinyl ether)s Synthesized via Ring-Expansion Cationic Polymerization. Macromolecules 2017, 50, 841–848. [Google Scholar] [CrossRef]
- Neitzel, A. Ring-Opening Polymerization of Cyclic Hemiacetal Esters for the Preparation of Hydrolytically and Thermally Degradable Polymers., University Of Minnesota, 2018. Available online: http://hdl.handle.net/11299/195385 (accessed on 29 October 2020).
- Kubota, H.; Yoshida, S.; Ouchi, M. Ring-expansion cationic cyclopolymerization for the construction of cyclic cyclopolymers. Polym. Chem. 2020, 11, 3964–3971. [Google Scholar] [CrossRef]
- Kohsaka, Y.; Yamashita, M.; Matsuhashi, Y.; Yamashita, S. Synthesis of poly(conjugated ester)s by ring-opening polymerization of cyclic hemiacetal ester bearing acryl skeleton. Eur. Polym. J. 2019, 120, 109185. [Google Scholar] [CrossRef]
- Hyoi, K.; Kanazawa, A.; Aoshima, S. Cationic Ring-Opening Co- and Terpolymerizations of Lactic Acid-Derived 1,3-Dioxolan-4-ones with Oxiranes and Vinyl Ethers: Nonhomopolymerizable Monomer for Degradable Co- and Terpolymers. ACS Macro Lett. 2019, 8, 128–133. [Google Scholar] [CrossRef]
- Ouchi, M.; Kammiyada, H.; Sawamoto, M. Ring-expansion cationic polymerization of vinyl ethers. Polym. Chem. 2017, 8, 4970–4977. [Google Scholar] [CrossRef]
- Neitzel, A.E.; Petersen, M.A.; Kokkoli, E.; Hillmyer, M.A. Divergent Mechanistic Avenues to an Aliphatic Polyesteracetal or Polyester from a Single Cyclic Esteracetal. ACS Macro Lett. 2014, 3, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Neitzel, A.E.; Haversang, T.J.; Hillmyer, M.A. Organocatalytic Cationic Ring-Opening Polymerization of a Cyclic Hemiacetal Ester. Ind. Eng. Chem. Res. 2016, 55, 11747–11755. [Google Scholar] [CrossRef]
- Nakane, Y.; Ishidoya, M. New crosslinking system using blocked carboxylic acid. Prog. Org. Coat. 1997, 31, 113–120. [Google Scholar] [CrossRef]
- Fiore, M. The synthesis of mono-alkyl phosphates and their derivatives: An overview of their nature, preparation and use, including synthesis under plausible prebiotic conditions. Org. Biomol. Chem. 2018, 16, 3068–3086. [Google Scholar] [CrossRef]
- Cho, C.G.; Feit, B.A.; Webster, O.W. Initiation of vinyl ether polymerization by trimethylsilyl triflate, dimethyl sulfide, and adventitious water. Macromolecules 1992, 25, 2081–2085. [Google Scholar] [CrossRef]
- Lide, D.R. CRC Handbook of Chemistry and Physics, 85th ed.; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Winne, J.M.; Leibler, L.; Du Prez, F.E. Dynamic covalent chemistry in polymer networks: A mechanistic perspective. Polym. Chem. 2019, 10, 6091–6108. [Google Scholar] [CrossRef]
- Lorke, S.; Müller, U.; Meissl, R.; Brüggemann, O. Covalent cross-linking of polymers at room temperature. Int. J. Adhes. Adhes. 2019, 91, 150–159. [Google Scholar] [CrossRef]
- Guerre, M.; Taplan, C.; Nicolaÿ, R.; Winne, J.; Du Prez, F.E. Fluorinated Vitrimer Elastomers with a Dual Temperature Response. J. Am. Chem. Soc. 2018, 140, 13272–13284. [Google Scholar] [CrossRef] [PubMed]
- Van Herck, N.; Maes, D.; Unal, K.; Guerre, M.; Winne, J.M.; Du Prez, F.E. Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol–yne Cross-Linking. Angew. Chem. Int. Ed. 2020, 59, 3609–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denissen, W.; Rivero, G.; Nicolaÿ, R.; Leibler, L.; Winne, J.M.; Du Prez, F.E. Vinylogous Urethane Vitrimers. Adv. Funct. Mater. 2015, 25, 2451–2457. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hemiacetal Ester | Acid, pKa [a] | [Acid]0/ [Vinyl Ether]0 | [Catalyst]0/[Acid Function]0 mol % | Conversion of the Acid (Limiting Reagent) in 6 h (%) |
|---|---|---|---|---|
| 1-butoxyethyl nonanoate (BEN) | Nonanoic acid, 4.96 | 1/2 | 0.4 | 87 |
| 1-butoxyethyl propionate (BEP) | Propionic acid, 4.87 | 1/2 | 0.4 | 96 |
| 1-butoxyethyl acrylate (BEA) | Acrylic acid, 4.26 | 1/2 | 0.4 | 83 |
| 1,10-dibutoxyethyl sebacate (DBES) | Sebacic acid, 4.59 | 1/4 | 2 | 95 |
| 1,4-dibutoxyethyl fumarate (DBEF) | Fumaric acid, 3.02 | 1/4 | 2 | 99 [b] |
| Reaction conditions | ||||
|---|---|---|---|---|
| [BEN]0/[acrylic acid]0 | [BEN]0 (mol.L−1) | [BEN]Eq (mol.L−1) | [BEA]Eq (mol.L−1) | |
| 1/1 | 1.6 | 9.1 10−1 | 6.9 10−1 | |
| 1/2 | 1.6 | 6.3 10−1 | 9.7 10−1 | |
| Exponential fit | ||||
| [BEN]0/[acrylic acid]0 | k1 (L.mol−1.s−1) | v0 (mol.L−1.h−1) | K | |
| 1/1 | 0.6 | 5.5 10−2 | 0.65 | |
| 1/2 | 1.8 | 1.7 10−1 | 0.74 | |
| Bimolecular reversible elementary reactions kinetic law | ||||
| [BEN]0/[acrylic acid]0 | k1 (L.mol−1.s−1) | k2 (L.mol−1.s−1) | K | |
| 1/1 | 0.037 | 0.010 | 3.5 | |
| 1/2 | 0.063 | −0.003 | −20.2 | |
| Order Calculation | ||||
| x [a] | y [a] | k1 (L2.mol−2.s−1) | k2 (L2.mol−2.s−1) | K |
| 1.65 | 1.35 | 0.015 | 0.028 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boucher, D.; Madsen, J.; Caussé, N.; Pébère, N.; Ladmiral, V.; Negrell, C. Hemiacetal Ester Exchanges, Study of Reaction Conditions and Mechanistic Pathway. Reactions 2020, 1, 89-101. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020008
Boucher D, Madsen J, Caussé N, Pébère N, Ladmiral V, Negrell C. Hemiacetal Ester Exchanges, Study of Reaction Conditions and Mechanistic Pathway. Reactions. 2020; 1(2):89-101. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020008
Chicago/Turabian StyleBoucher, David, Jeppe Madsen, Nicolas Caussé, Nadine Pébère, Vincent Ladmiral, and Claire Negrell. 2020. "Hemiacetal Ester Exchanges, Study of Reaction Conditions and Mechanistic Pathway" Reactions 1, no. 2: 89-101. https://0-doi-org.brum.beds.ac.uk/10.3390/reactions1020008