Solvent Effect on the Regulation of Urea Hydrolysis Reactions by Copper Complexes

 , , , and
, , , and
Abstract
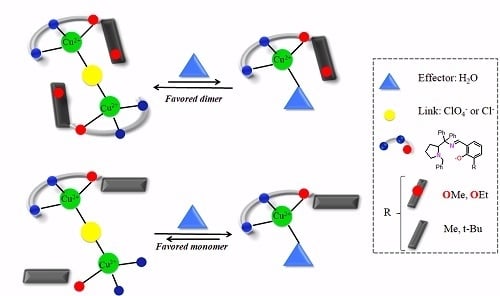
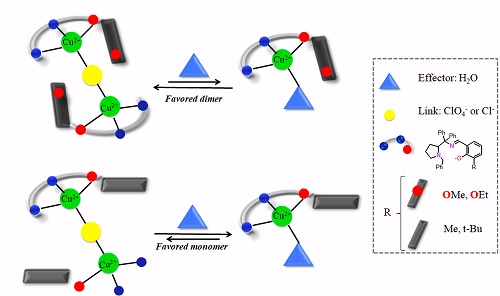
:
1. Introduction
2. Materials and Instruments
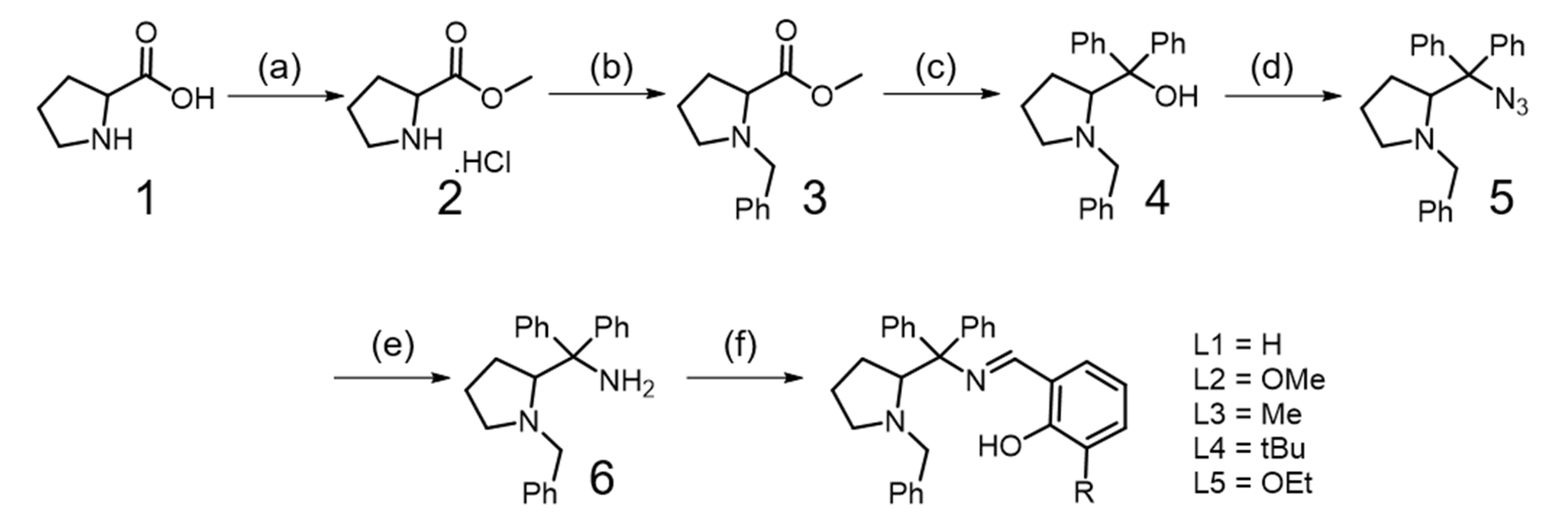
2.1. Synthesis of Ligands (L1–L5)
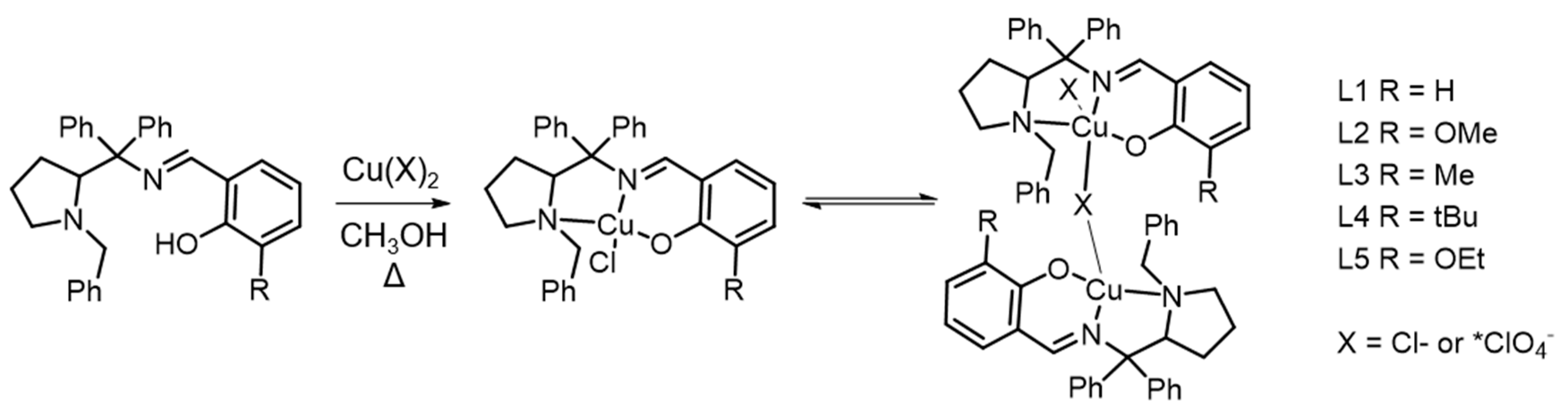
2.2. General Procedure of Synthesis of CuII Chlorido Complexes
2.3. Synthesis of CuI IPerchlorate Complex
2.4. Catalysis Protocol
2.5. Ammonia Quantification
2.6. Computer Simulations
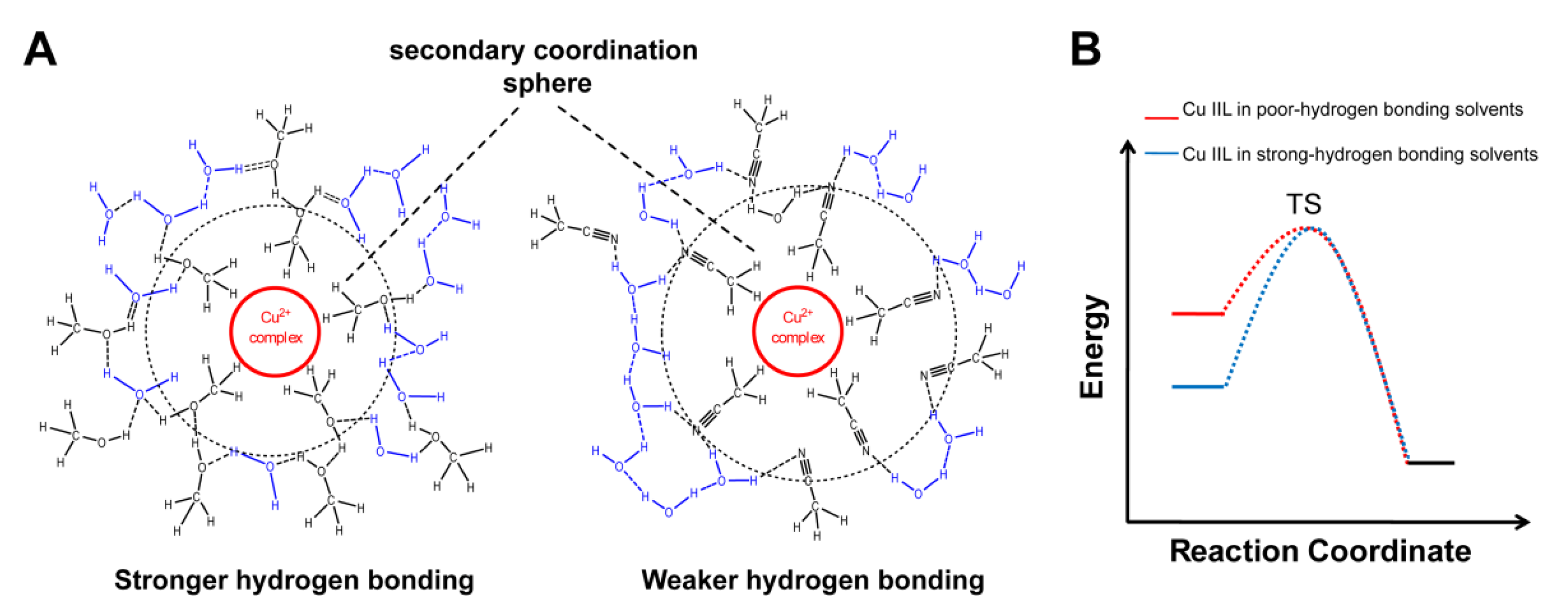
3. Results and Discussion
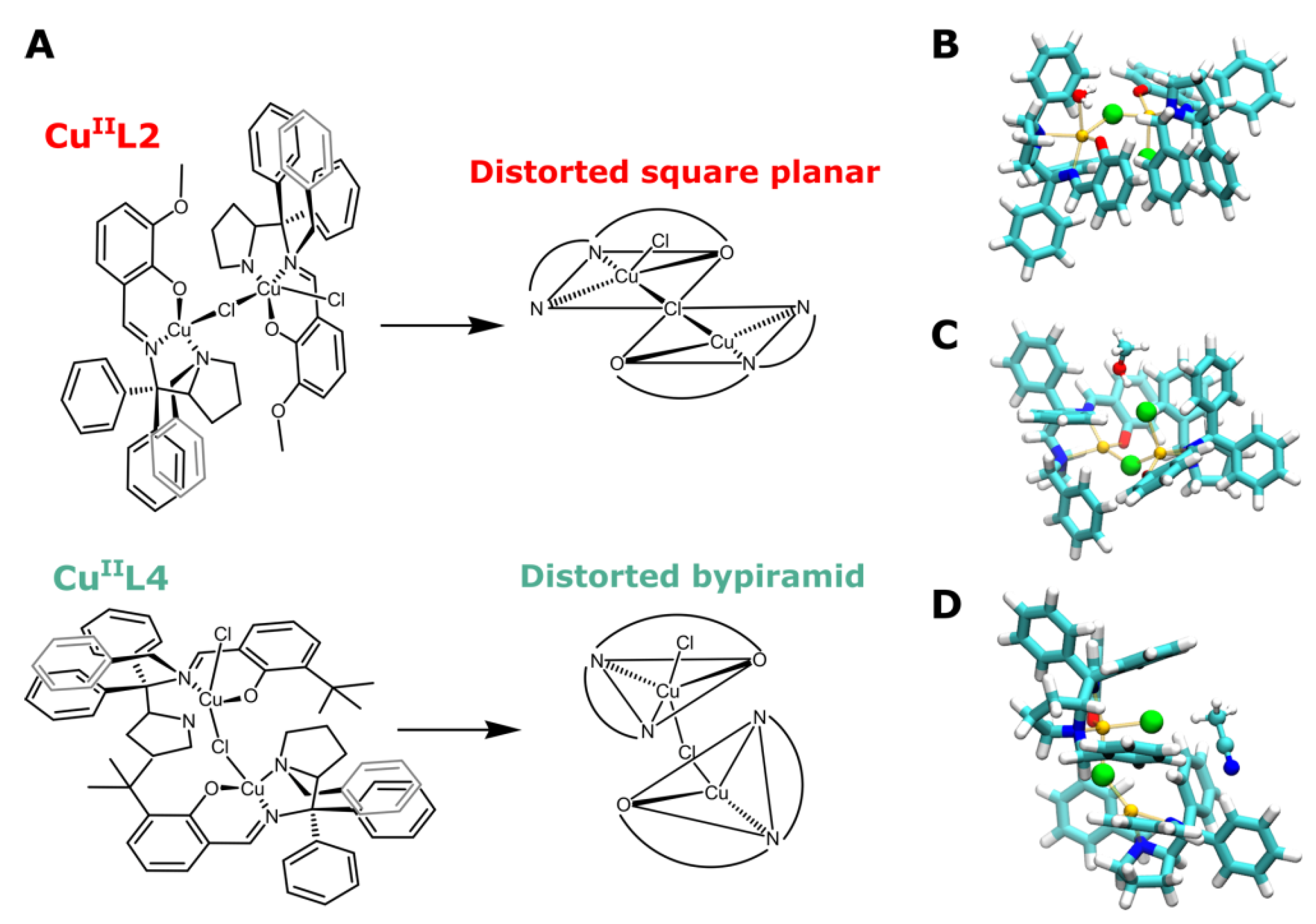
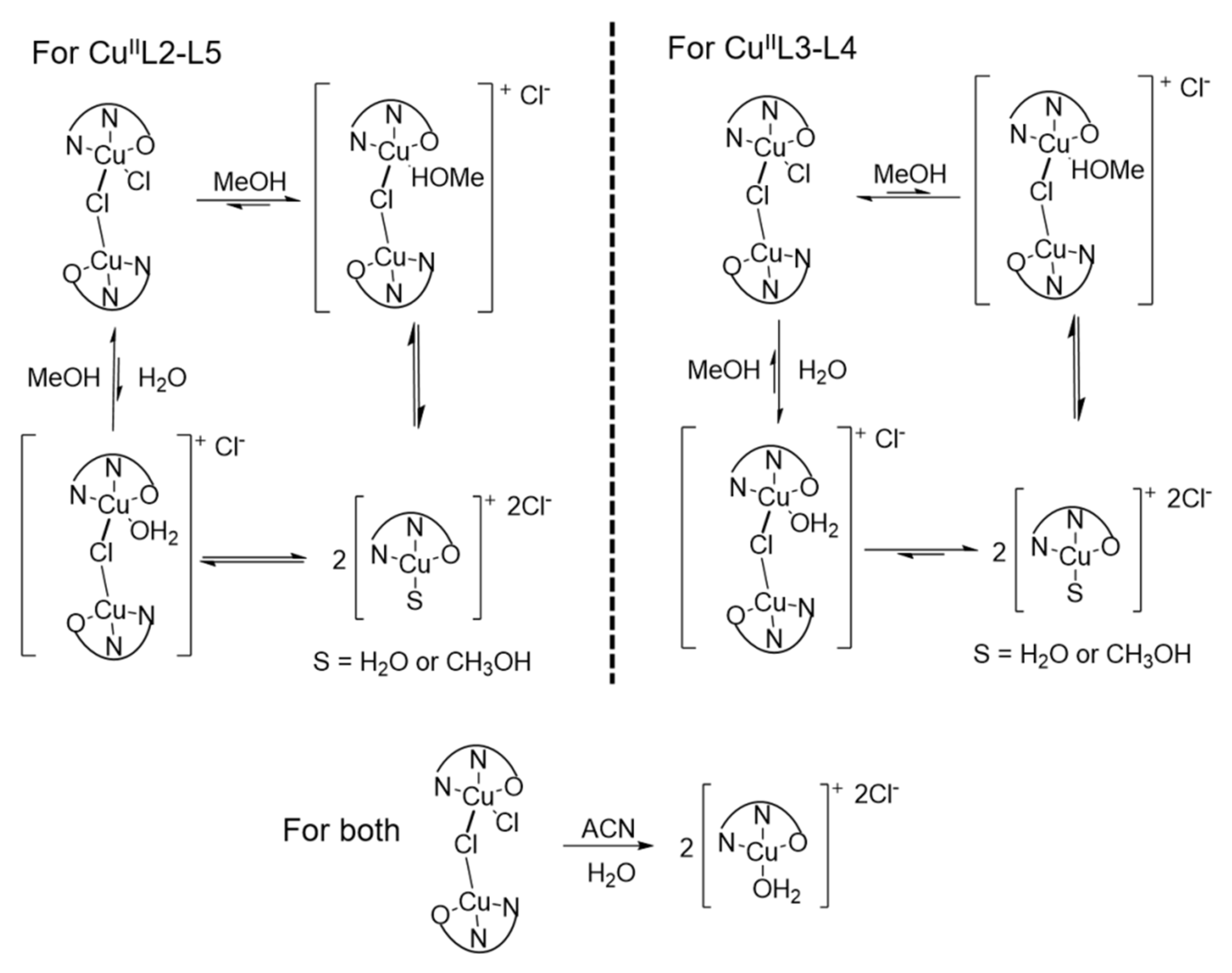
3.1. Probing Copper Coordination
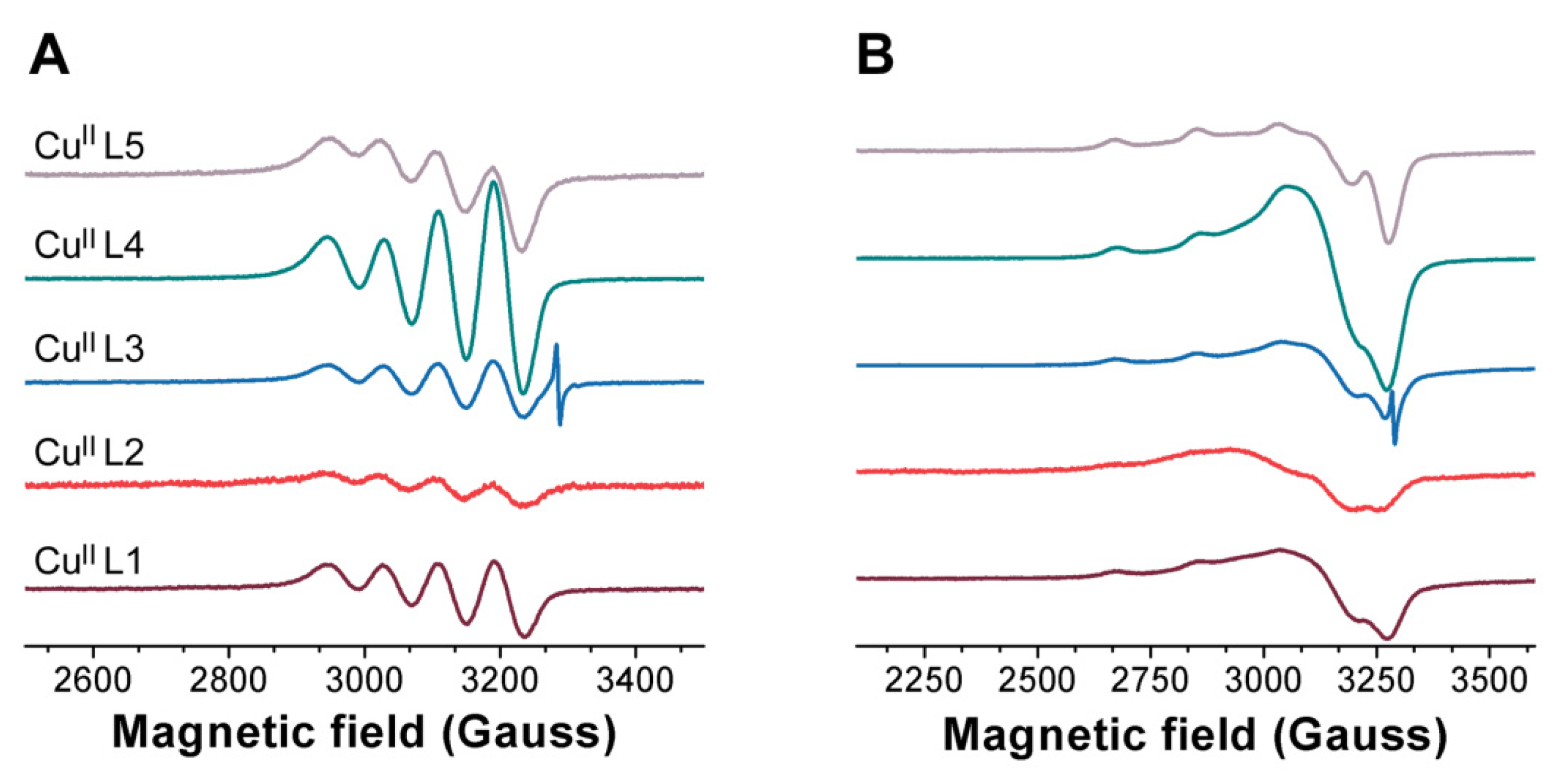
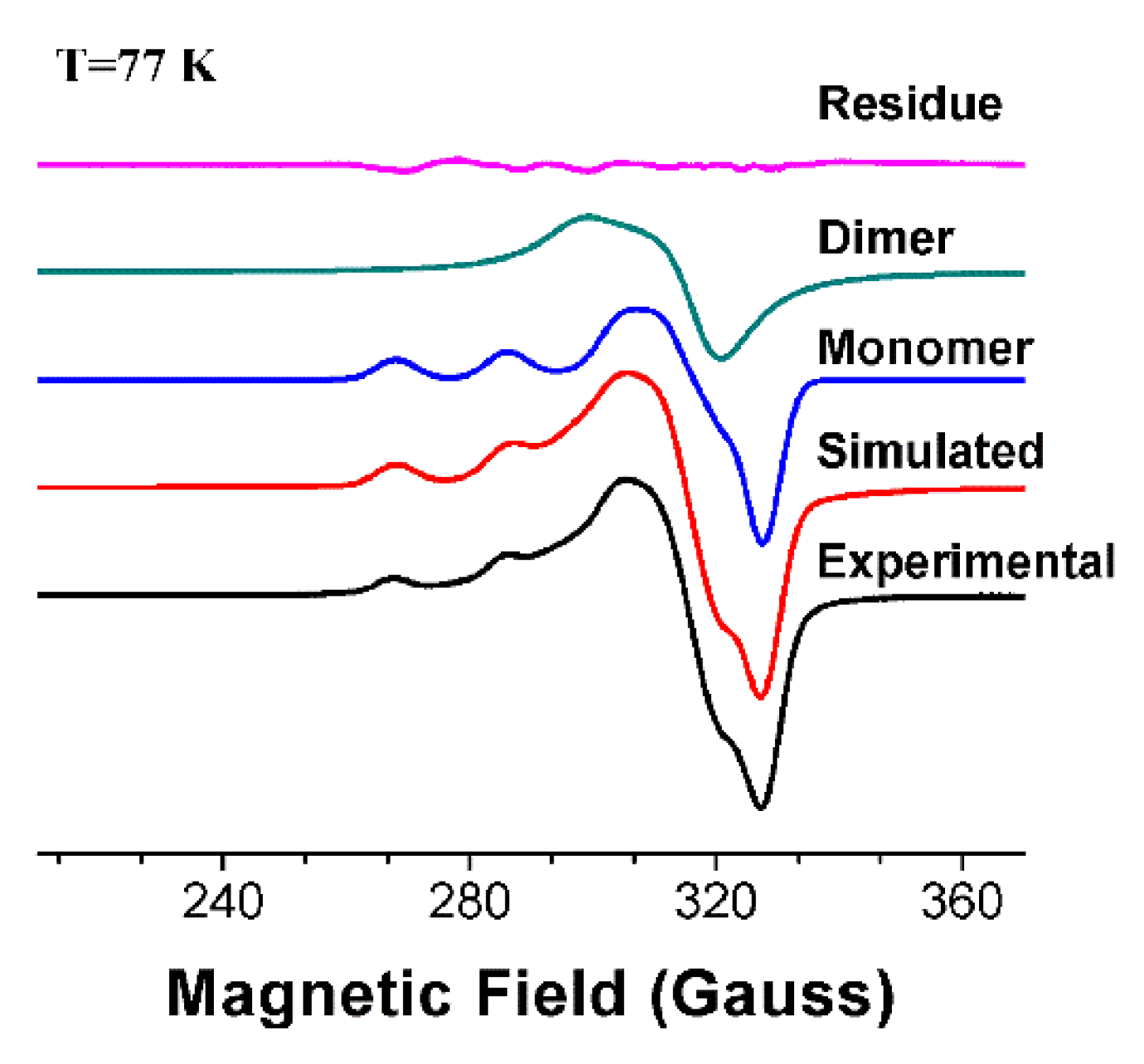
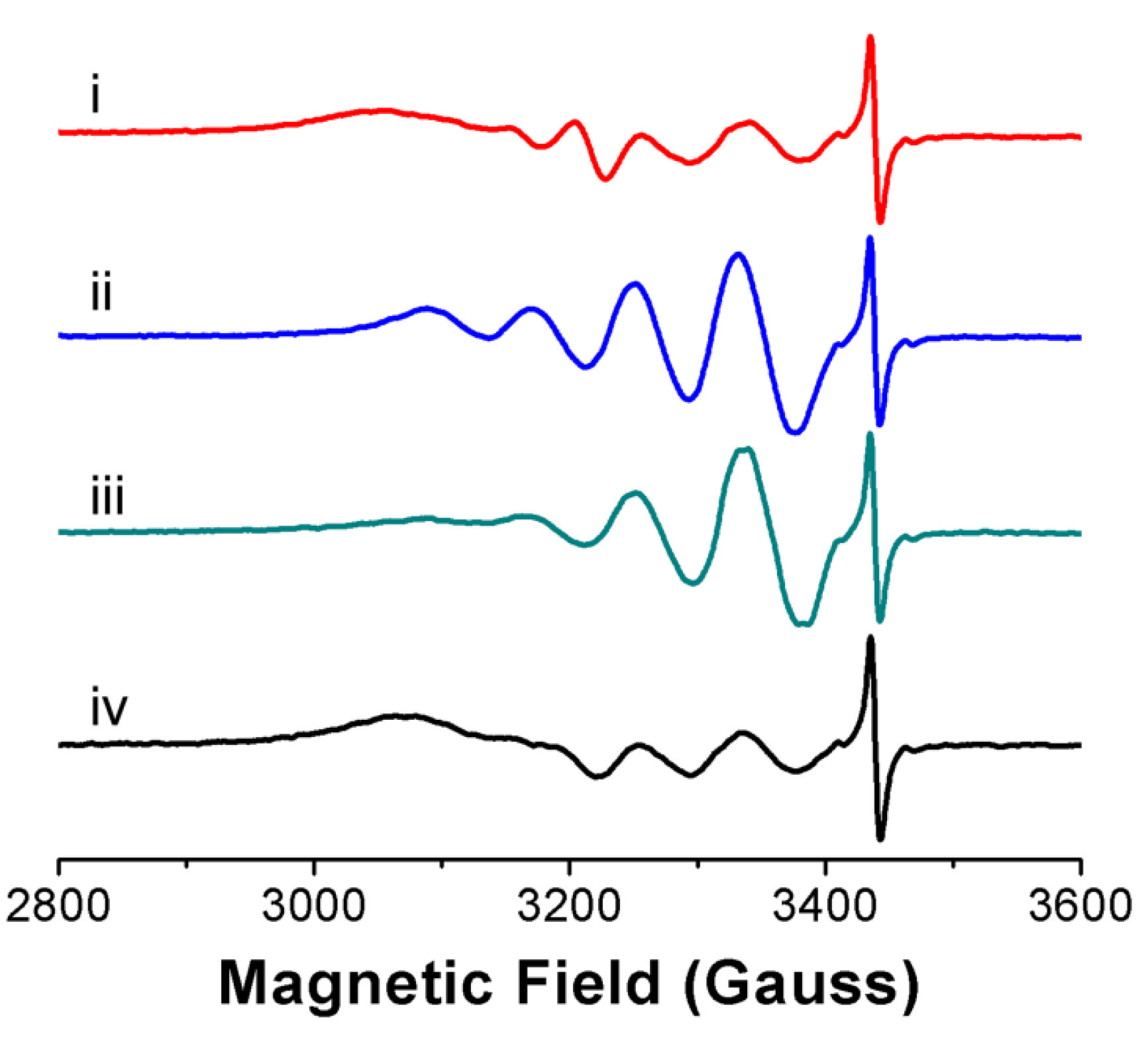
3.2. Ligand Substitution and Electron Paramagnetic Resonance Measurements
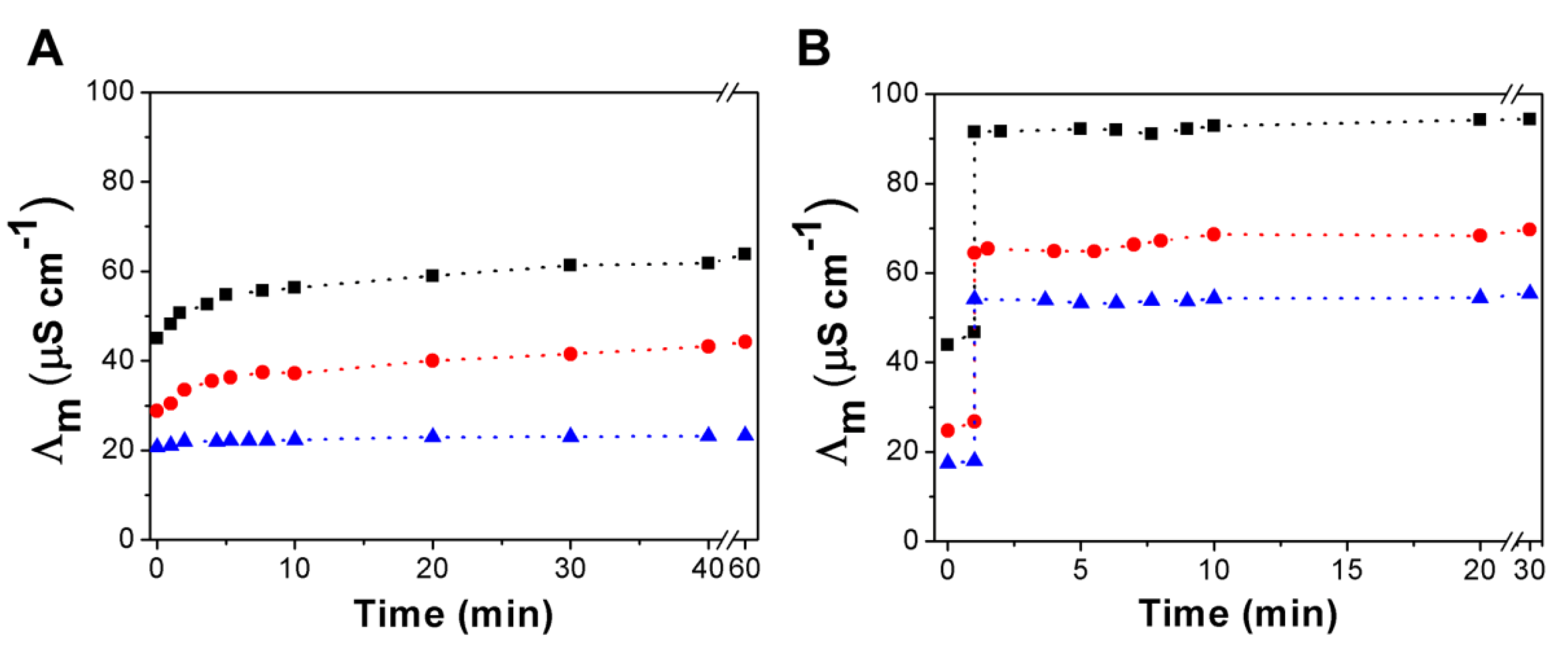
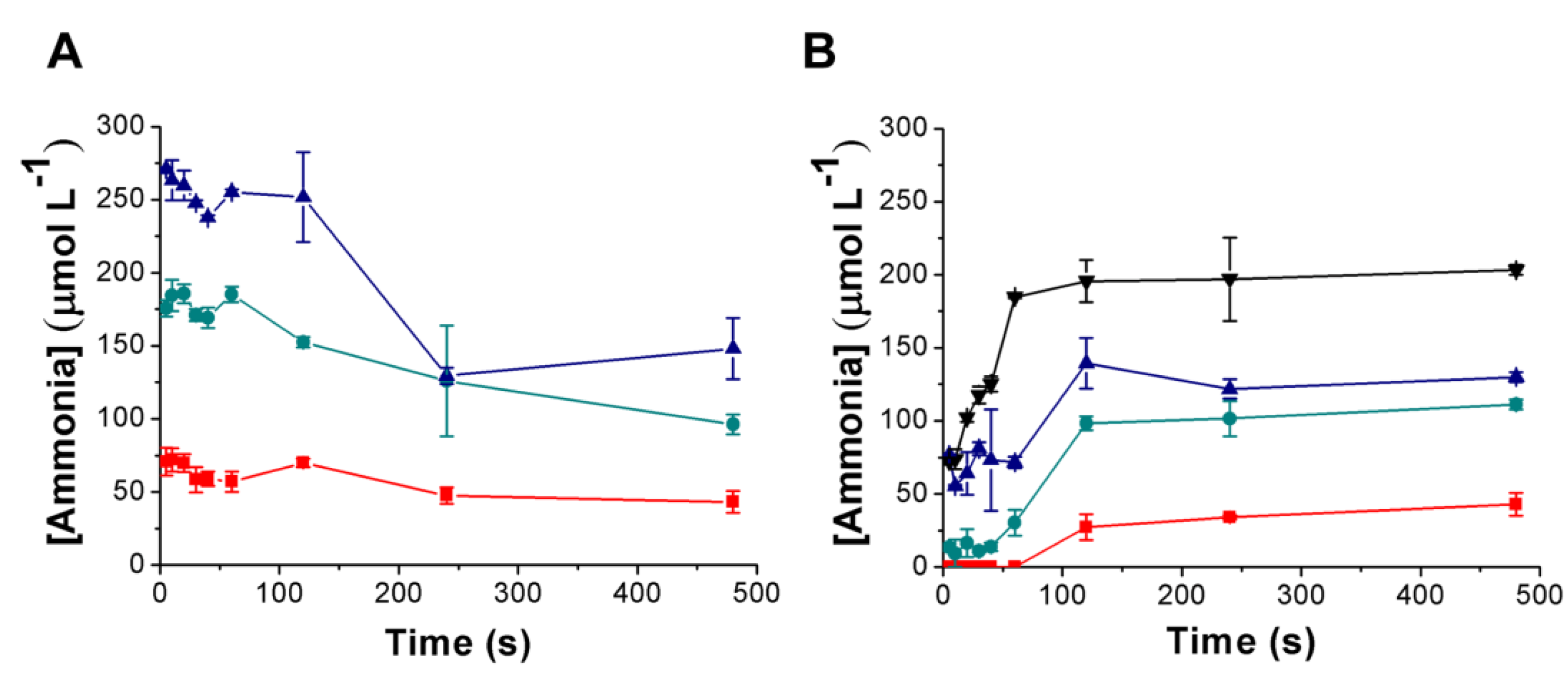
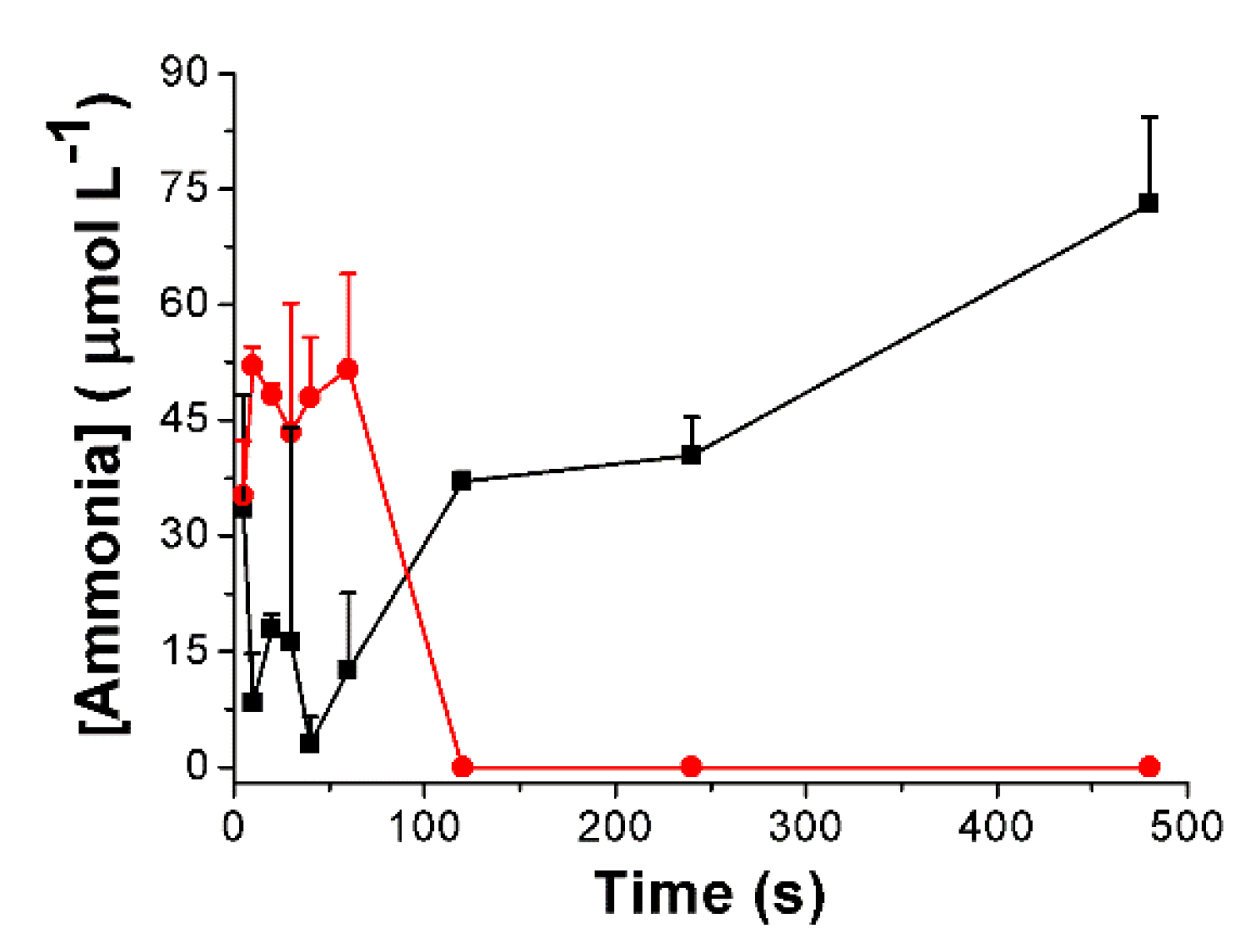
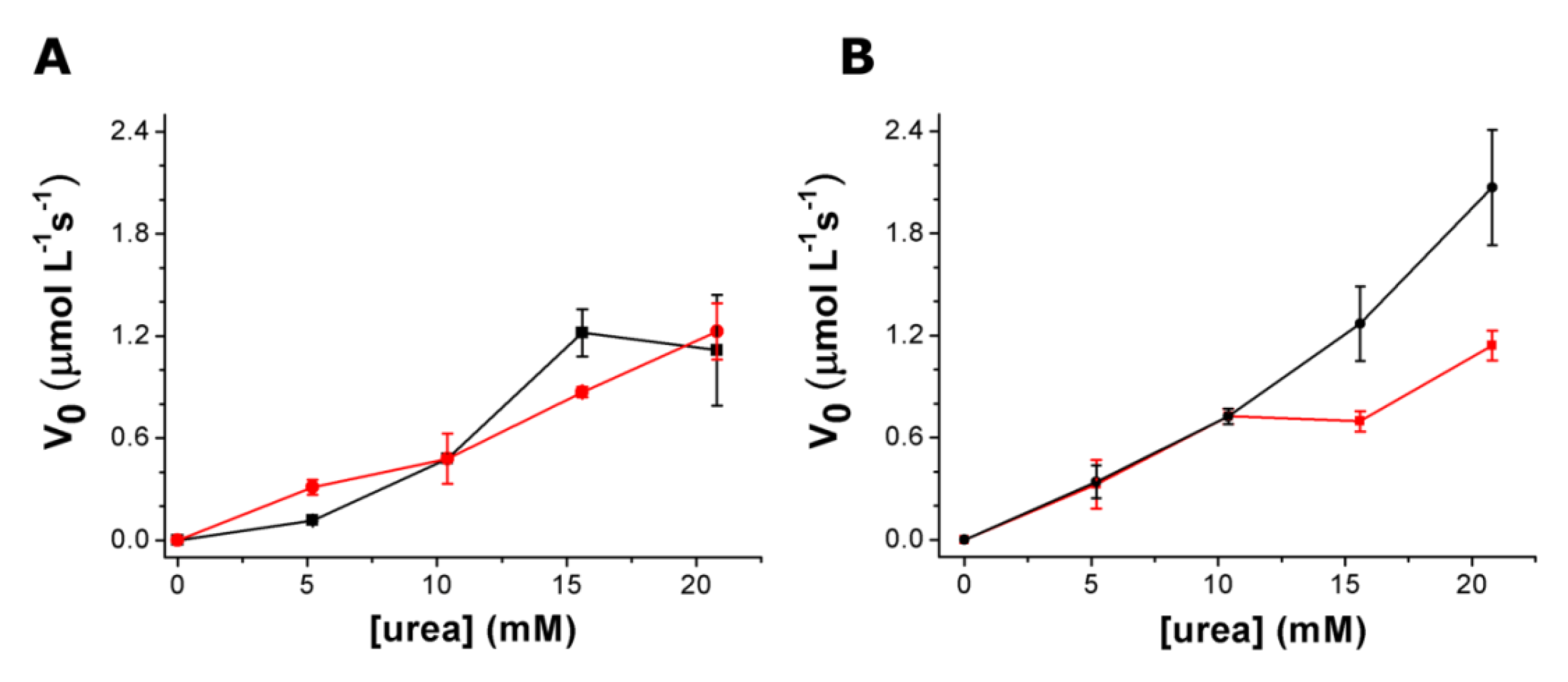
3.3. Urea Hydrolysis as a Model Reaction: Kinetics of NH3 Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abrusán, G.; Marsh, J.A.; Wilke, C. Ligand-Binding-Site Structure Shapes Allosteric Signal Transduction and the Evolution of Allostery in Protein Complexes. Mol. Biol. Evol. 2019, 36, 1711–1727. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Liu, J. Principles of Allosteric Interactions in Cell Signaling. J. Am. Chem. Soc. 2014, 136, 17692–17701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyonaka, S.; Kubota, R.; Michibata, Y.; Sakakura, M.; Takahashi, H.; Numata, T.; Inoue, R.; Yuzaki, M.; Hamachi, I. Allosteric Activation of Membrane-Bound Glutamate Receptors Using Coordination Chemistry within Living Cells. Nat. Chem. 2016, 8, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Takeya, M.; Osanai, T. Substrate Specificity and Allosteric Regulation of a D-Lactate Dehydrogenase from a Unicellular Cyanobacterium Are Altered by an Amino Acid Substitution. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarso, A.; Zaupa, G.; Houillon, F.B.; Prins, L.J.; Scrimin, P. Tripodal, Cooperative, and Allosteric Transphosphorylation Metallocatalysts. J. Org. Chem. 2007, 72, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Fritsky, I.O.; Ott, R.; Krämer, R. Allosteric Regulation of Artificial Phosphoesterase Activity by Metal Ions. Angew. Chem. Int. Ed. 2000, 39, 3255–3258. [Google Scholar] [CrossRef]
- Kovbasyuk, L.; Pritzkow, H.; Krämer, R.; Fritsky, I.O. On/off Regulation of Catalysis by Allosteric Control of Metal Complex Nuclearity. Chem. Commun. 2004, 4, 880–881. [Google Scholar] [CrossRef]
- Cheng, H.F.; D’Aquino, A.I.; Barroso-Flores, J.; Mirkin, C.A. A Redox-Switchable, Allosteric Coordination Complex. J. Am. Chem. Soc. 2018, 140, 14590–14594. [Google Scholar] [CrossRef]
- Kuwabara, J.; Yoon, H.J.; Mirkin, C.A.; Dipasquale, A.G.; Rheingold, A.L. Pseudo-Allosteric Regulation of the Anion Binding Affinity of a Macrocyclic Coordination Complex. Chem. Commun. 2009, 45, 4557–4559. [Google Scholar] [CrossRef]
- D’Aquino, A.I.; Cheng, H.F.; Barroso-Flores, J.; Kean, Z.S.; Mendez-Arroyo, J.; McGuirk, C.M.; Mirkin, C.A. An Allosterically Regulated, Four-State Macrocycle. Inorg. Chem. 2018, 57, 3568–3578. [Google Scholar] [CrossRef]
- McGuirk, C.M.; Mendez-Arroyo, J.; Lifschitz, A.M.; Mirkin, C.A. Allosteric Regulation of Supramolecular Oligomerization and Catalytic Activity via Coordination-Based Control of Competitive Hydrogen-Bonding Events. J. Am. Chem. Soc. 2014, 136, 16594–16601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiester, M.J.; Ulmann, P.A.; Mirkin, C.A. Enzyme Mimics Based upon Supramolecular Coordination Chemistry. Angew. Chem. Int. Ed. 2011, 50, 114–137. [Google Scholar] [CrossRef] [PubMed]
- Masar, M.S.; Gianneschi, N.C.; Oliveri, C.G.; Stern, C.L.; Nguyen, S.B.T.; Mirkin, C.A. Allosterically Regulated Supramolecular Catalysis of Acyl Transfer Reactions for Signal Amplification and Detection of Small Molecules. J. Am. Chem. Soc. 2007, 129, 10149–10158. [Google Scholar] [CrossRef] [PubMed]
- Gianneschi, N.C.; Nguyen, S.B.T.; Mirkin, C.A. Signal Amplification and Detection via a Supramolecular Allosteric Catalyst. J. Am. Chem. Soc. 2005, 127, 1644–1645. [Google Scholar] [CrossRef]
- Hyo, J.Y.; Mirkin, C.A. PCR-like Cascade Reactions in the Context of an Allosteric Enzyme Mimic. J. Am. Chem. Soc. 2008, 130, 11590–11591. [Google Scholar]
- Kennedy, R.D.; MacHan, C.W.; McGuirk, C.M.; Rosen, M.S.; Stern, C.L.; Sarjeant, A.A.; Mirkin, C.A. General Strategy for the Synthesis of Rigid Weak-Link Approach Platinum(II) Complexes: Tweezers, Triple-Layer Complexes, and Macrocycles. Inorg. Chem. 2013, 52, 5876–5888. [Google Scholar] [CrossRef]
- Jeon, Y.M.; Heo, J.; Brown, A.M.; Mirkin, C.A. Triple-Decker Complexes Formed via the Weak Link Approach. Organometallics 2006, 25, 2729–2732. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, J.; Stern, C.L.; Mirkin, C.A. A Coordination Chemistry Approach to a Multieffector Enzyme Mimic. J. Am. Chem. Soc. 2007, 129, 10074–10075. [Google Scholar] [CrossRef]
- Hyo, J.Y.; Heo, J.; Mirkin, C.A. Allosteric Regulation of Phosphate Diester Transesterification Based upon a Dinuclear Zinc Catalyst Assembled via the Weak-Link Approach. J. Am. Chem. Soc. 2007, 129, 14182–14183. [Google Scholar]
- Yoon, H.J.; Kuwabara, J.; Kim, J.H.; Mirkin, C.A. Allosteric Supramolecular Triple-Layer Catalysts. Science 2010, 330, 66–69. [Google Scholar] [CrossRef]
- Gianneschi, N.C.; Bertin, P.A.; Nguyen, S.B.T.; Mirkin, C.A.; Zakharov, L.N.; Rheingold, A.L. A Supramolecular Approach to an Allosteric Catalyst. J. Am. Chem. Soc. 2003, 125, 10508–10509. [Google Scholar] [CrossRef] [PubMed]
- Pankhurst, J.R.; Paul, S.; Zhu, Y.; Williams, C.K.; Love, J.B. PolynuclearAlkoxy-Zinc Complexes of Bowl-Shaped Macrocycles and Their Use in the Copolymerisation of Cyclohexene Oxide and CO2. Dalton Trans. 2019, 48, 4887–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, C.E.; Harding, L.P.; Hardie, M.; Riis-Johannessen, T.; Rice, C.R. Allosteric Effects in a Ditopic Ligand Containing Bipyridine and Tetra-Aza-Crown Donor Units. Chem. A Eur. J. 2012, 18, 3464–3467. [Google Scholar] [CrossRef] [PubMed]
- Shinkai, S.; Ikeda, M.; Sugasaki, A.; Takeuchi, M. Positive Allosteric Systems Designed on Dynamic Supramolecular Scaffolds: Toward Switching and Amplification of Guest Affinity and Selectivity. Acc. Chem. Res. 2001, 34, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Strotmeyer, K.P.; Fritsky, I.O.; Ott, R.; Pritzkow, H.; Krämer, R. Evaluating the Conformational Role of an Allosteric CuII Ion in Anion Recognition and Catalysis by a Tricopper Complex. Supramol. Chem. 2003, 15, 529–547. [Google Scholar] [CrossRef]
- Park, J.; Hong, S. Cooperative Bimetallic Catalysis in Asymmetric Transformations. Chem. Soc. Rev. 2012, 41, 6931–6943. [Google Scholar] [CrossRef]
- Park, J.; Lang, K.; Abboud, K.A.; Hong, S. Self-Assembly Approach toward Chiral Bimetallic Catalysts: Bis-Urea-Functionalized (Salen)Cobalt Complexes for the Hydrolytic Kinetic Resolution of Epoxides. Chem. A Eur. J. 2011, 17, 2236–2245. [Google Scholar] [CrossRef]
- Park, J.; Lang, K.; Abboud, K.A.; Hong, S. Self-Assembled Dinuclear Cobalt(II)-Salen Catalyst through Hydrogen-Bonding and Its Application to Enantioselective Nitro-Aldol (Henry) Reaction. J. Am. Chem. Soc. 2008, 130, 16484–16485. [Google Scholar] [CrossRef]
- Shirvan, A.; Golchoubian, H.; Bouwman, E. Syntheses and Chromotropic Behavior of Two Halo Bridged DinuclearCopper(II) Complexes Containing Pyridine-Based Bidentate Ligand. J. Mol. Struct. 2019, 1195, 769–777. [Google Scholar] [CrossRef]
- Albert, J.; Bosque, R.; D’Andrea, L.; Durán, J.A.; Granell, J.; Font-Bardia, M.; Calvet, T. Synthesis and Crystal Structure of the DinuclearCyclopalladated Compounds of Methyl (E)-4-(Benzylideneamino)Benzoate with Acetato and Chlorido Bridge Ligands: Study of Their Splitting Reactions with Pyridine. J. Organomet. Chem. 2016, 815–816, 44–52. [Google Scholar] [CrossRef]
- Sangeetha, N.R.; Pal, S. Dimeric and Polymeric Square-Pyramidal Copper(II) Complexes Containing Equatorial-Apical Chloride or Acetate Bridges. Polyhedron 2000, 19, 1593–1600. [Google Scholar] [CrossRef]
- Grosshauser, M.; Comba, P.; Kim, J.Y.; Ohto, K.; Thuéry, P.; Lee, Y.H.; Kim, Y.; Harrowfield, J. Ferro- and Antiferromagnetic Coupling in a Chlorido-Bridged, Tetranuclear Cu(II) Complex. Dalton Trans. 2014, 43, 5662–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, C.M.; Sun, X.X.; Gao, S.; Ma, S.L.; Yuan, D.Q.; Fan, C.H.; Huang, H.B.; Zhu, W.X. Chlorido-Bridged MnII Schiff-Base Complex with Ferromagnetic Exchange Interactions. Eur. J. Inorg. Chem. 2007, 2007, 3663–3668. [Google Scholar] [CrossRef]
- Luis Olivares-Romero, J.; Juaristi, E. Synthesis of Three Novel Chiral Diamines Derived from (S)-Proline and Their Evaluation as Precursors of Diazaborolidines for the Catalytic Borane-Mediated Enantioselective Reduction of Prochiral Ketones. Tetrahedron 2008, 64, 9992–9998. [Google Scholar] [CrossRef]
- Reyes-Rangel, G.; Vargas-Caporali, J.; Juaristi, E. In Search of Diamine Analogs of the α,α-Diphenyl Prolinol Privileged Chiral Organocatalyst. Synthesis of Diamine Derivatives of α,α-Diphenyl-(S)-Prolinol and Their Application as Organocatalysts in the Asymmetric Michael and Mannich Reactions. Tetrahedron 2016, 72, 379–391. [Google Scholar] [CrossRef]
- Sibi, M.P.; Zhang, R.; Manyem, S. A New Class of Modular Chiral Ligands with Fluxional Groups. J. Am. Chem. Soc. 2003, 125, 9306–9307. [Google Scholar] [CrossRef]
- Pracht, P.; Caldeweyher, E.; Ehlert, S.; Grimme, S. A Robust Non-Self-Consistent Tight-Binding Quantum Chemistry Method for Large Molecules. ChemRxiv 2019, 1–19. [Google Scholar]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-XTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All Spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- AtoholiSema, H.; Bez, G.; Karmakar, S. Asymmetric Henry Reaction Catalysed by L-Proline Derivatives in the Presence of Cu(OAc)2: Isolation and Characterization of an in Situ Formed Cu(II) Complex. Appl. Organomet. Chem. 2014, 28, 290–297. [Google Scholar] [CrossRef]
- Kalita, M.; Bhattacharjee, T.; Gogoi, P.; Barman, P.; Kalita, R.D.; Sarma, B.; Karmakar, S. Synthesis, Characterization, Crystal Structure and Bioactivities of a New Potential Tridentate (ONS) Schiff Base Ligand N-[2-(Benzylthio) Phenyl] Salicylaldimine and Its Ni(II), Cu(II) and Co(II) Complexes. Polyhedron 2013, 60, 47–53. [Google Scholar] [CrossRef]
- Balakrishnan, C.; Theetharappan, M.; Kowsalya, P.; Natarajan, S.; Neelakantan, M.A.; Mariappan, S.S. Biocatalysis, DNA–Protein Interactions, Cytotoxicity and Molecular Docking of Cu(II), Ni(II), Zn(II) and V(IV) Schiff Base Complexes. Appl. Organomet. Chem. 2017, 31, e3776. [Google Scholar] [CrossRef]
- Valentová, J.; Varényi, S.; Herich, P.; Baran, P.; Bilková, A.; Kožíšek, J.; Habala, L. Synthesis, Structures and Biological Activity of Copper(II) and Zinc(II) Schiff Base Complexes Derived from Aminocyclohexane-1-Carboxylic Acid. New Type of Geometrical Isomerism in Polynuclear Complexes. Inorg. Chim. Acta 2018, 480, 16–26. [Google Scholar] [CrossRef]
- Iftikhar, B.; Javed, K.; Khan, M.S.U.; Akhter, Z.; Mirza, B.; Mckee, V. Synthesis, Characterization and Biological Assay of Salicylaldehyde Schiff Base Cu(II) Complexes and Their Precursors. J. Mol. Struct. 2018, 1155, 337–348. [Google Scholar] [CrossRef]
- Rosu, T.; Pahontu, E.; Maxim, C.; Georgescu, R.; Stanica, N.; Gulea, A. Some New Cu(II) Complexes Containing an on Donor Schiff Base: Synthesis, Characterization and Antibacterial Activity. Polyhedron 2011, 30, 154–162. [Google Scholar] [CrossRef]
- Knoblauch, S.; Benedix, R.; Ecke, M.; Gelbrich, T.; Sieler, J.; Somoza, F.; Hennig, H. Synthesis, Crystal Structure, Spectroscopy, and Theoretical Investigations of Tetrahedrally Distorted Copper(II) Chelates with [CuN2S2] Coordination Sphere. Eur. J. Inorg. Chem. 1999, 1999, 1393–1403. [Google Scholar] [CrossRef]
- Rauf, A.; Shah, A.; Khan, A.A.; Shah, A.H.; Abbasi, R.; Qureshi, I.Z.; Ali, S. Synthesis, PH Dependent Photometric and Electrochemical Investigation, Redox Mechanism and Biological Applications of Novel Schiff Base and Its Metallic Derivatives. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 176, 155–167. [Google Scholar] [CrossRef]
- Butsch, K.; Günther, T.; Klein, A.; Stirnat, K.; Berkessel, A.; Neudörfl, J. Redox Chemistry of Copper Complexes with Various Salen Type Ligands. Inorg. Chim. Acta 2013, 394, 237–246. [Google Scholar] [CrossRef]
- Hosseinzadeh, P.; Lu, Y. Design and Fine-Tuning Redox Potentials of Metalloproteins Involved in Electron Transfer in Bioenergetics. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 557–581. [Google Scholar] [CrossRef]
- Banerjee, S.; Mazumdar, S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012, 2012, 1–40. [Google Scholar] [CrossRef] [Green Version]
- Geary, W.J. The Use of Conductivity Measurements in Organic Solvents for the Characterisation of Coordination Compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Fousiamol, M.M.; Sithambaresan, M.; Smolenski, V.A.; Jasinski, J.P.; Kurup, M.R.P. Halogen/Azide Bridged Box Dimer Copper(II) Complexes of 2-Benzoylpyridine-3-Methoxybenzhydrazone: Structural and Spectral Studies. Polyhedron 2018, 141, 60–68. [Google Scholar] [CrossRef]
- Kunnath, R.J.; Sithambaresan, M.; Aravindakshan, A.A.; Natarajan, A.; Kurup, M.R.P. The Ligating Versatility of Pseudohalides like Thiocyanate and Cyanate in Copper(II) Complexes of 2-Benzoylpyridine Semicarbazone: Monomer, Dimer and Polymer. Polyhedron 2016, 113, 73–80. [Google Scholar] [CrossRef]
- RecioDespaigne, A.A.; Da Costa, F.B.; Piro, O.E.; Castellano, E.E.; Louro, S.R.W.; Beraldo, H. Complexation of 2-Acetylpyridine- and 2-Benzoylpyridine-Derived Hydrazones to Copper(II) as an Effective Strategy for Antimicrobial Activity Improvement. Polyhedron 2012, 38, 285–290. [Google Scholar] [CrossRef]
- Hricovíni, M.; Mazúr, M.; Sîrbu, A.; Palamarciuc, O.; Arion, V.B.; Brezová, V. Copper(II) Thiosemicarbazone Complexes and Their Proligands upon Uva Irradiation: An Epr and Spectrophotometric Steady-State Study. Molecules 2018, 23, 721. [Google Scholar] [CrossRef] [Green Version]
- Hangan, A.C.; Borodi, G.; Stan, R.L.; Páll, E.; Cenariu, M.; Oprean, L.S.; Sevastre, B. Synthesis, Crystal Structure, DNA Cleavage and Antitumor Activity of Two Copper(II) Complexes with N-Sulfonamide Ligand. Inorg. Chim. Acta 2018, 482, 884–893. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Zablotny, S.; Bryce, D.L. Halogen Bonding as a Supramolecular Dynamics Catalyst. Nat. Commun. 2019, 10, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Selyutin, G.E.; Podrugina, T.A.; Gal’pern, M.G.; Luk, E.A. Aggregation Properties of Copper dibenzo-barrelenophthalocyanine. Chem. Heterocycl. Compd. 1995, 31, 146–149. [Google Scholar] [CrossRef]
- Moliner-Martínez, Y.; Herráez-Hernández, R.; Campíns-Falcó, P. Improved Detection Limit for Ammonium/Ammonia Achieved by Berthelot’s Reaction by Use of Solid-Phase Extraction Coupled to Diffuse Reflectance Spectroscopy. Anal. Chim. Acta 2005, 534, 327–334. [Google Scholar] [CrossRef]
- Stehle, S.; Braeuer, A.S. Hydrogen Bond Networks in Binary Mixtures of Water and Organic Solvents. J. Phys. Chem. B 2019, 123, 4425–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langford, C.H.; Tong, J.P.K. Preferential Solvation and the Role of Solvent in Kinetics. Examples from Ligand Substitution Reactions. Acc. Chem. Res. 1977, 10, 258–264. [Google Scholar] [CrossRef]
- Brown, R.S. Bio-Inspired Approaches to Accelerating Metal Ion-Promoted Reactions: Enzyme-like Rates for Metal Ion Mediated Phosphoryl and Acyl Transfer Processes. Pure Appl. Chem. 2015, 87, 601–614. [Google Scholar] [CrossRef]
- Blandamer, M.J.; Engberts, J.B.F.N.; Gleeson, P.T.; Reis, J.C.R. Activity of Water in Aqueous Systems; a Frequently Neglected Property. Chem. Soc. Rev. 2005, 34, 440–458. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Yuen, C.; Grant, D.J.W. Influence of Water Activity in Organic Solvent + Water Mixtures on the Nature of the Crystallizing Drug Phase. 1. Theophylline. Int. J. Pharm. 1996, 135, 151–160. [Google Scholar] [CrossRef]
- Subirats, X.; Rosés, M.; Bosch, E. On the Effect of Organic Solvent Composition on the PH of Buffered HPLC Mobile Phases and the PKa of Analytes—A Review. Sep. Purif. Rev. 2007, 36, 231–255. [Google Scholar] [CrossRef]
- Colombari, F.M.; Bernardino, K.; Gomes, W.R.; Lozada-Blanco, A.; Moura, A.F. Themis: A Software to Assess Association Free Energies Via Direct Estimative of Partition Functions. Unpublished work.
- Sun, M.; Xu, L.; Qu, A.; Zhao, P.; Hao, T.; Ma, W.; Hao, C.; Wen, X.; Colombari, F.M.; de Moura, A.F.; et al. Site-selective photoinduced cleavage and profiling of DNA by chiral semiconductor nanoparticles. Nature Chem. 2018, 10, 821–830. [Google Scholar] [CrossRef]
- Bernardino, K.; Lima, T.A.; Ribeiro, M.C.C. Low-Temperature Phase Transitions of the Ionic Liquid 1-Ethyl-3-methylimidazolium Dicyanamide. J. Phys. Chem. B 2019, 123, 9418–9427. [Google Scholar] [CrossRef]
- Bernardino, K.; Goloviznina, K.; Gomes, M.C.; Pádua, A.A.H.; Ribeiro, M.C.C. Ion pair free energy surface as a probe of ionic liquid structure. J. Chem. Phys. 2020, 152, 014103. [Google Scholar] [CrossRef]
- Colombari, F.M.; da Silva, M.A.R.; Homsi, M.; Souza, B.R.L.; Araujo, M.; Francisco, J.L.; Silva, G.T.S.T.; Silva, I.F.; de Moura, A.F.; Teixeira, I.F. Graphitic Carbon Nitrides asPlatforms for Single-Atom Photocatalysis. Faraday Discuss 2020. In Press. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | G | tcorr, ps | A, cm−1 (×10−4) | ||||
|---|---|---|---|---|---|---|---|
| gx | gy | gz | Ax | Ay | Az | ||
| CuIIL1 | 2.0501 | 2.0932 | 2.1626 | 71.9 | 14.53 | 11.51 | 200.2 |
| CuIIL2 | 2.0584 | 2.0932 | 2.1626 | 114.5 | 11.88 | 11.51 | 198.8 |
| CuIIL3 | 2.0515 | 2.0932 | 2.1626 | 75.3 | 11.88 | 11.51 | 201.0 |
| CuIIL4 | 2.0494 | 2.0518 | 2.2063 | 44.2 | 14.53 | 14.06 | 199.5 |
| CuIIL5 | 2.0504 | 2.0932 | 2.1626 | 116.7 | 11.88 | 11.51 | 188.6 |
| Compounds | a, Å | V, Å3 (×102) | ||||
|---|---|---|---|---|---|---|
| DCM | ACN | ACN/H2O | DCM | ACN | ACN/H2O | |
| CuIIL1 | 5.55 | 4.20 | 4.38 | 7.16 | 3.10 | 3.51 |
| CuIIL2 | 6.49 | 5.14 | 4.19 | 11.4 | 5.68 | 3.08 |
| CuIIL3 | 5.64 | 4.69 | 4.14 | 7.51 | 4.32 | 2.97 |
| CuIIL4 | 4.72 | 4.69 | 4.34 | 4.40 | 4.32 | 3.42 |
| CuIIL5 | 6.53 | 4.68 | 4.48 | 11.7 | 4.29 | 3.76 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, C.B.; Silveira, R.G.; Colombari, F.M.; de Moura, A.F.; Nascimento, O.R.; Marques Netto, C.G.C. Solvent Effect on the Regulation of Urea Hydrolysis Reactions by Copper Complexes. Chemistry 2020, 2, 525-544. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2020032
Castro CB, Silveira RG, Colombari FM, de Moura AF, Nascimento OR, Marques Netto CGC. Solvent Effect on the Regulation of Urea Hydrolysis Reactions by Copper Complexes. Chemistry. 2020; 2(2):525-544. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2020032
Chicago/Turabian StyleCastro, Caio B., Rafael G. Silveira, Felippe M. Colombari, André Farias de Moura, Otaciro R. Nascimento, and Caterina G. C. Marques Netto. 2020. "Solvent Effect on the Regulation of Urea Hydrolysis Reactions by Copper Complexes" Chemistry 2, no. 2: 525-544. https://0-doi-org.brum.beds.ac.uk/10.3390/chemistry2020032