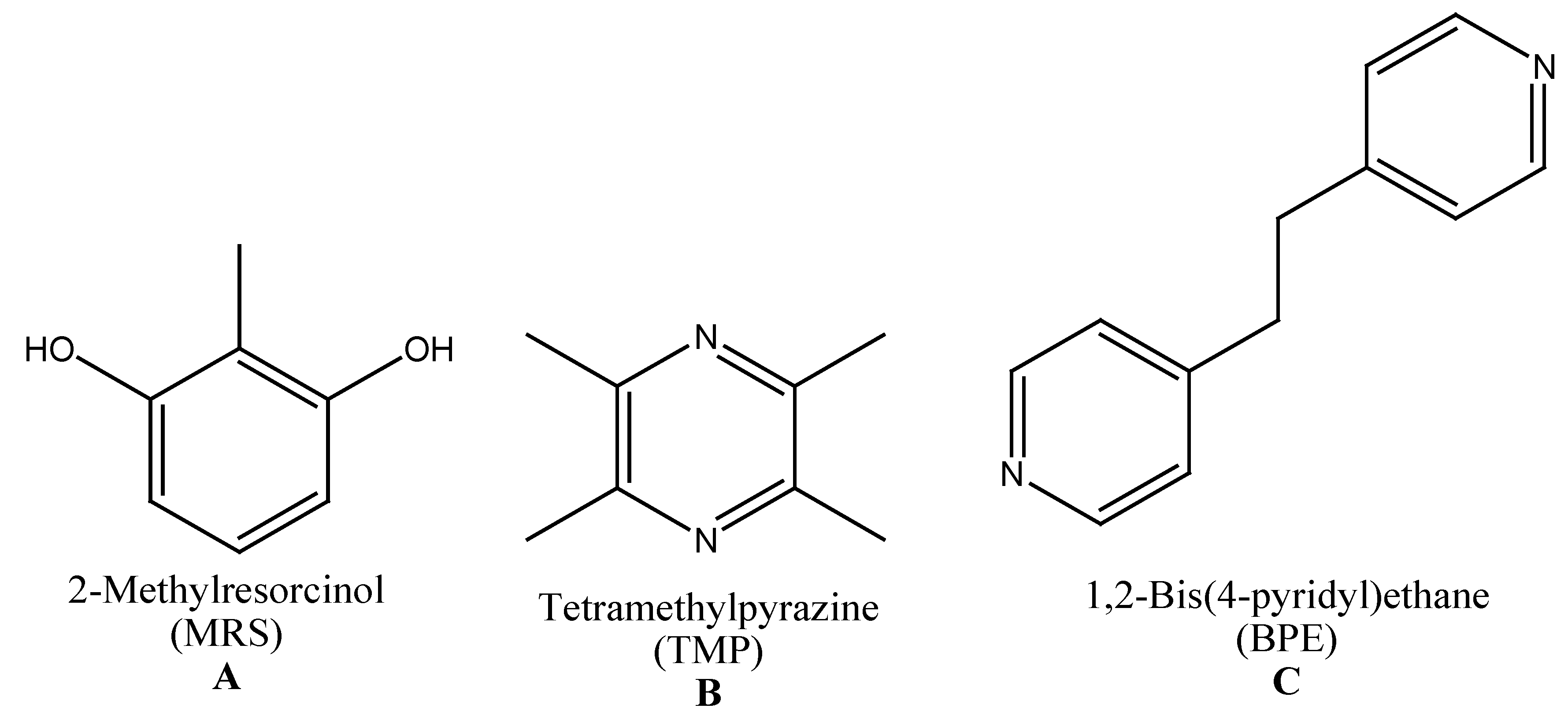
Controlled Stepwise Synthesis and Characterization of a Ternary Multicomponent Crystal with 2-Methylresorcinol

1
Universitaet Duesseldorf, Inorganic Chemistry, Universitaetsstrasse 1, 40225 Düsseldorf, Germany
2
Solid-Chem GmbH, Universitätstrasse 136, 44799 Bochum, Germany
3
Inorganic Chemistry, Ruhr-University Bochum, Inorganic Chemistry 1, Universitaetstrasse 150, 44801 Bochum, Germany
*
Author to whom correspondence should be addressed.
Chemistry 2020, 2(1), 93-100; https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010009
Submission received: 6 February 2020
/
Revised: 27 February 2020
/
Accepted: 28 February 2020
/
Published: 4 March 2020
(This article belongs to the Special Issue Supramolecular Chemistry in the 3rd Millennium)
Abstract
:A typical approach of a multicomponent crystal design starts with a retrosynthetic analysis of the target molecule followed by a one-pot reaction of all components. To develop protocols for multicomponent crystal syntheses, controlled stepwise syntheses of a selected crystalline ternary multicomponent system 1 involving 2-methylresorcinol (MRS), tetramethyl-pyrazine (TMP), and 1,2-bis(4-pyridyl)ethane (BPE) are presented. The obtained binary cocrystals 2 (involving MRS and TMP) and 3 (involving MRS and BPE) as well as the final resulting ternary multicomponent system 1 were characterized by X-ray analysis.

1. Introduction
Polymorphism and cocrystallization are tools to add or enhance desirable properties of crystalline products [1,2,3]. The developments in this field in recent years have enabled more carefully tunable properties [4]. The tunability of the properties of (co)crystalline systems is of much importance, especially in the fields of pharmacy and medicine [5,6,7]. By introducing various coformers, factors like solubility, stability, bioavailability, or tolerability of active pharmaceutical ingredients can be influenced [8,9,10]. A better understanding of the driving force behind the genesis of molecular aggregation processes in the solid state can therefore be helpful to increase the efficiency of the synthesis of such crystalline products. Steed et al. have explained that asymmetry of crystalline structures can be exploited to purposefully produce medically relevant cocrystals [11]. While binary cocrystals have been focused on more commonly, less attention has been imposed on higher multicomponent systems like ternary [12], or even quaternary and quintenary [13] structures. In contrast to a well thought out stepwise multistage organic synthesis, a typical approach of a multicomponent crystal design starts with a retrosynthetic analysis of the target molecule followed by a one-pot reaction of all components in solution or solid state. To develop sophisticated and reliable protocols for multicomponent crystal syntheses, the complex modular assembly processes must be investigated by analysis of a hierarchy of intermolecular interactions and the molecular environment of involved components on each aggregation step [14].
In recent years, the group of Desiraju proposed that differing structural environments in the (n − 1)-multicomponent systems could lead to a state in which the incorporation of a new coformer would be more favourable for the overall structure [13]. Furthermore, Nangia and Bolla have confirmed that when using geometrically similar (in regards to size, planarity, etc.) heterosynthons, the formation of multicomponent crystals could simply be predicted by the strongest hydrogen bond former interactions, showing the importance of the kinetic effect [15]. Results presented by Aakeröy and Gunawardana also lay an emphasis on the geometric environment of (n − 1)-crystal systems as crucial for the formation of higher component products [16].
Hence, in this contribution, in contrast to a one-pot synthesis, the controlled stepwise syntheses of a selected crystalline ternary multicomponent system is presented. The mechanism of the formation of multicomponent cocrystals could be figured out based on the analysis of molecular arrangement in solid state of the selected components. The binary cocrystals as well as the final resulting ternary multicomponent system were characterized by X-ray analysis.
For this, 2-methylresorcinol (MRS) formation capabilities in conjunction with the N-bases tetramethylpyrazine (TMP) and 1,2-bis(4-pyridyl)ethane (BPE) (Scheme 1) were investigated, using neat, drop-, or liquid-assisted grinding techniques. Additionally, sequential and one-pot reactions were performed. The obtained crystalline products were characterized using Powder X-ray Diffraction (PXRD) and single crystal structure analysis.
2. Materials and Methods
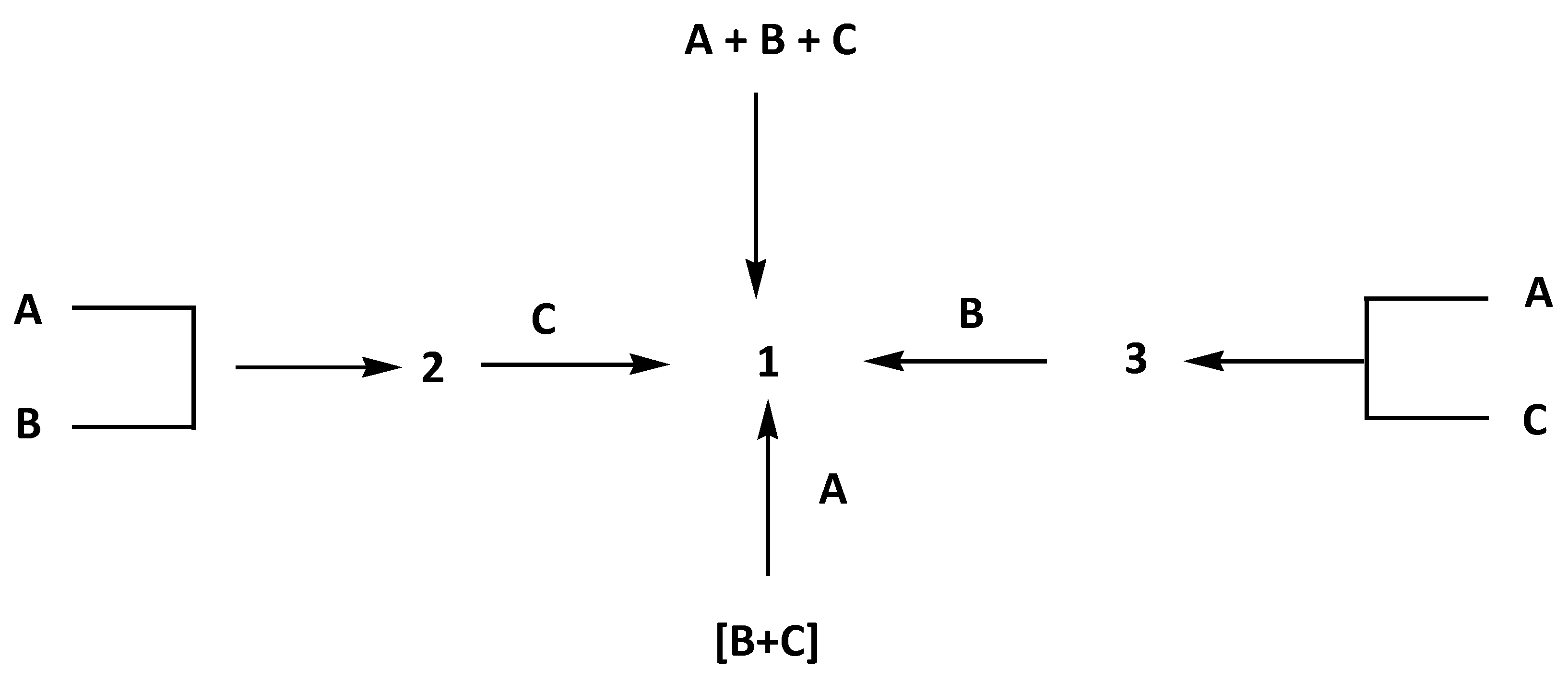
By varying the component ratios or the solvent during the grinding experiments, it was possible to access cocrystals of A:2B 3 and A:C 2 as well as the ternary cocrystal 2A:2B:C 1. Modified parameters include the ratio of the used components; grinding conditions like neat, drop-, or liquid-assisted grinding; and the choice of the solvents tetrahydrofuran vs. n-hexane. Only liquid-assisted grinding experiments in the presence of tetrahydrofuran were successful. Modification of the grinding conditions or the usage of n-hexane resulted in the observation of physical component mixtures. The experiments were either done as one-pot reactions of three components or by adding a component to sequentially synthesized and characterized binary cocrystals (Scheme 2).
PXRD was performed on each of the received crystalline products. Furthermore, single crystalline entities suitable for X-ray structure analysis of these multicomponent crystals was grown from solution (Table 1).
Preparation of 2: 1 mmol of A and 2 mmol of B were placed in a mortar. Ten drops of tetrahydrofuran (THF) were added, and the resulting mixture was ground for 5 min. The resulting powder was characterized by PXRD.
Preparation of 3: 1 mmol of A and C, respectively, were placed in a mortar. Ten drops of THF were added, and the resulting mixture was ground for 5 min. The resulting powder was characterized by PXRD.
Preparation of 1:
- One-pot synthesis: 2 mmol of A, 2 mmol B, and 1 mmol C were placed in a mortar and pestle. Ten drops of THF were added, and the resulting mixture was ground for 5 min. The resulting powder was characterized by PXRD.
- From 2: The latter was prepared in the previously mentioned way and identified as 2 by PXRD. The powder of 2 was placed in a mortar, and 1 mmol of A and 1 mmol of C and 10 drops of THF were added to the mixture. It was then ground for 5 min. The resulting powder was characterized by PXRD.
- From 3: The latter was prepared in the previously mentioned way and identified as 3 by PXRD. The powder of 3 was placed in a mortar, and 1 mmol of A and 2 mmol of B and 10 drops of THF were added to the mixture. It was then ground for 5 min. The resulting powder was characterized by PXRD.
- From the physical mixture of B and C: 1 mmol of each were put in a mortar. 10 drops of THF were added, and the mixture was ground for 5 min. The resulting powder was characterized by PXRD. Only reflections of the physical mixture (B and C) were observed in this PXRD. Thereafter, the powder was placed in a mortar and 2 mmol of A and 1 mmol of B, and 10 drops of THF were added. The mixture was ground for 5 min, and the resulting powder was characterized by PXRD.
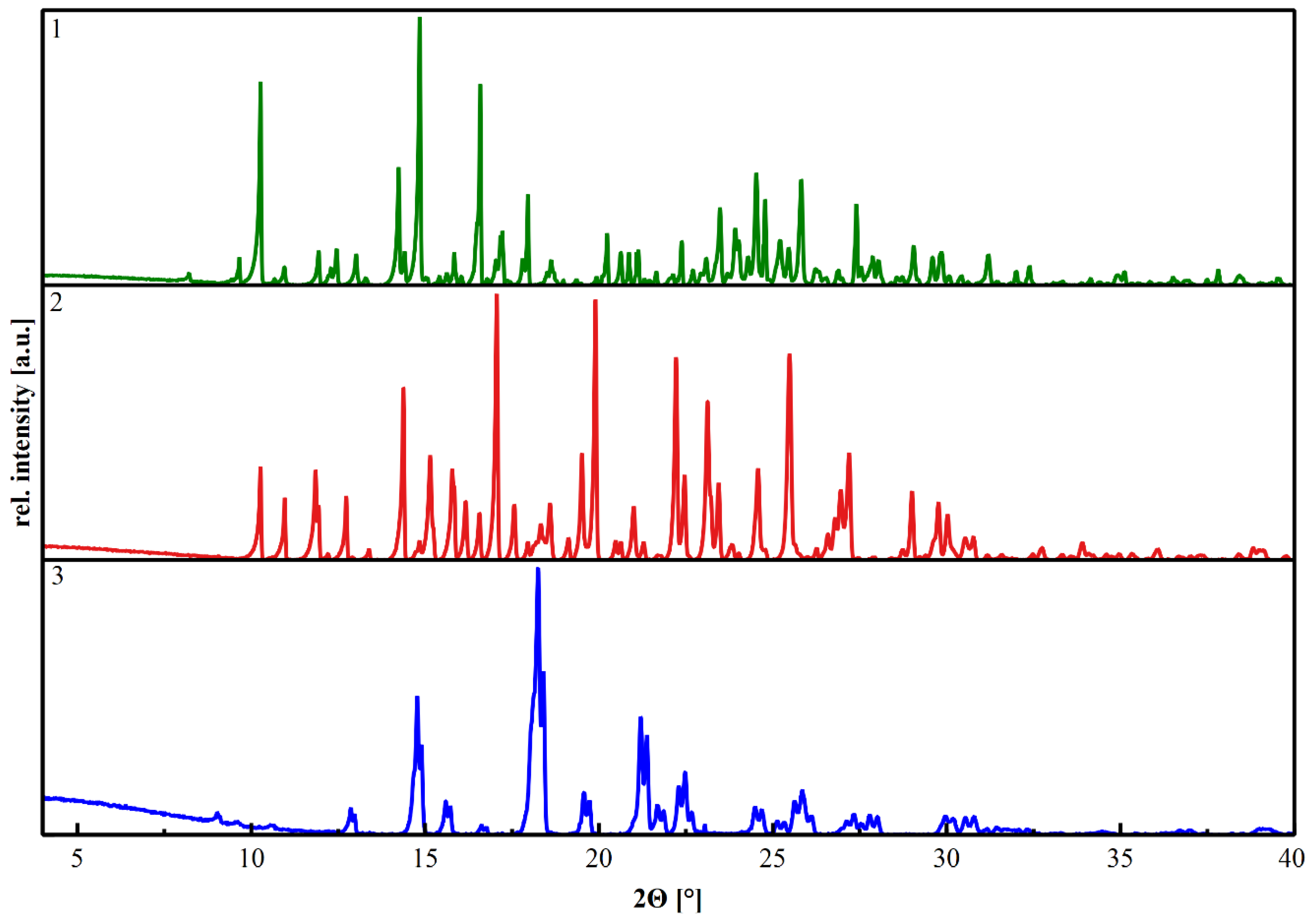
All obtained crystalline materials were characterized by powder X-ray diffraction (PXRD) (Figure 1), measured on a Bruker D2 PHASER diffractometer in flat mode and Bragg–Brentano geometry using filtered CuKα and CuKβ radiation.
Single-crystal X-ray diffraction measurements of 2, 3 were carried out on a Rigaku Synergy diffractometer using monochromated Mo Kα radiation (λ = 0.71073). Single-crystal X-ray diffraction measurement of 1 was carried out on a Rigaku Supernova using monochromated Cu Kα radiation (λ = 1.54184). Structures were solved by direct methods, and all nonhydrogen atoms were refined anisotropically on F2 (program SHELXTL-97, G.M. Sheldrick, University of Göttingen, Göttingen, Germany). Crystallographic crystal data and processing parameters are shown in Table 1. CIF-files giving X-ray data with details of refinement procedures for 1, 2, and 3 CCDC Nr. 1981507-1981509 are available free of charge via the Internet at http://pubs.acs.org.
The UNI Force Field Calculations of Mercury crystallography package were used to calculate inter-molecular potentials of the crystal structure 2 with normalized hydrogens (kJ/mol).
3. Results
The crystalline product obtained by liquid-assisted grinding of a mixture of A, B, and C in the presence of THF shows reflections in the PXRD that cannot be attributed to the educts. The crystalline product was dissolved in nitromethane, and by cooling crystallization a single crystal was obtained (Table 1). The X-ray structure analysis confirms the successful crystallization of a ternary 2:2:1 A:B:C multicomponent system 1 (Figure 2).
The central 1,2-bis(4-pyridyl)ethane building block C forms hydrogen bonds via its two aromatic nitrogen centers to hydroxy groups from each of the two neighbouring 2-methylresorcinol molecules A. The remaining hydroxy groups of A coordinate via hydrogen bonds to one nitrogen center each of the two terminal tetramethylprazine molecules. Surprisingly, the second nitrogen centers of B are not involved in any further intermolecular interactions. Consequently, the 2:2:1 A:B:C multicomponent system 1 does not form infinite chains. This behavior is observed for B in only few selected multicomponent systems. Examples are cocrystalline systems with Br-C6F4-OH12 or Br-C6F4-COOH12 [16]. The analysis of the coordination behavior of A shows that the hydrogen bonds from A to B, in comparison to A to C, have no significant differences in the distances O…N 2750 Å and O…N 2755 Å as well as in the angles of the ring planes A:B 75° and A:C 69°.
As mentioned, the geometric environment of the involved building blocks and their intermolecular interactions are crucial parameters for the formation of higher multicomponent crystals. The one-pot reaction of all components (A, B, and C) leads to the ternary 2A:2B:C system 1. The 2A:2B:C motif is discretely isolated by the unusual coordination behavior of the tailored B molecules. The hypothesis is that the geometric molecular environment is responsible in each aggregation step in the design of 1. Based on differing structural environments in the crystalline products obtained by first aggregation steps, A with B and A with C, the crystal packing of 2A:2B:C—starting from A—can be built up by successive substitution of B by C or C by B. To investigate this hypothesis and to gain a deeper understanding of the formation of the multicomponent system, the cocrystals involving A and B as well A and C were crystallized and the crystal packing analyzed.
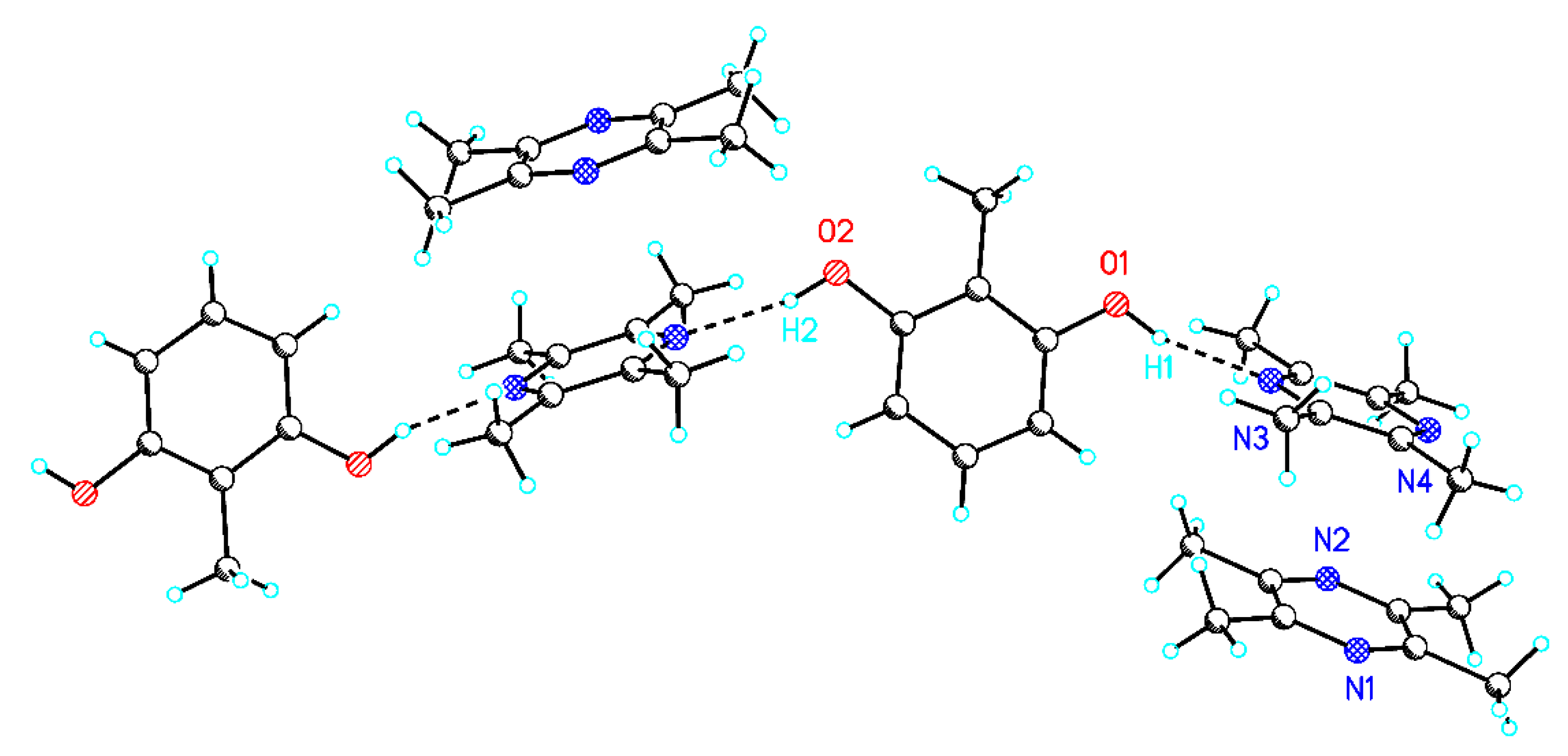
Cocrystals of 2 (A:2B) and 3 (A:C) could be obtained by grinding a 1:2 ratio (2) and 1:1 ratio (3). After that, suitable single crystals for XRD were formed after recrystallization of the crystalline powder from THF (2) and nitromethane (3). In contrast to the known crystal structure of the A:B cocrystal [8], the crystal structure of 2 represents a 2:1 cocrystal with one A and two B molecules (Figure 3). Due to the fact that the two molecules of B in 2 show explicit different structural environments, the description A:B:B’ seems more appropriate for this system. The central A unit forms chains via hydrogen bonds (O1…N1 2.84 Å, O2…N2 2.85 Å) to two neighbouring B molecules. While the D distances of the hydrogen bonds are practically identical, the dihedral angles between the planes of the arene ring of A and the neighboring pyrazine rings of B show slight variations (62°/68°). Additionally, present B’ molecules form columns with B by π-π stacking interactions. A comparison of the intermolecular potentials in the molecular arrangement of 2 pointed out the dominating role of the π-π stacking motif (−42 and −43 kJ/mol) in contrast to the described hydrogen bonds (−32 and −33 kJ/mol).
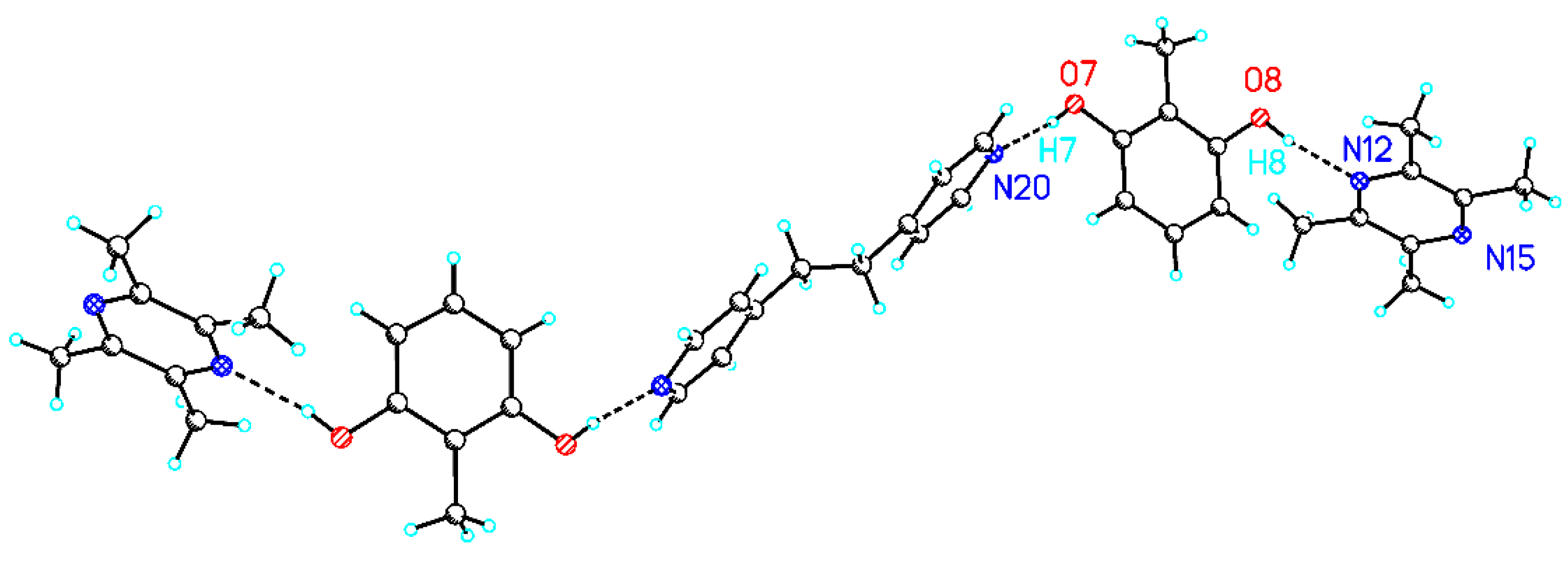
The crystal structure 3 is built up by chains of linked 1:1 A:C cocrystals (Figure 4). Both hydroxyl groups of the central A molecule form hydrogen bonds to nitrogen centers of the two neighbouring C molecules. A comparison of both hydrogen bonds that indicates slight differences is be remarkable. In contrast to the hydrogen bond O1-H1…N1, O2-H2…N2 shows enlarged covalent O1-H2 and O2…N2 hydrogen bonds. Besides the slightly different hydrogen bonds to 2, differences in dihedral angles between the planes of the arene ring of A and the neighboring pyridyl rings of B are observed (68°/75°).
4. Discussion
If the formation of the described ternary multicomponent crystal 1 in a one-pot-synthesis is due to the different environments during the molecular recognition of A, B, and C in the crystallization process, 1 should also be synthesizable from the cocrystals 2 and 3. In both cocrystals (2 and 3), the building block A shows significant differences in the molecular environment (Figure 5). In order to verify the assumption, appropriate crystallization experiments were carried out. Cocrystal 2 was grinded with C, and cocrystal 3 was grinded with B. Additionally, a physical mixture of B and C was grinded with A. The powder diffractograms of all three crystallization experiments show exclusively the reflections of the expected ternary multicomponent system 1. It seems, when attempting to design a multicomponent crystal of nth degree, it is helpful to take a closer look at the (n − 1)th degree predecessors.
The hypothesis that differences in the molecular environment during the molecular recognition in the crystallization process are responsible for the formation of multicomponent crystal has been demonstrated in this study by using the building blocks 2-methylresorcinol (A), tetramethylpyrazine B and 1,2-bis(4- pyridyl)ethane C. The carefully designed cocrystals 2 (A:2B) and 3 (A:C) with differences in their molecular environment are suitable starting materials for successfully synthesizing the ternary multicomponent crystal 1 (2A:2B:C). Furthermore, the results indicate that Ostwald’s rule of stages can be applied to solid state synthesis just as well as it can be applied to crystallization from solution.
Author Contributions
Project administration, supervision, and single crystal structure determination, K.M.; investigation and original draft preparation, D.K.; X-ray powder diffraction and solid state analysis, C.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the DFG Open Access Publication Funds of the Ruhr-Universität Bochum.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bhowal, R.; Biswas, S.; Tumbarathil, A.; Koner, A.L.; Chopra, D. Exploring the Relationship between Intermolecular Interactions and Solid-State Photophysical Properties of Organic CoCrystals. J. Phys. Chem. C 2019, 123, 9311–9322. [Google Scholar] [CrossRef]
- Soliman, I.I.; Kandil, S.M.; Abdou, E.M. Gabapentin-saccharin cocrystals with enhanced physicochemical properties and in vivo absorption formulated as oro-dispersible tablets. Pharm. Dev. Technol. 2020, 2, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Fondren, N.S.; Fondren, Z.T.; Unruh, D.K.; Maiti, A.; Cozzolino, A.; Lee, Y.J.; Hope-Weeks, L.; Weeks, B.L. Study of Physicochemical and Explosive Properties of a 2,4,6-Trinitrotoluene/Aniline Cocrystal Solvate. Cryst. Growth Des. 2020, 1, 116–129. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: Structure, property and beyond. IUCr 2017, 4, 710–711. [Google Scholar] [CrossRef] [PubMed]
- Sathisaran, I.; Dalvi, S.V. Engineering Cocrystals of Poorly Water-Soluble Drugs to Enhance Dissolution in Aqueous Medium. Pharmaceutics 2018, 10, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangia, A.K.; Desiraju, G.R. Crystal Engineering: An Outlook for the Future. Angew. Chem. Int. Ed. 2019, 58, 4100–4107. [Google Scholar] [CrossRef] [PubMed]
- Cains, P.W. Classical methods of preparation of polymorphs and alternative solid forms. In Drugs and the Pharmaceutical Sciences, 2nd ed.; Brittain, H.G., Ed.; CRC Press: New York, NY, USA, 2009; Volume 192, pp. 76–138. [Google Scholar]
- Berry, D.J.; Steed, J.W. Pharmaceutical cocrystals, salts and multicomponent systems; intermolecular interactions and property based design. Adv. Drug Deliv. Rev. 2017, 117, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, N.K.; Perry, M.L.; Almarsson, O.; Zaworotko, M.J. Pharmaceutical cocrystals: Along the path to improved medicines. Chem. Commun. 2016, 52, 640–655. [Google Scholar] [CrossRef] [PubMed]
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Probert, M.R.; Whiteley, C.N.; Rowland, A.M.; Goeta, A.E.; Steed, J.W. Designing CoCrystals of Pharmaceutically Relevant Compounds That Crystallize with Z′ > 1. Cryst. Growth Des. 2008, 9, 1082–1087. [Google Scholar] [CrossRef]
- Mir, N.A.; Dubey, R.; Tothadi, S.; Desiraju, G.R. Combinatorial crystal synthesis of ternary solids based on 2-methylresorcinol. CrystEngComm 2015, 17, 7866–7869. [Google Scholar] [CrossRef]
- Mir, N.A.; Dubey, R.; Desiraju, G.R. Four- and five-component molecular solids: Crystal engineering strategies based on structural inequivalence. IUCr 2016, 3, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, C.A.; Aakeröy, C.B. Cocrystal synthesis: Fact, fancy, and great expectations. Chem. Commun. 2018, 54, 14047–14060. [Google Scholar] [CrossRef]
- Bolla, G.; Nangia, A. Multicomponent ternary cocrystals of the sulfonamide group with pyridine-amides and lactams. Chem. Commun. 2015, 51, 15578–15581. [Google Scholar] [CrossRef] [PubMed]
- Aakeroy, C.B.; Spartz, C.L.; Dembowski, S.; Dwyre, S.; Desper, J. A systematic structural study of halogen bonding versus hydrogen bonding within competitive supramolecular systems. IUCrJ 2015, 2, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Scheme 1.
Selected compounds.

Figure 1.
The diffractograms show the diffraction patterns of the obtained multicomponent crystals 1 (2A:2B:C) green graph, 2 (A:2B) red graph, and 3 (A:C) blue graph.
Figure 1.
The diffractograms show the diffraction patterns of the obtained multicomponent crystals 1 (2A:2B:C) green graph, 2 (A:2B) red graph, and 3 (A:C) blue graph.

Scheme 2.
Performed synthesis routes for the multicomponent crystal 1 (2A:2B:C).

Figure 2.
Packing view of ternary 2A:2B:C multicomponent system 1.

Figure 3.
Packing view of (a) 2 (A:2B) O1-H1: 0.900 Å, O1…N3 2.84 Å, O1-H1…N3 161°, O2-H2: 0.883 Å, O2…N2 2.85 Å, and O2-H2…N4 167°.
Figure 3.
Packing view of (a) 2 (A:2B) O1-H1: 0.900 Å, O1…N3 2.84 Å, O1-H1…N3 161°, O2-H2: 0.883 Å, O2…N2 2.85 Å, and O2-H2…N4 167°.

Figure 4.
Packing view of (b) 3 (A:C). O1-H1: 0.908 Å, O1…N1 2.716 Å, O1-H1…N1 175°, O2-H2: 1.041 Å, O2…N2 2.743 Å, and O2-H2…N2 171°.
Figure 4.
Packing view of (b) 3 (A:C). O1-H1: 0.908 Å, O1…N1 2.716 Å, O1-H1…N1 175°, O2-H2: 1.041 Å, O2…N2 2.743 Å, and O2-H2…N2 171°.

Figure 5.
General proposed formation process of a ternary multicomponent crystal.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
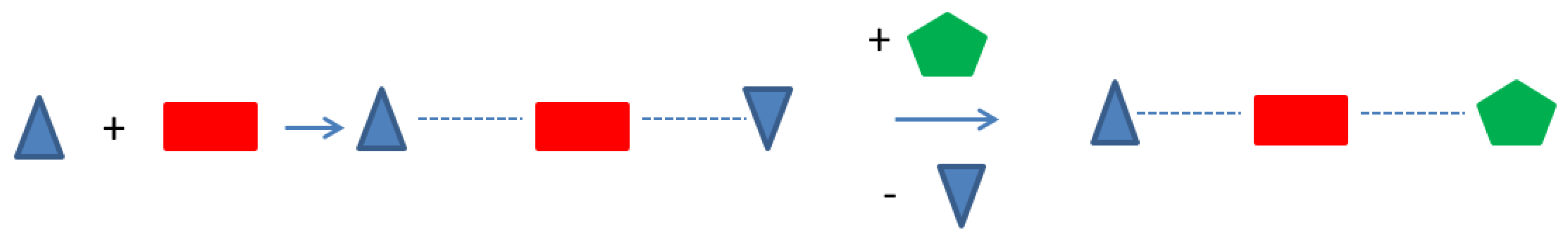{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of crystal data, data collection, and refinement parameters for 1, 2, and 3.
| 1 2A:2B:C | 2 A:2B | 3 A:C | |
|---|---|---|---|
| formula | 2(C7H8O2): 2(C8H12N2): C12H12N2 | C7H8O2: 2(C8H12N2) | C7H8O2: C12H12N2 |
| formula weight | 704.90 | 396.53 | 308.37 |
| temperature [K] | 173 | 173 | 173 |
| crystal system | triclinic | monoclinic | monoclinic |
| space group | P-1 | P21/c | P21/c |
| a | 7.5145(2) | 7.2801(2) | 8.7006(9) |
| b | 8.6750(2) | 24.9768(8) | 12.026(1) |
| c | 15.0243(4) | 12.0692(4) | 17.180(2) |
| α | 100.440(2) | 90 | 90 |
| β | 101.969(2) | 102.504(3) | 103.78(1) |
| γ | 92.396(2) | 90 | 90 |
| V [Å3] | 939.04(4) | 2142.5(1) | 1745.9(3) |
| Z | 1 | 4 | 4 |
| F (000) | 378 | 856 | 656 |
| no. of rflns. measured | 16,811 | 16,224 | 23,798 |
| no. of unique rflns. | 3699 | 5440 | 3074 |
| μ [mm−1] | 0.646 | 0.080 | 0.077 |
| parameters | 242 | 270 | 291 |
| S (F2) | 1.035 | 1.086 | 1.022 |
| R1 [I > 2σ(I)] | 0.0547 | 0.0440 | 0.0545 |
| wR2 (all rflns.) | 0.1363 | 0.1280 | 0.1713 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Komisarek, D.; Schauerte, C.; Merz, K. Controlled Stepwise Synthesis and Characterization of a Ternary Multicomponent Crystal with 2-Methylresorcinol. Chemistry 2020, 2, 93-100. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010009
AMA Style
Komisarek D, Schauerte C, Merz K. Controlled Stepwise Synthesis and Characterization of a Ternary Multicomponent Crystal with 2-Methylresorcinol. Chemistry. 2020; 2(1):93-100. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010009
Chicago/Turabian StyleKomisarek, Daniel, Carsten Schauerte, and Klaus Merz. 2020. "Controlled Stepwise Synthesis and Characterization of a Ternary Multicomponent Crystal with 2-Methylresorcinol" Chemistry 2, no. 1: 93-100. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2010009